Antimicrobial Drugs: Discovery, design & Resistance
1/86
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
87 Terms
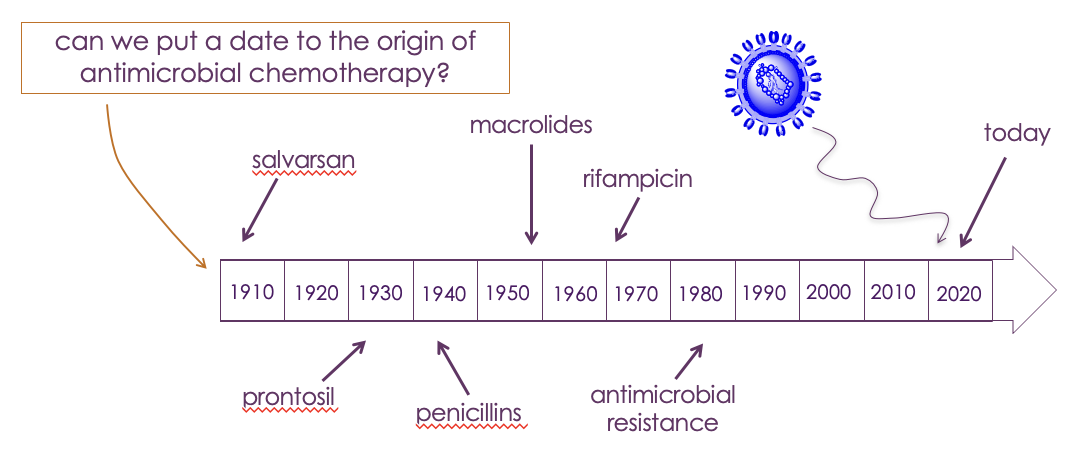
timeline of modern antimicrobial chemotherapy
what is the germ theory (discovery of infective diseases)
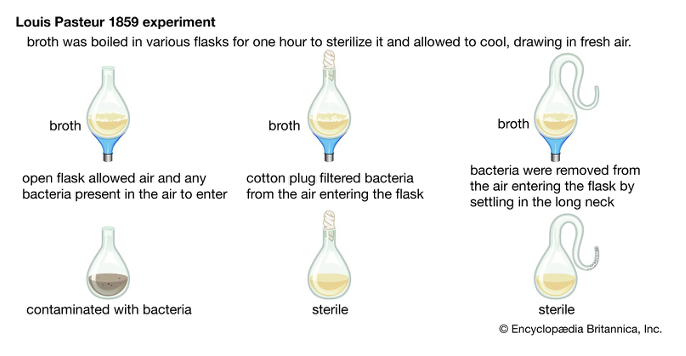
Louis Pasteur showed that germs cause the fermentation/decay of organic substances and proved that germs caused diseases
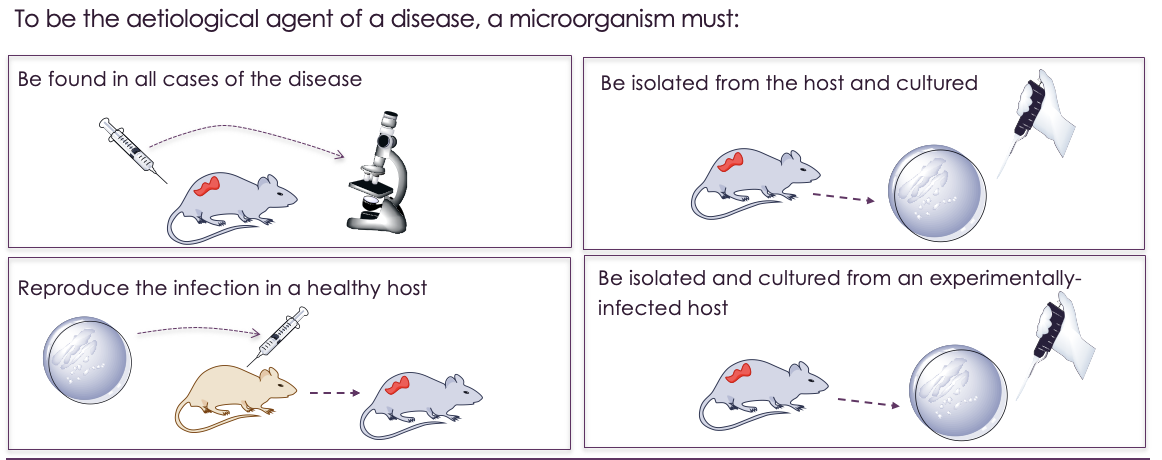
what is the Koch’s postuates
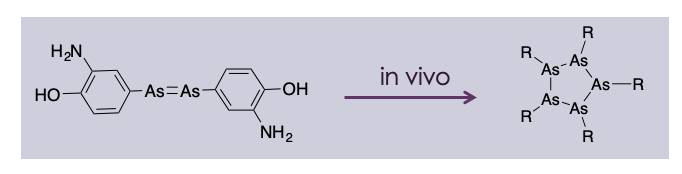
what was the first antimicrobial agent
Salvarsan was introduced against T. pallidum (syphillis) by Paul Ehrlich
Ehrlich introduced the concept of selective therapy using compounds that selectively target the disease, whilst having no effect on healthy tissues
how were sulfonamides developed
by Gerhardt Dogmagk
Prontosil is the first drug effective against systemic bacterial infection
Pontosil is a prodrug of sulfonamide, which must be activated by gut microflora
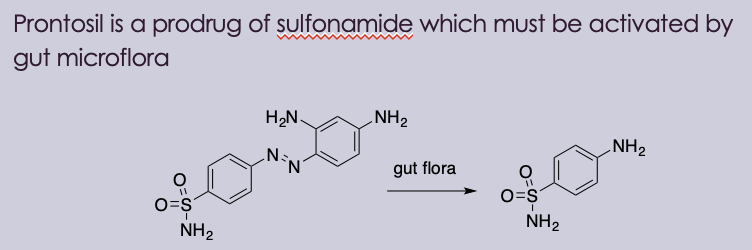
what was the first antibiotic discovered
penicillin
Alexander Fleming noticed that bacterial growth was inhibited around a mould contamination → identified penicillin G
6-APA became the building block for semisynthetic penicillins
what is an antibiotic
a chemical produced by microorganisms (natural products) able to affect the growth of other microorganisms
what is an antibacterial
a chemical that kill bacterial cells (bactericidal) or inhibit their growth (bacteriostatic)
what is an antimicrobial
General term for any chemotherapeutic agent active against microbial infection (bacteria, viruses, fungi…..)
what is anti-infective
General term for any chemotherapeutic agent active against an infective disease
how were antibiotics developed from systematic screening
Selman Waksman screened soil bacteria and fungi as a source of antibiotics.
This led to the discovery of streptomycin and the class of aminoglycosides
Streptomycin was the first agent active against tuberculosis
what is the golden age of antibiotic discovery
in the mid 40s to mid 60s, antibiotics were discovered by systematic screening → 7000 antibiotics discovered
the improvement era (till 80s) → New antibiotics mainly introduced via semi-synthesis (improvement of spectrum, ADME, etc)
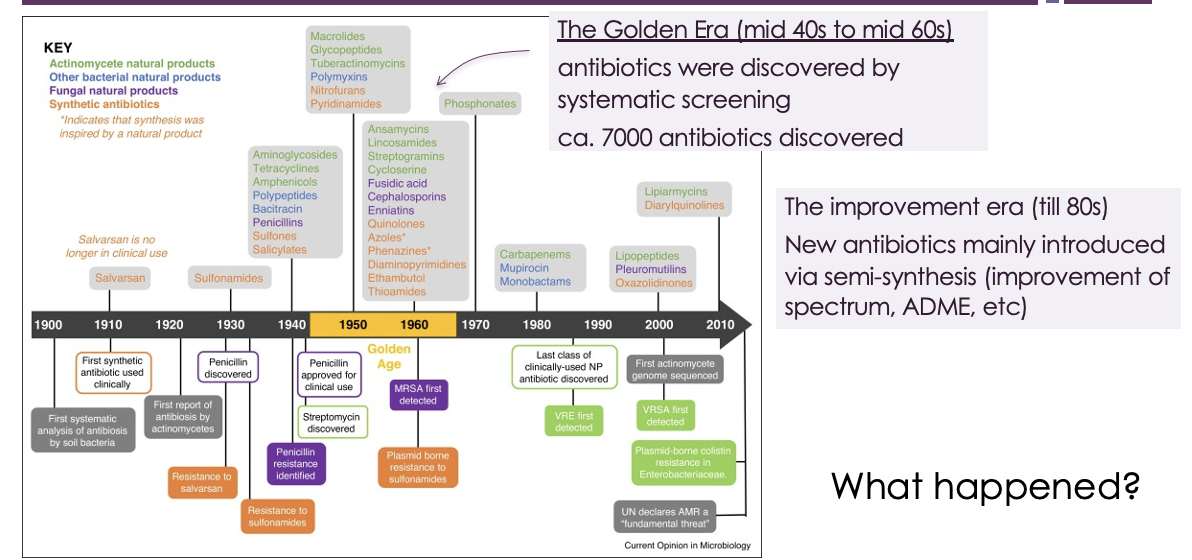
how is antimicrobial resistance an issue
Resistance to antimicrobial agents became a wide-scale issue.
Use of antimicrobials HAD to be limited
Minor improvements of current structures inadequate to circumvent the problem
It becomes progressively harder to identify new structures against a background of known but not useful species
Pharmaceutical companies stopped investing because of low revenues
how do we discover antibiotics nowadays
Genomics (and other –omics) to identify new targets
Target-based high-throughput screening (target validation or deconvolution)
Structure-based discovery
how are natural products and ethnopharmacology a prolific source of (potential) active principles

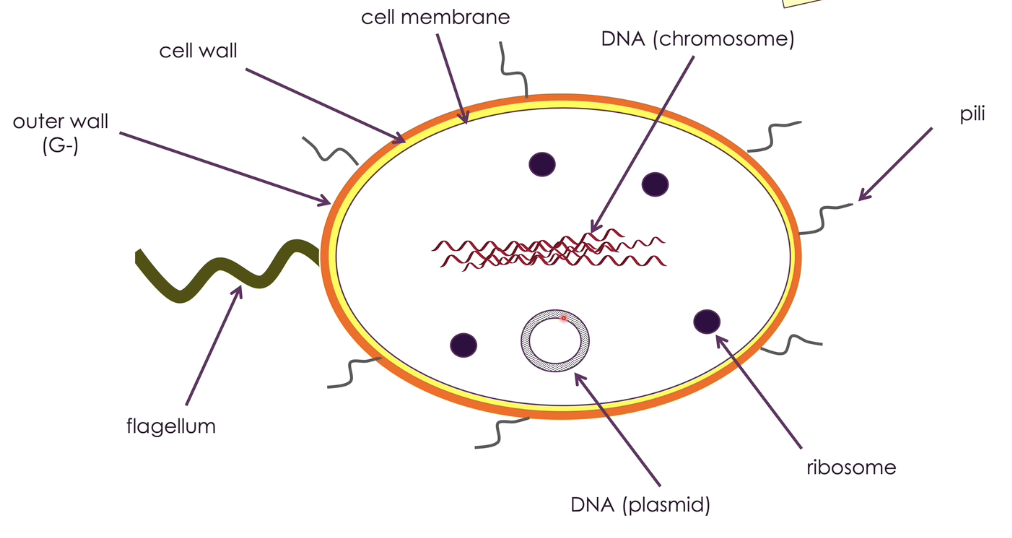
bacterial cell
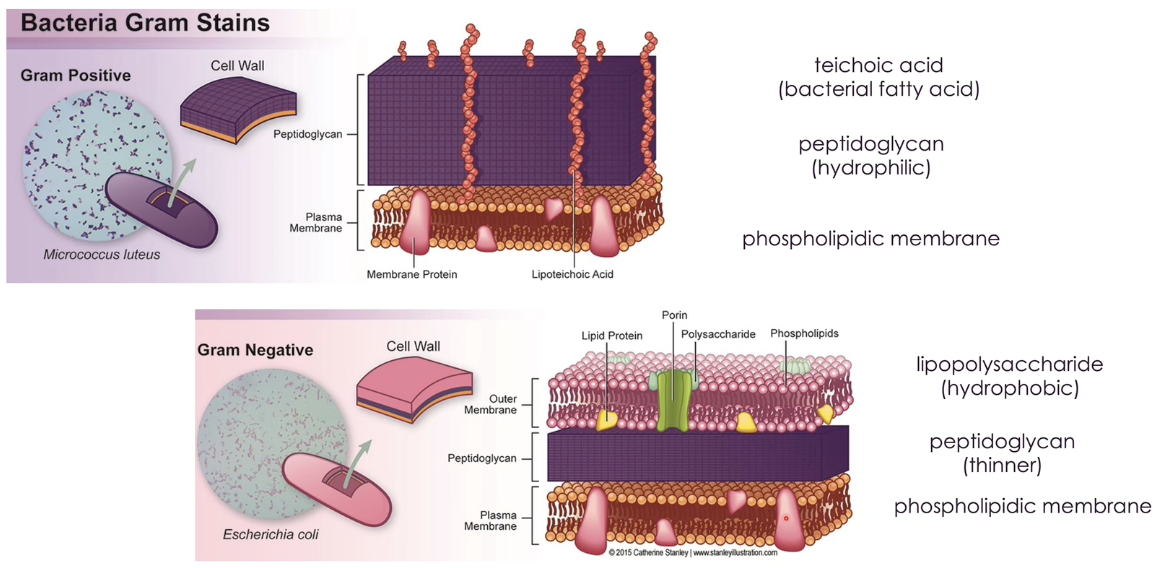
what is the gram staining method
application of purple dye
application of iodine mordant to help dye stay
wash with alcohol (some bacteria retain purple colour → gram positive)
application of safranin counterstain (bacteria that waswashed away stains pink → gram negative)
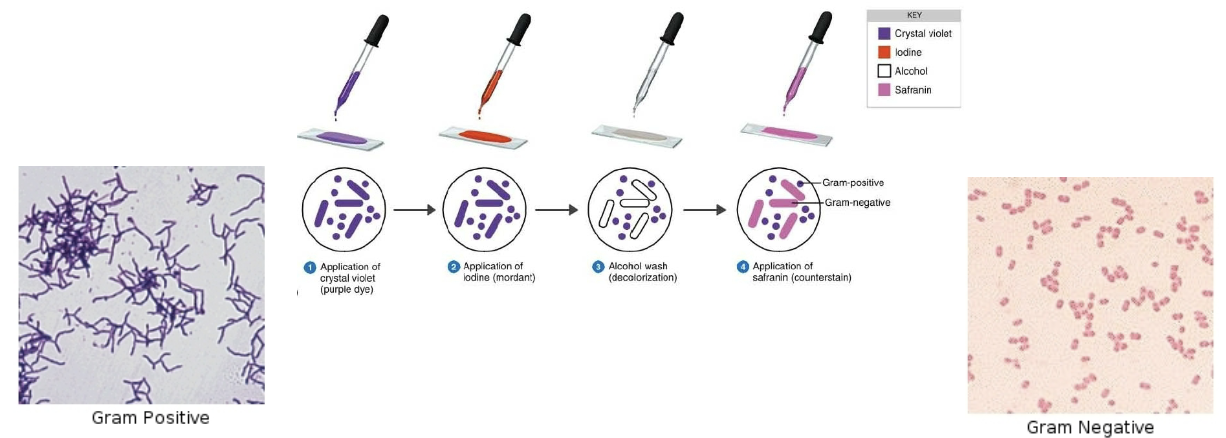
why do bacteria react differently to the gram-staining dyes
due to the chemistry of the bacteria cell wall
gram positive cell wall → has a plasma membrane and thick peptidoglycan layer outside of its membrane (purple)
gram negative cell wall → has a plasma membrane, a thin peptidoglycan layer outside of its membrane and an outer hydrophobic membrane (red/pink)
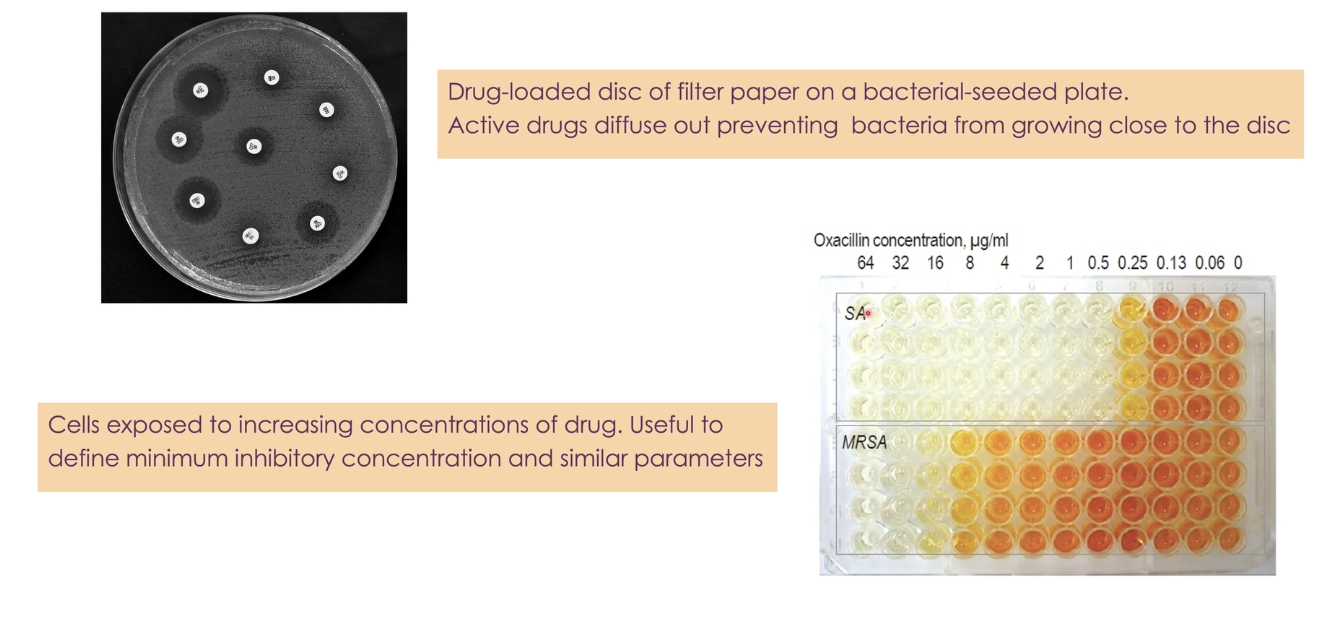
how are antibacterial agents tested
1. Disk Diffusion Test (Kirby–Bauer Test)
Each disc is soaked with an antibiotic.
Effective drug diffuses out → bacteria fails to grow near discs
2. Broth Microdilution Test (MIC Determination)
Bacterial growth is monitored across the gradient of drug concentrations.
Used to determine the Minimum Inhibitory Concentration (MIC) → the lowest concentration that prevents visible growth.
darker colour → higher number of bacteria
what are the three main approaches (targets) for antibacterial agents
interfere with cell wall synthesis or maintenance
interferes with nucleic acid synthesis
interferes with protein synthesis
what classes of antibiotics inhibit the synthesis of the cell wall
β-lactams
penicilllins
cephalosporins
carbapenems
monobactams
glycopeptides: vancomycin
lipopeptides: daptomycin
polypeptides
bacitracin
cholestin
how do antibiotics inhibit nucleic acid synthesis
inhibit DNA gyrase and/or topoisomerase IV e.g. quinolones
Inhibit folate synthesis e.g. sulfonamides and trimethoprim
create free radicals e.g. nitroimidazoles and nitrofurans
what antibiotics inhibit protein synthesis
on 50 sub unit
macrolides
clindamycin
linezolid
streptogramins
chloramphenicol
on 30s sub unit
aminoglycosides
tetracylines
tigecyline
what are the main agents that impair cell wall synthesis
β-lactams
Glycopeptides
Peptides
Cycloserin
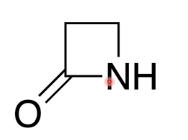
what are β-lactams (impair cell wall synthesis)
first antibiotic discovered
Wide class (antibiotic and semisynthetic products)
Generally bactericidal (kills)
includes penicillins, carbapenems, cephalosporins, monobactams
structure → four member ring
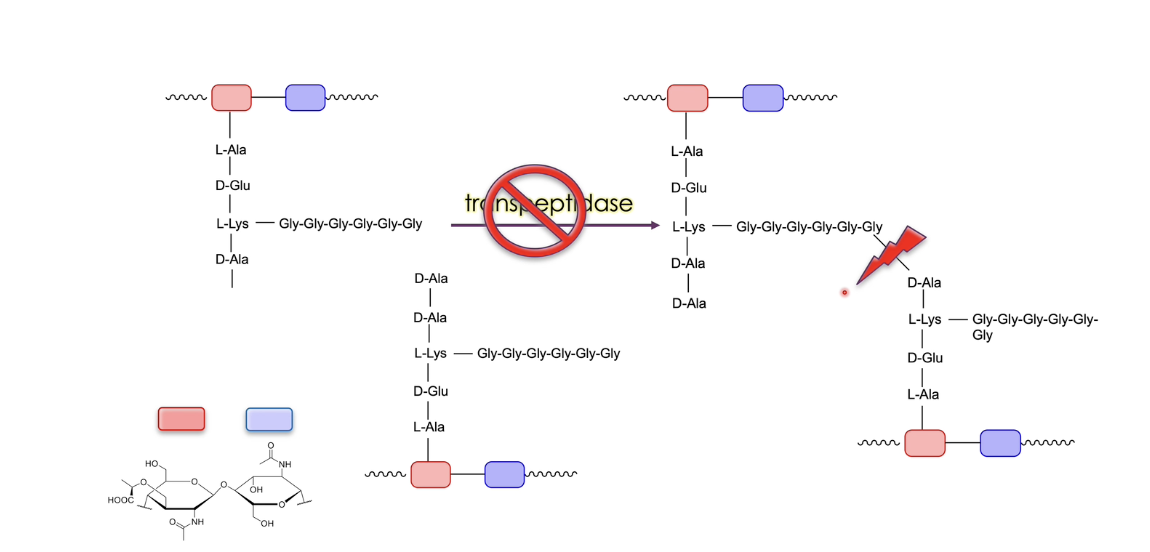
how is the peptidoglycan layer formed in bacterium
bacterium synthesises a dimer
red and purple squares represent sugars (N-acetylglucosamine and N-acetylmuramic acid), which form the base of the peptidoglycan layer
NAM has a short peptide chain, branches on one side with 5/6 glycines
enzymes polymerize repeating NAM–NAG–NAM–NAG units into long carbohydrate strands → proteoglycan layer is formed by layers of these species
the bacterium has an enzyme called transpeptidase to form cross links between two different chains → one amino acid from a layer is removed → formation of a bond between one layer and the next

what is the mechanism of action of β-lactams, carbepenems, cephalosporin, penicillin etc against the proteoglycan/peptidoglycan wall
inhibition of the last step of the biosynthesis of the proteoglycan (peptidoglycan) layer of the cell wall by inhibiting transpeptidase
this prevents formation of the cross-linking → cell wall is unstable and not functional → bacterium dies
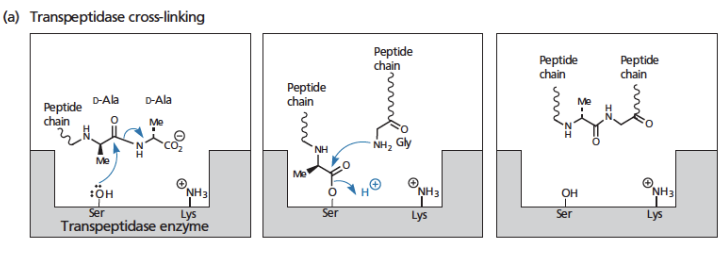
how does transpeptidase cross linking occur (mechanism of action)
transpeptidase has a serine amino acid and lysine with a positive charge in its active site
when peptide chain arrives, the OH on serine reacts with D-alanine residue on peptide → D-Alanine is released
peptide chain is attached temporarily to the active site of the enzyme, to serine
when another peptide chain arrives to make the cross link, its glycine amino group reacts with the carbonyl group on the peptide that is attached to serine
this promotes detachment of the original peptide chain species from serine
the enzyme’s serine is now restored to its free Ser–OH state → new peptide links form the final peptidoglycan cross-link between the two glycan strands.
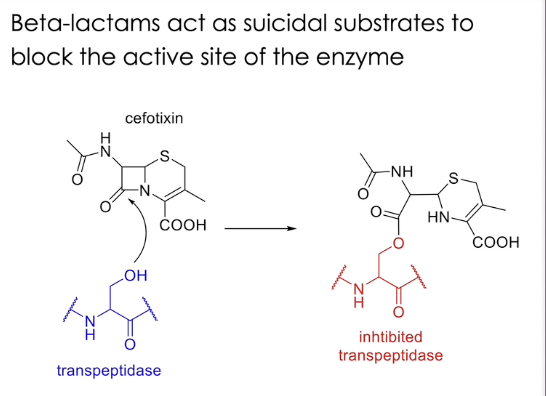
how does beta lactam inhibition occur
beta lactam has a free carboxylic acid, its pH is negatively charged due loss of proton
negative charge on beta lactam and positive charge of lysine on the enzyme
four ring structure of beta lactam opens up and is attacked by the OH of serine
incoming peptide chain can’t react anymore as enzyme is blocked
Driving force of the reaction is the ring strain of the lactam (4-member rings have strained bond angles = unstable)

what kind of inhibiton is beta lactam
suicide inhibition
beta lactams act as suicidal substrates to block the active site of the enzyme
what are penicillins
the first beta-lactams to be discovered
hard to isolate it as it is acid labile → easily destroyed in acidic environment
what are the different penicillins
penicillin G
6-amino penicillin
penicillin V
what is penicillin G
obtained from Penicillum sp.
ACID-LABILE → not stable in gastric environment → has to be administered parenterally, not orally

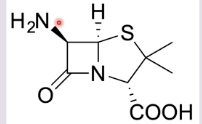
what is 6-amino penicillin
obtained by hydrolysis from penicillin G
starting material for semi-synthesis of penicillin as you are able to obtain a large amount of 6-amino penicillin and synthesise it
similar structure to penicillin G but chain ends at the NH group
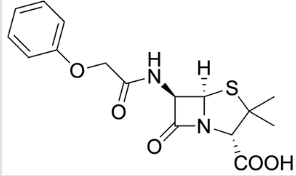
what is penicillin V
obtained from Penicillum sp. with phenoxyacetic acid in culture medium
similar structure to penicillin G, but has an extra O → increased stability means it is stable at gastric pH → can be taken orally
what happens when you have two fused rings for penicillin
for penicillin to work, you need two fused rings: beta lactam and thioazolidine (five member ring)
by changing the substituent on position 6, you can alter activity
if we want penicillin to be active against gram-negative bacteria, R group needs to be very hydrophilic
for gram positive, you need very hydrophobic substituents
electron-withdrawing groups increase acidic stability of the species → drug can be administered orally
bulky group increase activity of β-lactam against bacteria (β-lactamase resistance)
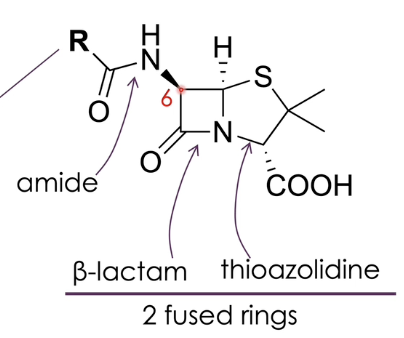
what is β-lactamase
an enzyme that bacteria make to break down and inactivate β-lactam
what are cephalosporins
• Cephalosporium acremonium identified as antibiotic-producing microorganism
• Cephalosporin C isolated and identified first
• Contain a β-lactam ring → same family as penicillin
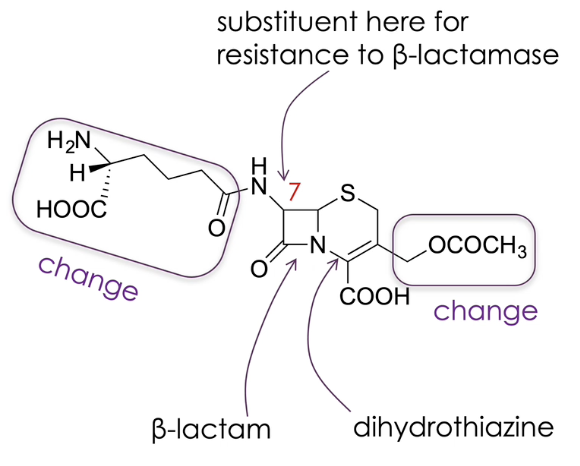
what is the structure of cephalosporins
similar to penicillin with two fused rings, containing a beta lactam however, other ring is a six member ring with a double bond called dihydrothiazine
substituents can be changed (as seen in diagram)
altering group on position 7 creates a drug that is less susceptible to beta lactamase
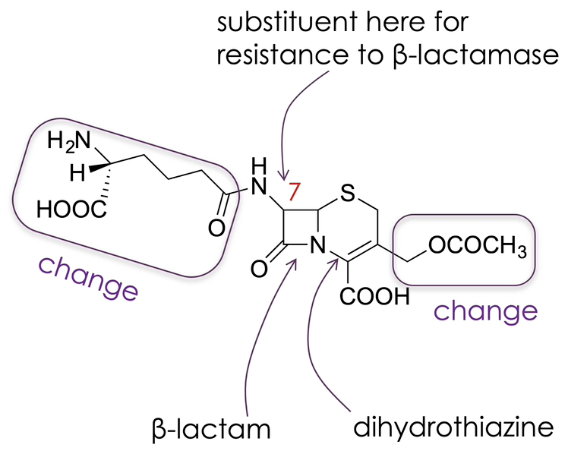
what are the different generations of cephalosporins
people have worked a lot on this drug
1st generation → 1000 less active than penicillin but broader spectrum of action: active against both both G+ and G-
2nd generation → methoxy group (OCH3) in position 7 causes resistance to β-lactamase. Broad spectrum
3rd generation → methoxy group (OCH3) in position 7 causes resistance to β-lactamase and change to left chain on diagram, more active against G-
4th generation → hydrophilic/heterocyclic substituent in 7 to increase penetration in G- cells
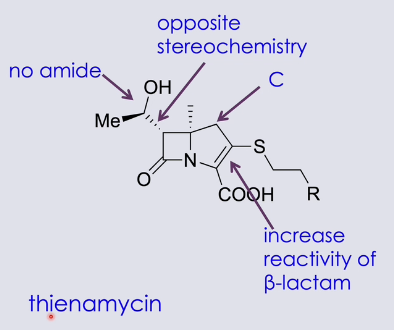
what are carbapenems
resemble cephalosporins and penicillin with the beta lactam ring, but five-member ring has a C and double bond
double bond makes beta lactam ring more reactive → more prone to reacting with active site of transpeptidase enzyme
no amide
very broad spectrum of action
active against P. aeruginosa
e.g.: thienamycin
what are monobactams
single beta lactam ring
negative charge given by the sulphonic group
the only class of beta lactams that doesn’t have a fused ring
narrow spectrum but active against P. aeruginosa
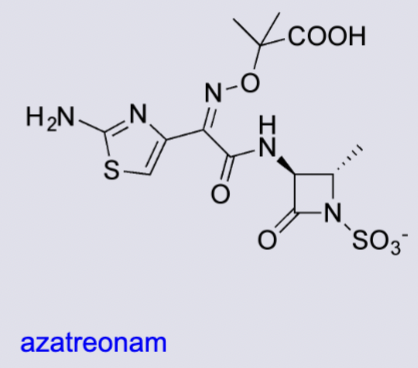
what are polypeptides and glycopeptides
examples are vancomycin, cycloserin, bacitracin
impair cell wall synthesis, but in a different way compared to beta lactams
what is vancomycin
• Glycopeptide isolated from S. orientalis
• Very effective against Staphylococcus aureus infections (gram positive)
• Replaced by methicillin, but back in use against MRSA → only used as LAST resort
what is bacitracin
• Polypeptide complex produced by Bacillus subtilis
• Dispensed as a powder (highly unstable in solution)
• Not absorbed in GI tract and hard to penetrate through tissues → Active principle in TOPICAL OTC preparations
• Wide spectrum
what is cylcoserin
Isolated from Streptomyces garyphalus
active site inhibitor → inhibits two key enzymes by mimicking their natural substrate (D-alanine)
Alanine racemase
D-Ala–D-Ala ligase
Second line treatment of tuberculosis
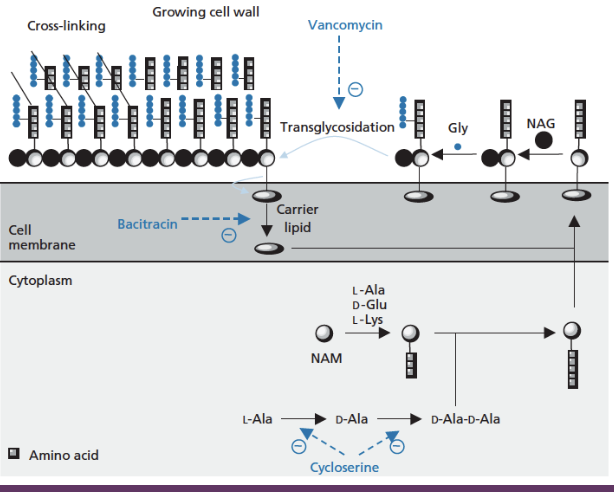
mechanism of action of vancomycin, cycloserine and bacitracin
vancomycin prevents monomer attaching to cell wall → prevents transglycosidation. works in the periplasmic space (space between cell membrane and cell wall), similar to beta lactams
bacitracin blocks recycling of carrier lipid, which helps assemble the membrane. has to get inside the cell wall
cycloserin prevents synthesis of double alanine
what agents impair protein synthesis
Aminoglycosides
Tetracyclines
Oxazolidinones
Macrolides
Chloramphenicol
ATOM-C
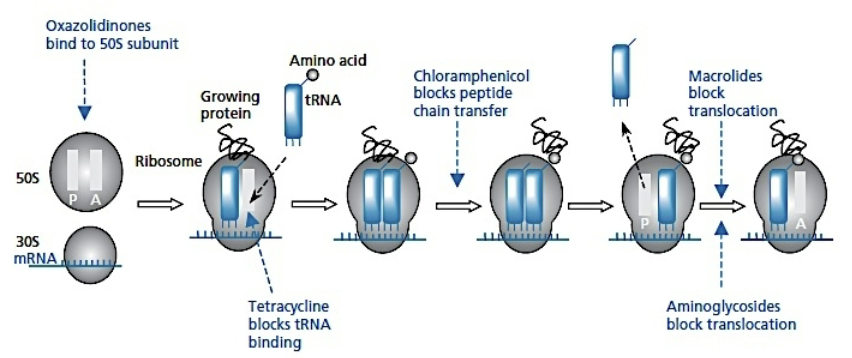
how is protein synthesis inhibited in bacteria
act on the ribosome
Oxazolidinones → bind to 50s subunit
tetracylcine → blocks tRNA binding
chloramphenicol → blocks peptide chain transfer
macrolides and aminoglycosides → block translocation
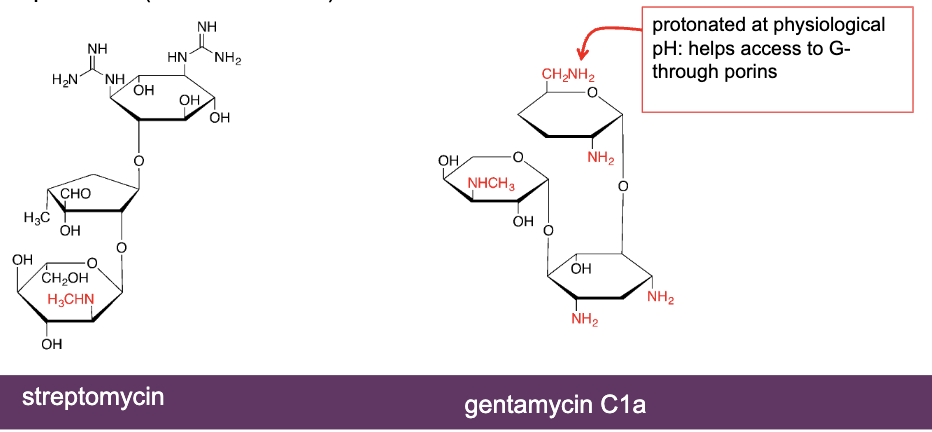
what are aminoglycosides (bactericidal)
Isolated from Streptomyces species during a systematic screening
Good activity against aerobic Gram- (systemic infections)
Poorly absorbed (< 1%), used to treat GI tract infections
Ototoxic (ear) and nephrotoxic (kidney) → limited their use
protonated (positively charged) at physiological pH → outer cell wall of gram-negative is hydrophobic, but porins are hydrophilic to allow them through
has many basic groups
examples: streptomycin and gentamycin C1a
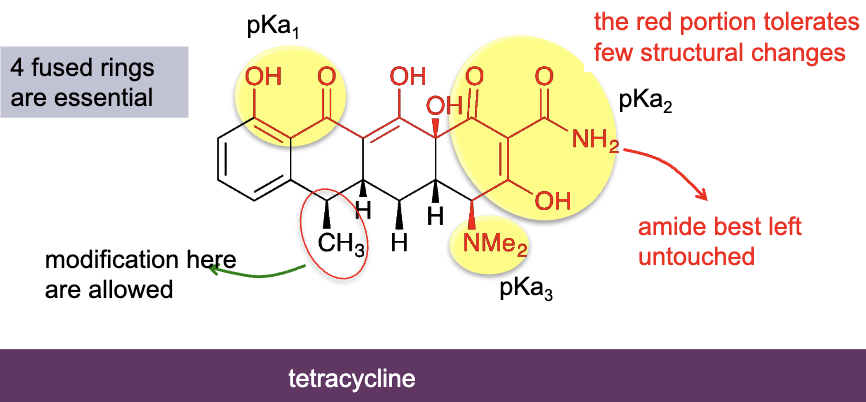
what are tetracyclines (bacteriostatic)
first one was Aureomycin isolated from Streptomyces aureofaciens during a systematic screening
Inhibit the attachment of aminoacyl-tRNA to 30s subunit of ribosome
Most prescribed class after penicillins
Very broad spectrum of action (G+, G-, mycoplasma…)
4 fused rings, can't remove/modify certain groups, some need a basic group due to PKA
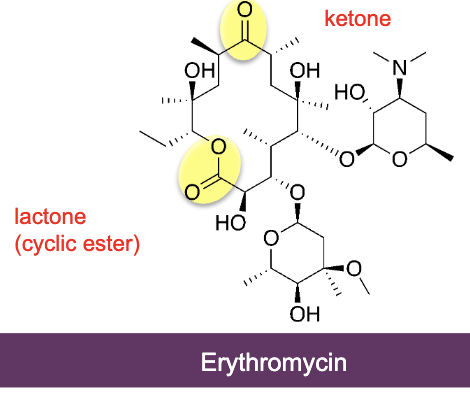
what are macrolides (bacteriostatic)
Erythromycin isolated from Streptomyces erythreus
Inhibit translocation by binding to 50s subunit of ribosome
Class has >40 compounds
broad spectrum of action resembles that of penicillin (G+ cocci and bacilli, G- cocci) but also mycoplasma. Not active on G- bacilli
many 6-member rings, macro rings, ketone and lactone function
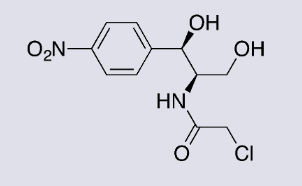
what is Chloramphenicol (bacteriostatic)
Originally isolated from S. venezuela now synthetic.
Binds to 50s subunit preventing the elongation of the peptide chain.
Potent and broad spectrum. Used in typhoid and for eye infections
Very toxic to the bone marrow
Toxic metabolism in babies (grey baby syndrome)
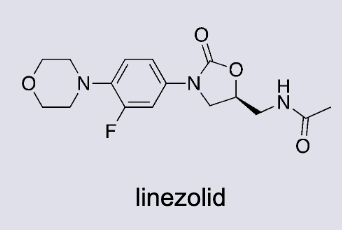
what are Oxazolidinones (bacteriostatic)
New class of synthetic antibacterial agents
Bind to the 50s subunit and prevent the ribosome from assembling (50s to 30s)
Active orally against G+ (including MRSA)
what agents damage or impair the synthesis of DNA
Sulfonamides
Sulfones
Trimethoprim
Rifamycins
Quinolones
Nitroimidazole
SSTRQN
how do agents inhibit nucleic acid synthesis
Either inhibit enzymes involved in DNA/RNA synthesis or cause direct damage to DNA
polymerase/gyrase/topoisomerase
direct DNA damage
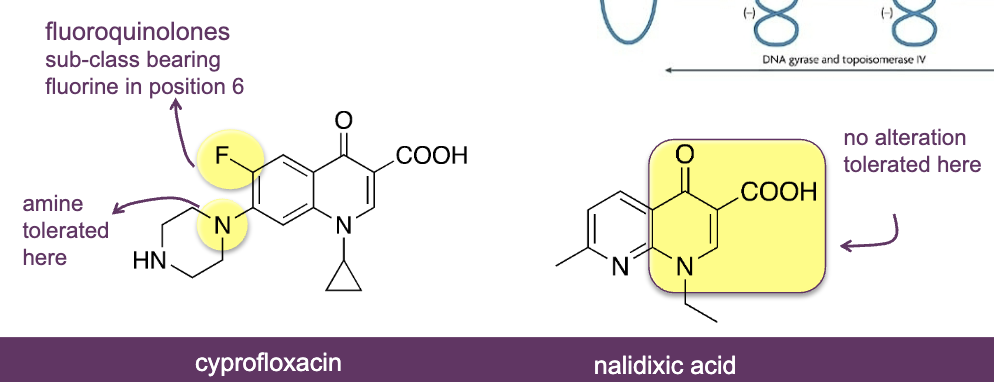
what are Quinolones (bactericidal)
Very wide class of synthetic antibacterial agents
MOA: bind to topoisomerase IV and gyrase, preventing supercoiling of bacterial DNA
Active against G+ and G-
Used in urinary tract infections
two fused ring, C=O group, COOH group, double bond in ring, N atom
examples: ciprofloxacin and nalidixic acid
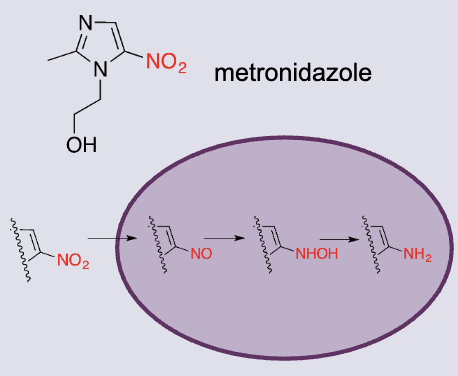
what are Nitroimidazoles (bactericidal)
Synthetic antibacterial agents (originally antiprotozoal for parasites)
causes direct damage to DNA
Nitro group essential for activity, can’t modify
nitro group undergoes reduction steps to form amino group → Form oxygen radical species toxic to DNA
what are rifamycins (bactericidal)
Isolated from S. mediterranei
Active on Gram positives
Binds to DNA-dependent RNA polymerases
Used against bacterial diarrhoea and E. coli infection
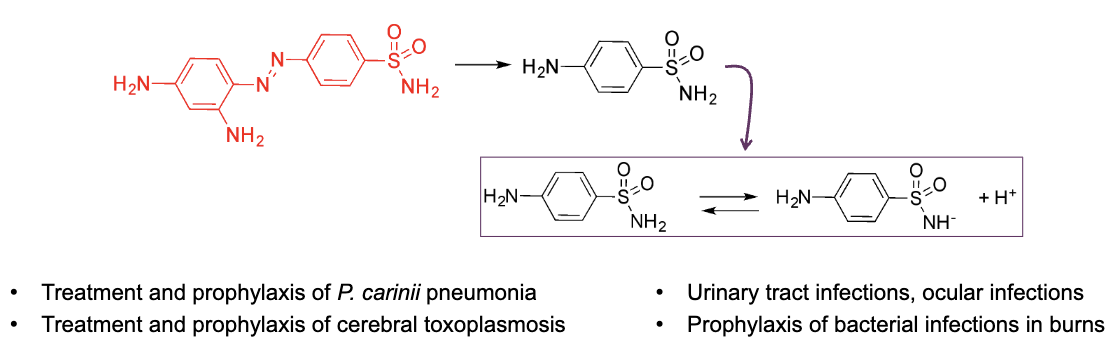
what are sulfonamides (bacteriostatic)
Class of antibacterials derived from Prontosil (red structure)
inhibit dna synthesis without interfering with the enzymes directly
First effective chemotherapeutics for systemic infections
Big reduction in mortality caused by bacterial infections
Superseded by penicillins as they are more active
prontosil when adminstered systemically releases sulfonamide
acidic group on right loses proton due to presence of electron attractive group
mechanism of action of sulfonamides
bacteria synthesise their own dihydropteorate, a prescursor of tetrahydrofolate (co-factor of the enzyme that allows antibiotic to pass into bacterial cells to produce DNA bases from RNA)
first start with species with two phosphate groups
dehydropteorate synthetase removes the phosphate groups and attaches para-aminobenzoic acid to make dihydropteorate
glutamic acid residue attached
reduction → double bond on second ring is lost to tetrahydrofolate, co enzyme F
sulfonamide interferes with removing phosphate groups and attaches para-aminobenzoic acid to make dihydropteorate → compete against para-aminobenzoic acid as they are structurally similar → need specific groups, but can modify
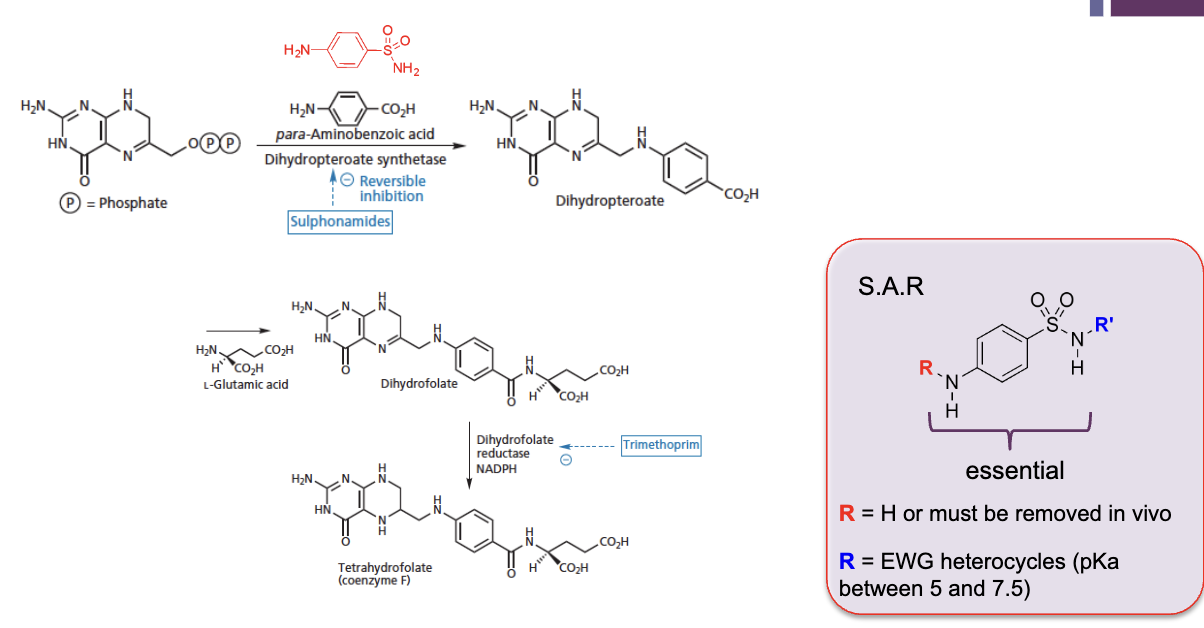
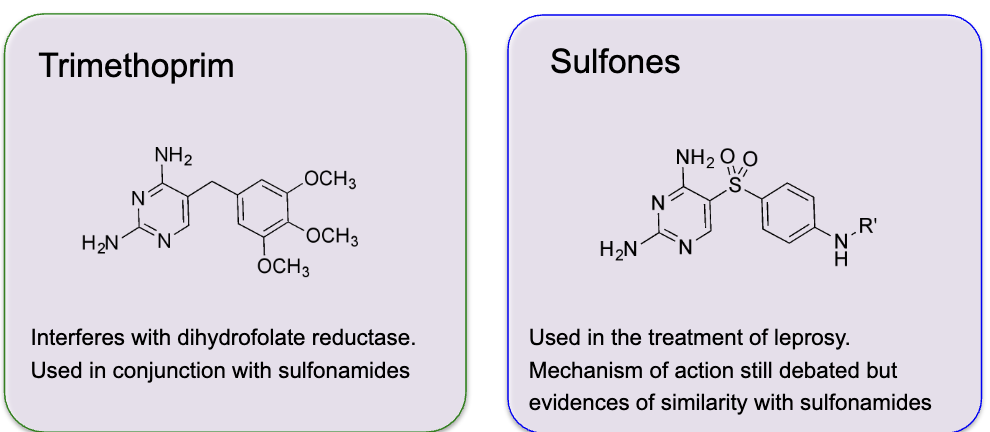
what are Trimethoprim and Sulfones (bacteriostatic)
Main antimicrobial agents that interfere with CoF synthesis → antimetabolites
trimethoprim → Interferes with dihydrofolate reductase. Used in conjunction with sulfonamides
sulfones → Used in the treatment of leprosy.Mechanism of action still debated but, evidences of similarity with sulfonamides
what is a virus
genetic material packed in proteins (reproduction occurs only via colonisation of a host cell)
hides from the immune system → harder to single out with therapeutic agent
there is a long lag time between time of infection and symptoms (e.g., HIV, rabies)
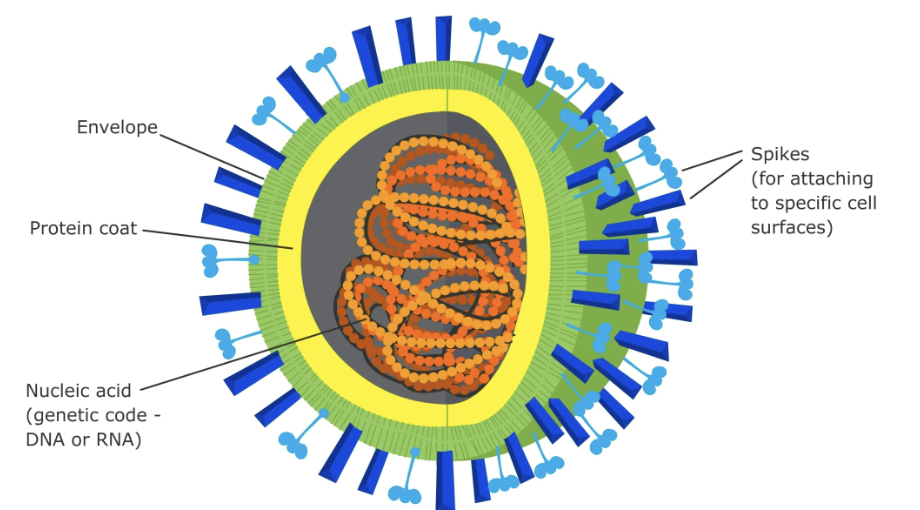
what is the structure of viruses
spikes are proteins
function of genetic material → propagates the virus and gives the target cell instructions to make more viruses dentinal to the entered virus
what happens when humans meet viruses
Some can pass from animals to humans (zoonoses)
Virus can be air-, food-, water-borne, by direct contact or through vectors (e.g., ticks)
Humankind has been challenged by several viral epidemics/pandemic
Best approach = vaccination (less effective for rapidly mutating viruses)
what are the characteristics of antiviral therapy
• disrupt stage of virus life cycle
• bear little resemblance to human proteins (+ selectivity)
• common to a variety of viruses (+ broad spectrum)
• important for early stages of viral life (- symptoms/spreading)
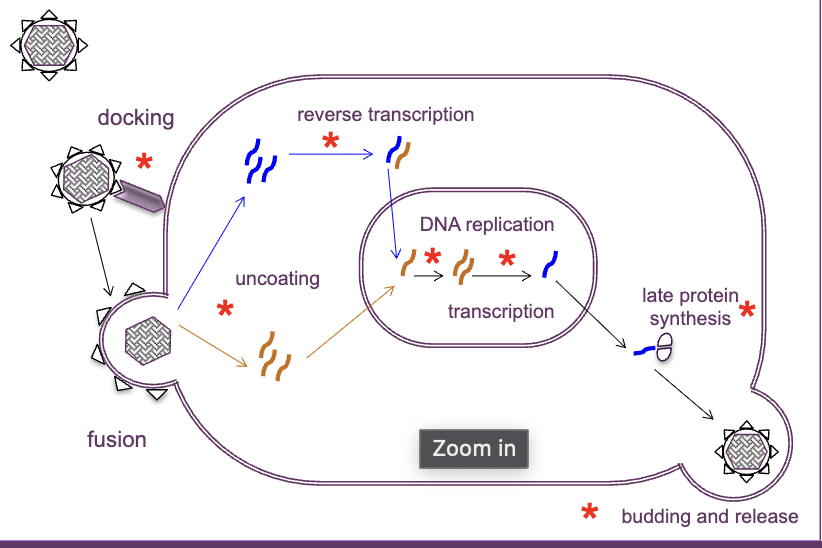
what is the cycle of viral infection
virus docks through cell wall structure with help of spike protein
fuse with membrane with help of spike protein
virus uncoats and releases genetic material into nucelus, if DNA reverse transcription occurs to become DNA
DNA replication occurs → transcription to make viral proteins
protein synthesis → virus ressembled with the copied DNA, spikes etc
budding and released to infect new cells
antiviral agents can block any of these steps in the cycle
what are the classes of antiviral agents
Inhibitors of DNA polymerase
Inhibitor of reverse transcriptase (RNA into DNA)
Nucleoside reverse transcriptase inhibitors
Non-nucleoside reverse transcriptase inhibitors
Protease inhibitors
Aspartyl protease inhibitors (HIV)
Neuraminidase inhibitors (influenza)
NS3-4A protease inhibitors (hepatitis C)
Interferon
Uncoating/fusion inhibitors
what are inhibitors of DNA polymerase
Nucleoside-like structures which are phosphorylated, incorporated in growing DNA chain to lead to chain interruption
MOA; they resemble the nuceloside structure → phosphorylated with a kinase enzyme(three phospahte groups) → becomes part of DNA chain → their structure prevents the chain from growing
example: aciclovir, red bit is not included in the drug, but is in deoxyguanosine
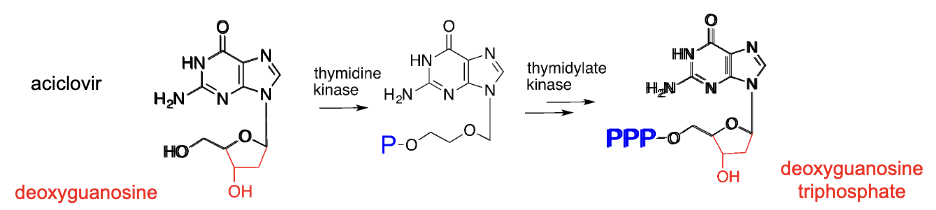
how is aciclovir modified
has poor oral bioavailabilty → given topically
valociclivir has improved oral bioavailabiltiy
more hydrophobic → faster onset and longer duration

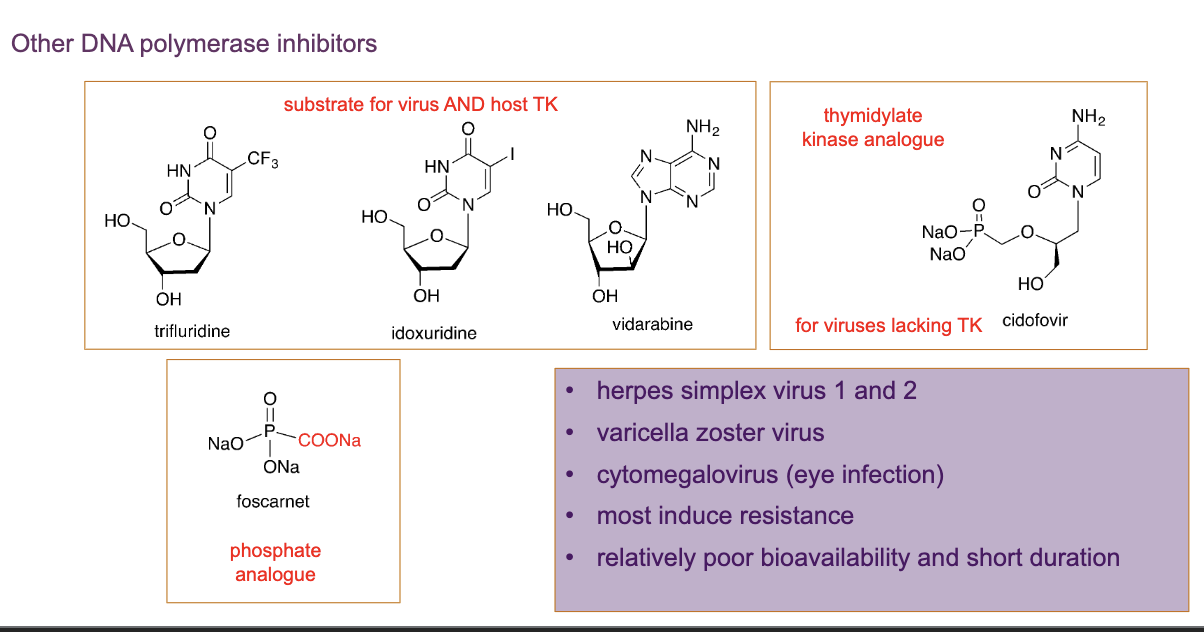
examples of other DNA polymerase inhibitors
contain the OH group, unlike aciclovir
they have the OH group that is needed for growing the chain
however, the structures alter pairing of complementary bases → more prone to damage and errors
they are substrates for the virus and host thymidylate/thymidine kinase → high possibility of toxicity to host enzyme → the DNA that is synthesised by the host gets affected
some viruses don’t produce thymidine kinase → use an analogue of the next step, thymidylate kinase analogue
foscarnet → phosphate analogue that prevents the attachment of phosphates on thre molecule. however it can’t distinguish between the viral enzyme and host enzyme
what are inhibitors of reverse transcriptase
inhibit reverse transcriptase from translating RNA into DNA, includes
Nucleoside reverse transcriptase inhibitors
Non-nucleoside reverse transcriptase inhibitors
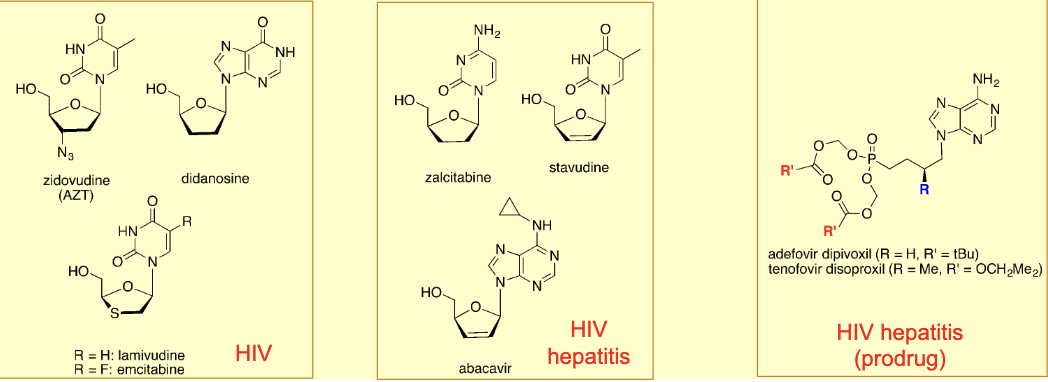
what are Nucleoside reverse transcriptase inhibitors (NRTI)
Nucleoside structures: phosphorylated, incorporated in growing DNA chain (chain interruption), same as DNA polymerase inhibitors
PROBLEM: in some cases (HIV) the virus does not produce TK → Host enzymes is used to activate NRTI instead→ less selectivity as all cells are affected
caution: reverse transcriptase is still a DNA polymerase, which humans have → Selective affinity for RT vs. DP of the host is vital
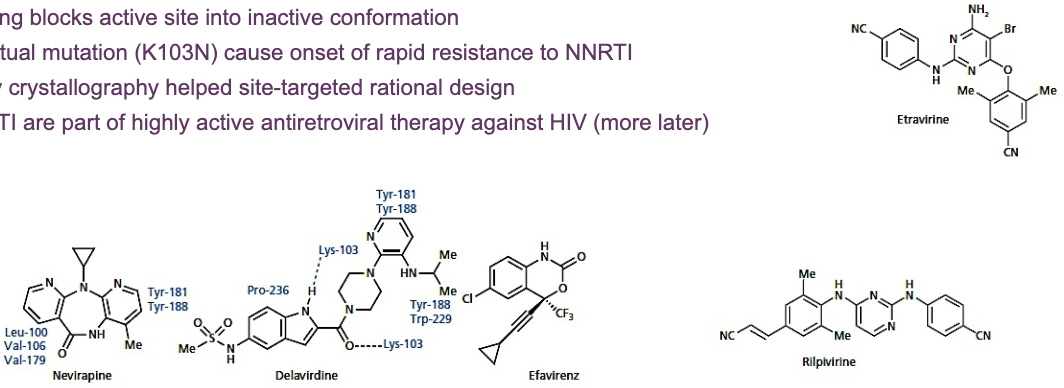
what are Non-nucleoside reverse transcriptase inhibitors (NNRTI)
Hydrophobic molecules that bind to allosteric binding in reverse transcriptase, a site other than the active site
Binding blocks active site into inactive conformation
Punctual mutation (K103N) cause onset of rapid resistance to NNRTI
X-ray crystallography helped site-targeted rational design
NNRTI are part of highly active antiretroviral therapy against HIV
what are protease inhibitors
Series of derivatives active against viral proteases, including
Aspartyl protease inhibitors (HIV)
Neuraminidase inhibitors (influenza)
NS3-4A protease inhibitors (hepatitis C)
virus produces protease to help enter the cell
what are inhibitors of HIV protease
No need to be metabolised (unlike NRTI), hence easier to evaluate activity in vitro
Target enzyme is aspartyl protease, which cleaves viral pro-enzymes to release mature, active viral enzymes
Designed based on the structure of the substrate (peptides) to block protease so proteins don’t get cut
however, Instability and poor bioavailability (typical of peptide drugs)
Rapid onset of resistance to these drugs as virus easily mutates
example: saquinavir (Roche)
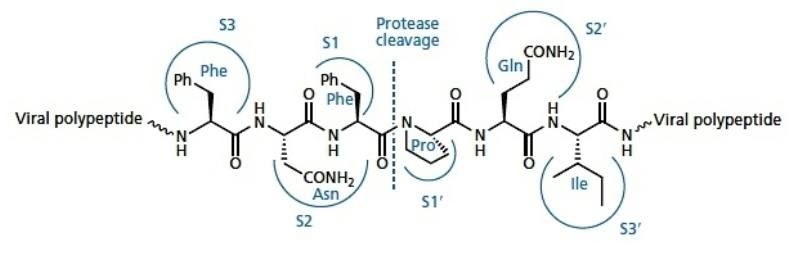
how were HIV protease inhibitors designed to look similar to the natural substrate for the enzyme aspartyl protease, but different enough to then prevent enzyme from working
Major medicinal chemistry effort aimed at
1) Improving affinity for enzyme (resemble the substrate transition state)
2) Improving stability to proteases (eliminate peptide bonds)
3) Improving oral bioavailability
4) Reduce molecular weight
5) Avoid resistance
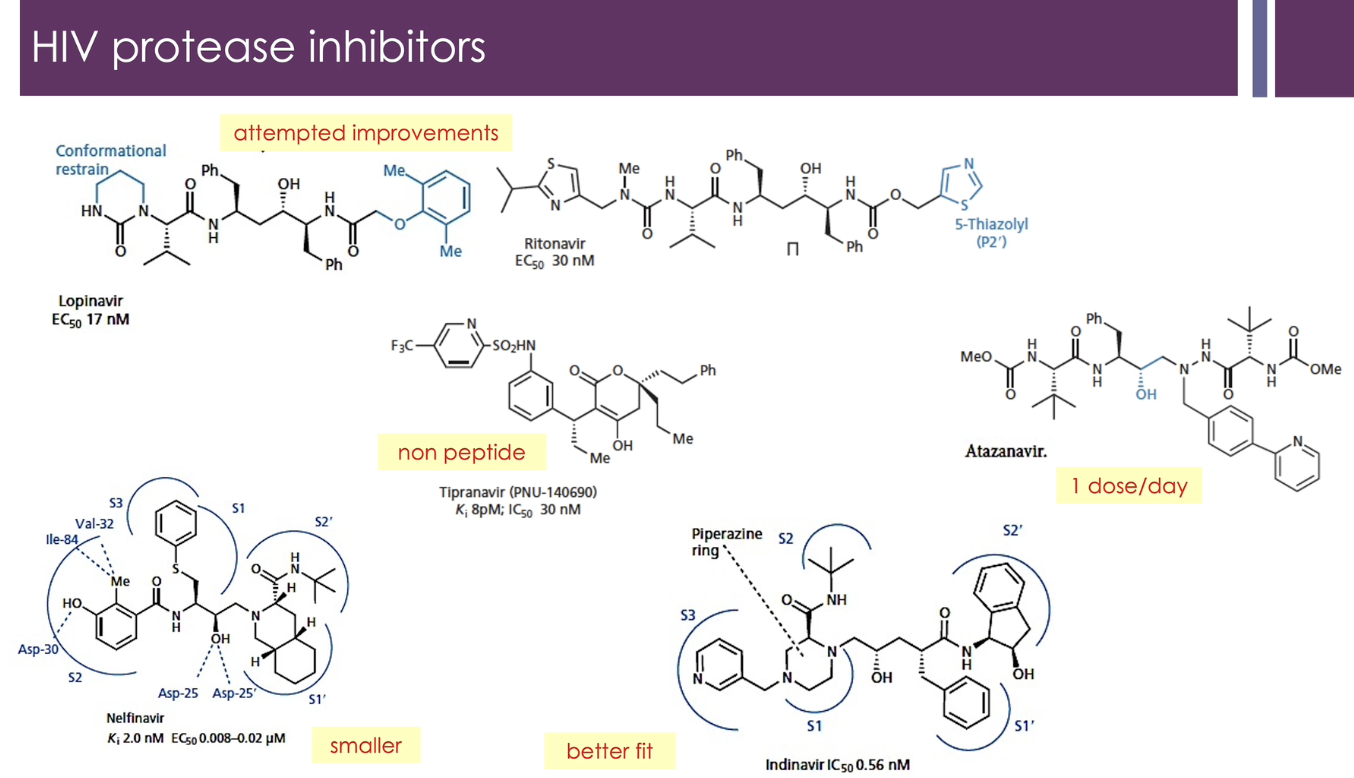
the first protease inhibitor was Saquinavir
examples of attempted improvements for HIV protease inhibitors, compared to Saquinavir
restraining its conformation (to the "active" shape to bind more strongly) → Lopinavir and Ritonavir
non-peptide species (to prevent hydrolysis) → Tipranavir
smaller → Nelfinavir
Better fit to enzyme aspartyl protease → Indinavir
1 dose/day → Atazanavir
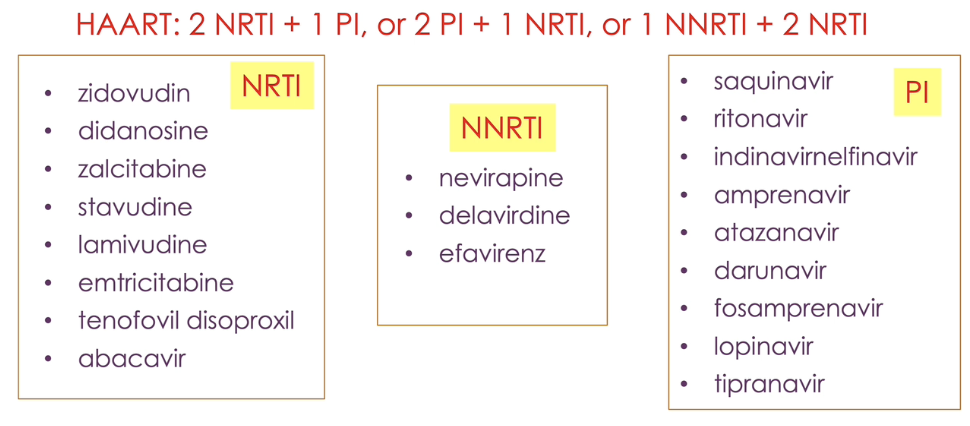
what is HAART
highly active anti-retroviral therapy → combination therapy to treat HIV
HIV mutates and becomes resistance to drugs quickly
HIV has no cure → anti-HIV drugs are for lifetime and have toxic side effects
examples of HAART are:
2 nucleoside reverse transcriptase inhibitors (NRTI) + 1 protease inhibitor (PI)
1 nucleoside reverse transcriptase inhibitor (NRTI) + 2 protease inhibitor (PI)
2 nucleoside reverse transcriptase inhibitors (NRTI) + 1 non-nucleoside reverse transcriptase inhibitors (NNRTI)
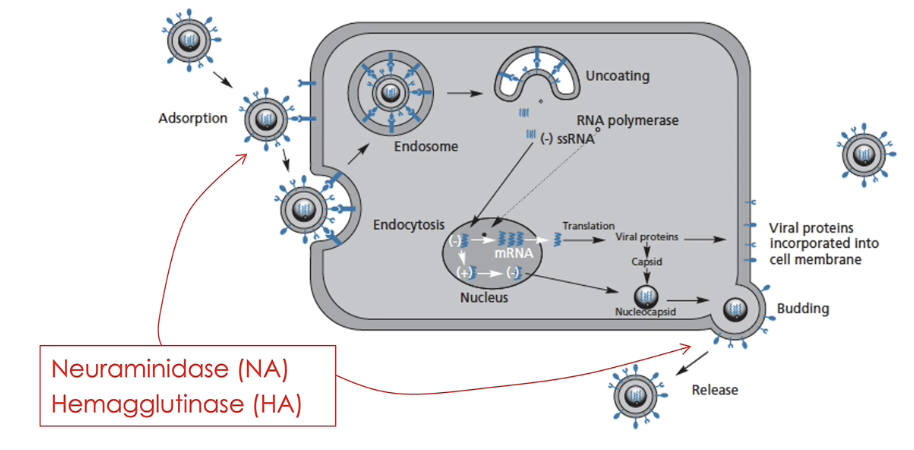
what are the protease inhibitors for influenza
influenza → airborne virus causing respiratory disease
mechanism of action
prevents docking, cellular uptake and spreading of the virus
blocks protease enzymes of the virus: neuraminidase (NA) and hemagglutinase (HA), which are responsible for the lysis of mucus, allowing the virus to reach epithelial cells (NA), access epithelial cells (HA) and budding (NA)
what are neuraminidase inhibitors (influenza)
MOA:
they mimic the structure of the transition state (the protease-binding sialic acid unit on glycoproteins and glycolipids
before virus cuts the glycosidic bond connecting sialic acid to a glycoprotein or glycolipid, drugs bind very strongly to neuraminidase as they mimic that distorted transition-state structure → allowing the drug to bind very tightly to the enzyme and prevent it from cleaving the real sialic acid-glycoprotein bond.
examples: Osletamavir, Zanamivir and Laninamivir
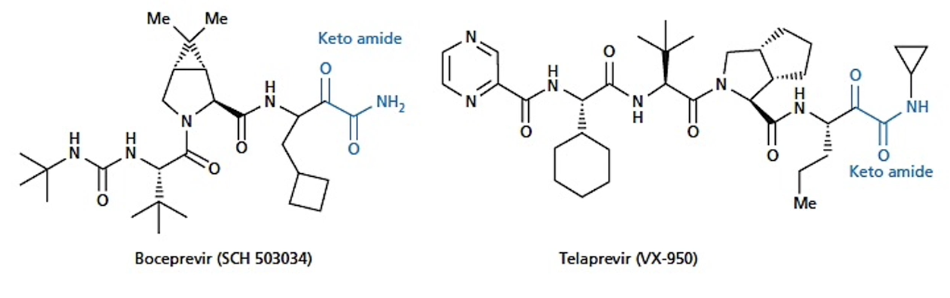
what are the protease inhibitors for hepatitis C
hepatitis C → blood-borne virus that causes an asymptomatic infection
MOA: block a protease that releases mature virus proteins from pro-peptides
exampkes: Boceprevir and Telaprevir
what are other antiviral agents
ion channel disrupters
capsid-binding agents
ribavirin
interferone
what are ion channel disrupters
Adamantanes are a class of antiviral drugs that act as ion channel disruptors against influenza A viruses
MOA for low dose: blocks an ion channel protein (M2 protein) on the virus → prevents exchange of electrolytes between virus
MOA for high dose: lowers the pH of the endosome to delay uncoating (virus removes its protective coat (capsid) and release genetic material into host cell)
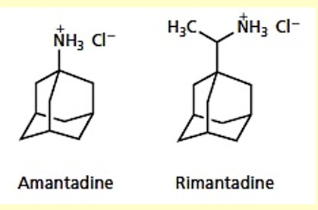
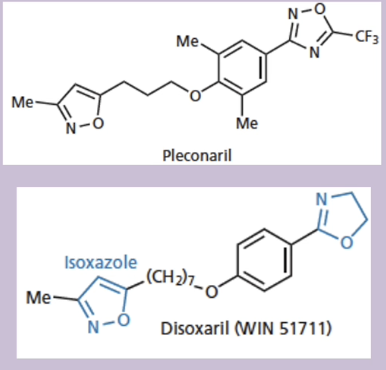
what are capsid-binding agents
used against picornaviruses (polio, hepatitis A, FMD)
MOA: bind to a pocked in the capsid of the virus to prevent release of viral RNA
examples: pleconaril and disoxaril
what is ribavirin
a broad-spectrum antiviral (works against RNA and DNA virusea)
only licensed for hepatitis C
MOA: induces mutations in the virus
what is interferone
a family of peptides involved in cell response to viral infections
examples of interferone: alpha, beta and gamma
they are released by infected cells to alert neighbouring cells of infection