BIMM 100 Final Exam (not cumulative)
1/39
Earn XP
Description and Tags
covers LE 14-18, week 9-10 DI slides, final exam review slides (+podcast, watch if have time), problem sets 7+extra, MB questions from BioConnect final exam review slides
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
40 Terms
def. epigenetics (← give 2 def.)
describe (2)
can be affected by __ and the __
name the 3 predominant forms of epigenetics
changes in the genome (gene expression & genome) that don’t change the gene/DNA sequence
specific/some epigenetic modifications can be passed down from parent to daughter cell & can affect more than one generation
changes are reversible
can be affected by diet and the environment
how your behaviors and environment can cause changes that affect how your genes work (← other definition)
__
epigenetics predominantly appears as:
histone modifications
non-coding RNAs
DNA methylation
(^ histone mod. talked about in previous midterm LEs, other 2 are new)
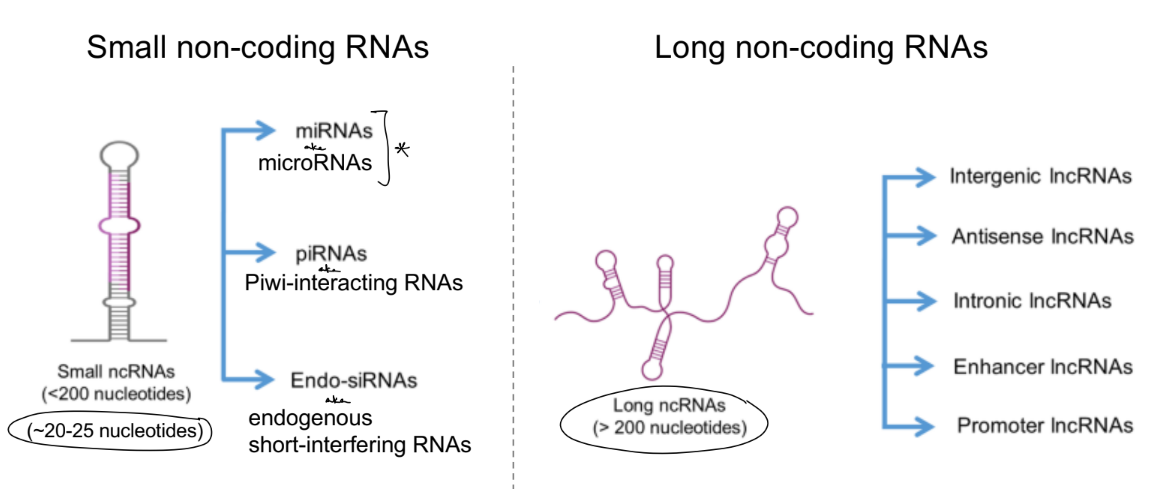
most RNAs in eukaryotic cells are __, specifically what 2 kinds?
in terms of non-coding RNAs, what are the 2 general categories of ncRNAs?
function of each category ncRNA (1) (2)
there are many types of long non-coding & small non-coding RNAs, where __ is a type of small non-coding RNAs
what deter. if small or long RNAs? aka give length of each
regulatory ncRNAs form different structures, ex: __ __ / they don’t exist as a straight line
most RNAs in eukaryotic cells are non-coding RNAs (ncRNAs), specifically what long non-coding RNAs (lncRNAs) & micro RNAs (miRNAs)
__
2 general categories of ncRNAs:
housekeeping ncRNAs — regulate translation
regulatory ncRNAs — regulate gene expression & mRNA stability
__
there are many types of long non-coding & small non-coding RNAs, where miRNAs is a type of small non-coding RNAs
small RNA if 20-25 nucleotides
long RNAs if >200 nucleotides
regulatory ncRNAs form different structures, ex: hairpin structure / they don’t exist as a straight line
describe micro RNAs / miRNAs (3 ← incl. function/result)← type of small non-coding RNA
t/f: miRNAs are processed in the nucleus & cytoplasm, and then in the cytoplasm, miRNA will interact w/ its target mRNA to determine the outcome
what is it called when miRNA interacts with its mRNA target?
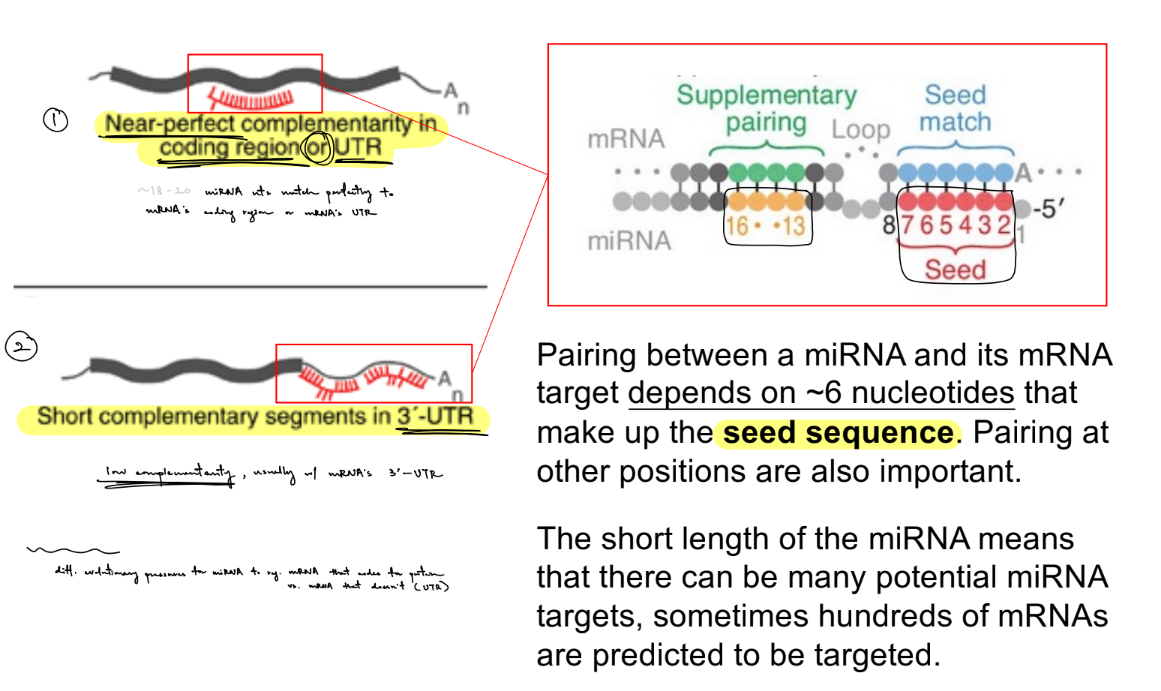
name 2 ways this can occur & what part(s) of the mRNA target is involved in each
which is exact/precise vs imprecise pairing?
pairing b/w a miRNA and its mRNA target depends on the __ __ (← is how long?)
miRNAs usually __ mRNAs from undergoing translation, so there is __ protein expressed
are encoded in the genome
are small RNAs of 20-25 nucleotides long
cause degradation (through cleavage) to inhibit translation of the mRNA target
__
true: miRNAs are processed in the nucleus & cytoplasm, and then in the cytoplasm, miRNA will interact w/ its target mRNA to determine the outcome
miRNA interacts with its mRNA target via standard base-pairing by:
near perfect/perfect complementarity in the coding region OR UTR (of the target mRNA) (that does exact pairing)
short complementarity in the 3’ UTR (that does imprecise pairing)
pairing b/w a miRNA and its mRNA target depends on the seed sequence ← ~6 nucleotides
_
miRNAs usually inhibit mRNAs from undergoing translation (so less protein is expression)
for exact pairing / near perfect complementarity in coding region or UTR
what is the outcome?
this will affect the __ of an organism ← explain w/ ex. of HOXB8 gene and miR-196 (general statement)
what happens to HOXB8 when miR-196 is overexpressed?
what happens to HOXB8 when miR-196 is reduced?
exact pairing extends beyond the __ sequence
____
for imprecise pairing / short complementarity in 3’ UTR
outcome (2)
mRNA degradation via target mRNA cleavage
this will affect the traits of an organism
miR-196 cleaves the HOXB8 mRNA, which limits the expression of the HOXB8 protein
when miR-196 is overexpressed → cleaved & degraded target mRNA → HOXB8 decreases → so shorter fish (than wt)
when miR-196 is reduced → increased mRNA → HOXB8 increases → so longer fish (than wt)
exact pairing extends beyond the seed sequence
____
for imprecise pairing
inhibit translation
(b/c reduced ribosomal activity)
destabilization b/c degradation via deadenylation (remove 3’ polyA tail)
(where mRNA will be degraded, so there’s less mRNA to translate)
____________________
^^ shows how the outcomes of both miRNA-mRNA pairings result in less protein (due to inhibition of certain processes)
describe lncRNAs (3 ← incl. function/result)
for the 2 lncRNA functions/results above, describe the 2 effects/functions for each
are encoded in the genome
long RNAs w/ >200 nucleotides
many activities, including reg. gene expression by/via affecting chromatin & post-transcriptional reg.
reg. of gene expression:
promote open chromatin
promote closed chromatin
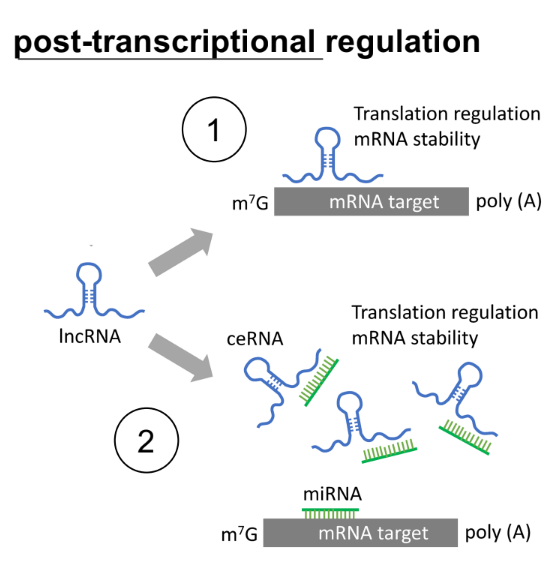
post-transcriptional reg.:
direct reg. of mRNA stability
indirect reg. of mRNA stability by involving miRNAs
for post-transcriptional lncRNA to reg. mRNAs
lncRNAs can directly stabilize mRNAs by binding to __
lncRNAs can indirectly stabilize mRNAs by involving __, where these lncRNAs are called __ (← stands for)
how does this work?
lncRNAs can directly stabilize mRNAs by binding to mRNA
lncRNAs can indirectly stabilize mRNAs by involving miRNAs, where these lncRNAs are called ceRNAs (← competing endogenous RNAs)
ceRNAs have complementary seq. to the miRNAs, so they bind together to move miRNA away from the target mRNA (SO less miRNA will cleave and degrade the target mRNA)
for reg. gene expression of lncRNA by affecting chromatin
some lncRNAs can promote __ & some can facilitate/cause __ chromatin
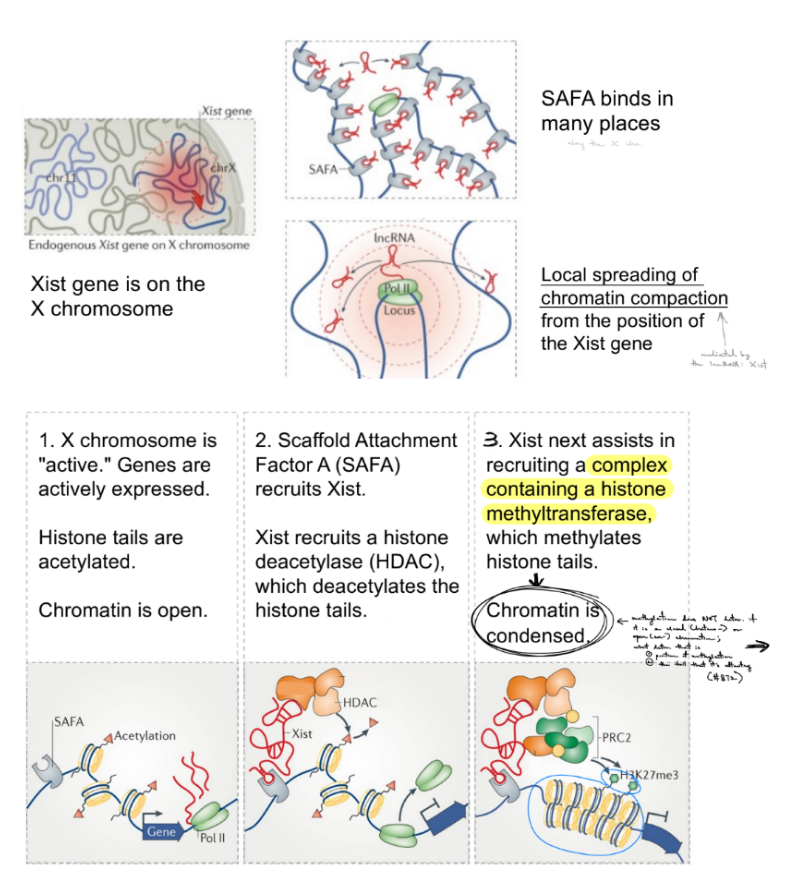
this is seen w/ X-inactivation (← def.)
state the 3 steps
though keep in mind that methylation does NOT determine if chromatin is closed (hetero-) or open (eu-). It is just that most condensed chromatin (heterochromatin) has __ histone proteins
^ Xist is a __
^ spec. how does Xist work?
_____
review:
3 main histone tail modifications
transition b/w chromatin states is reg. by (1), where lncRNA tends to turn __ chromatin into __ chromatin
some lncRNAs can promote open chromatin & some can cause closed chromatin
seen w/ X-inactivation — ONE X chromosome is inactivated by a lncRNA in females, while the other X chr. is active
BOTH X chr. are active, so genes are expressed. There is also acetylated histone tails, so the chromatin is open.
SAFA (scaffold attachment factor A) recruits Xist, where Xist will inactivate ONE X chr. in female cells; AND Xist will recruit HDAC (histone deacetylase), which deacetylates histone tails
Xist also recruits a complex containing histone methyltransferase, which methylates histone tails → SO chromatin is condensed
though keep in mind that methylation does NOT determine if chromatin is closed (hetero-) or open (eu-). It is just that most condensed chromatin (heterochromatin) has methylated histone proteins
^ Xist is a lncRNA (lncRNA that promotes closed chromatin in reg. of gene expression)
^ spec. Xist that inactivates 1 X chr. in female cells will spec. inactivate genes that are close to the Xist gene, b/c SAFA binds in many places & the recruited HDAC will cause chromatin compaction/condensed/closed that locally spreads where SAFAs exist
____________
review:
3 main histone tail modifications: acetylation, methylation, phosphorylation
transition b/w chromatin states is reg. by histone tail modifications, where lncRNA tends to turn active/open chromatin into inactive/condensed/closed chromatin
def. coding vs. noncoding strand
lncRNAs are encoded in the genome in many places, so there are many types of lncRNAs
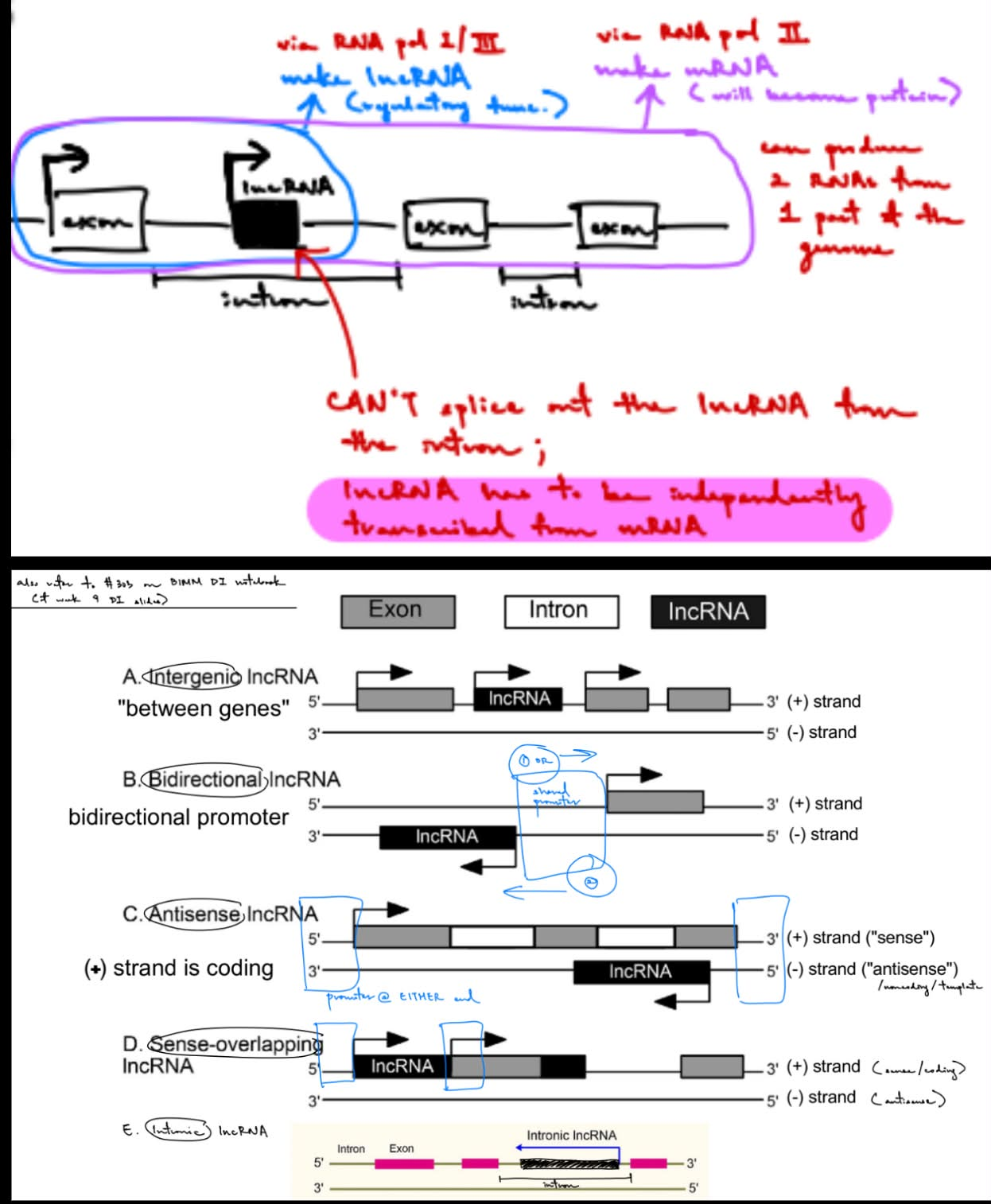
name 7 types (& describe 5 of them)
coding strand (sense strand) — DNA strand whose sequences matches the transcribed mRNA
(mRNA and DNA coding/sense strand have the same 5’ and 3’ end directions)
noncoding strand (antisense strand) — DNA strand where lncRNA is transcribed from / complementary to coding strand
(mRNA and DNA noncoding/antisense strand have the opposite 5’ and 3’ end directions)
__
intergenic
b/w 2 (coding) genes
bidirectional
made from a bidirectional promoter, so can transcribe RNA in either direction (direction of exon OR direction of lncRNA)
antisense
antisense lncRNA is on the antisense strand aka complementary to the coding strand that produces mRNA (aka comp. to strand that makes mRNA)
sense-overlapping
is on the sense/coding strand, BUT the lncRNA’s TSS is different from the coding gene’s TSS
intronic
is w/in an intron, but intronic lncRNAs can’t be spliced out & has to be independently translated from mRNA
enhancer lncRNA (lncRNA in enhancer)
promoter lncRNA (lncRNA in promoter)
for epigenetics: DNA methylation
the most common type of DNA methylation is (1)
is it mutagenic? explain
cytosine (and DNA in general) is methylated by __ (← stands for?) to form this common DNA methylation product
while DEmethylation of DNA involves the enzyme __ (← stands for?)
____
review:
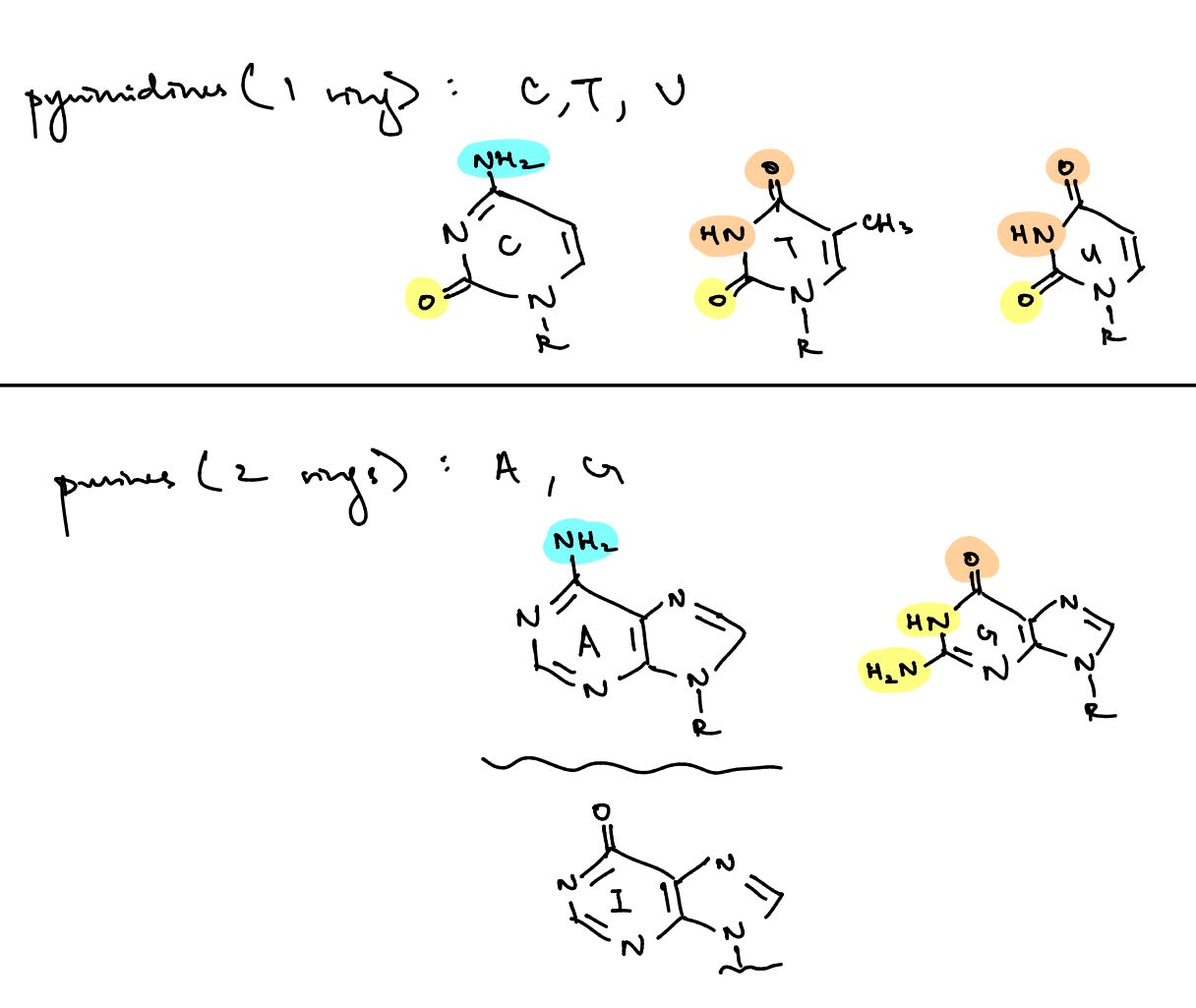
draw the 4 types of nucleotide bases & state if purine/pyrimidine
the most common type of DNA methylation is 5-methylcytosine
not mutagenic b/c doesn’t disrupt H bonding b/w C-G base pairs, so no change in DNA sequence/ not mutagenic
cytosine (and DNA in general) is methylated by DNMT (DNA methyltransferase) to form this common DNA methylation product: 5-methylcytosine
while DEmethylation of DNA involves the enzyme TET (ten eleven translocation enzyme)
for epigenetics: DNA methylation
2 parts of the genome that are usually methylated / have methylated CpG islands
1 part of the genome that is usually NOT methylated ← why?
def. islands
in general, DNA methylation is usually ass. w/ __ gene expression, but is sometimes ass. w/ __ gene expression
_
loss of transposon methylation can cause (2)
____
review:
t/f: more than half of human genome is made of transposable elements
usually methylated / have methylated CpG islands:
transposons/transposable elements
transcription unit of genes
usually NOT methylated:
CpG island promoters
b/c methylation of CpG island promoters will silence gene expression
islands — UNmethylated CpG sequences
in general, DNA methylation is usually ass. w/ reduced gene expression, but is sometimes ass. w/ increased gene expression
_
loss of transposon methylation can cause
transposon activity that destabilizes the genome
mis-reg. of nearby gene expression
(review: true)
for epigenetics: DNA methylation
for loss of transposon methylation that can cause misreg. of gene expression, which affects traits:
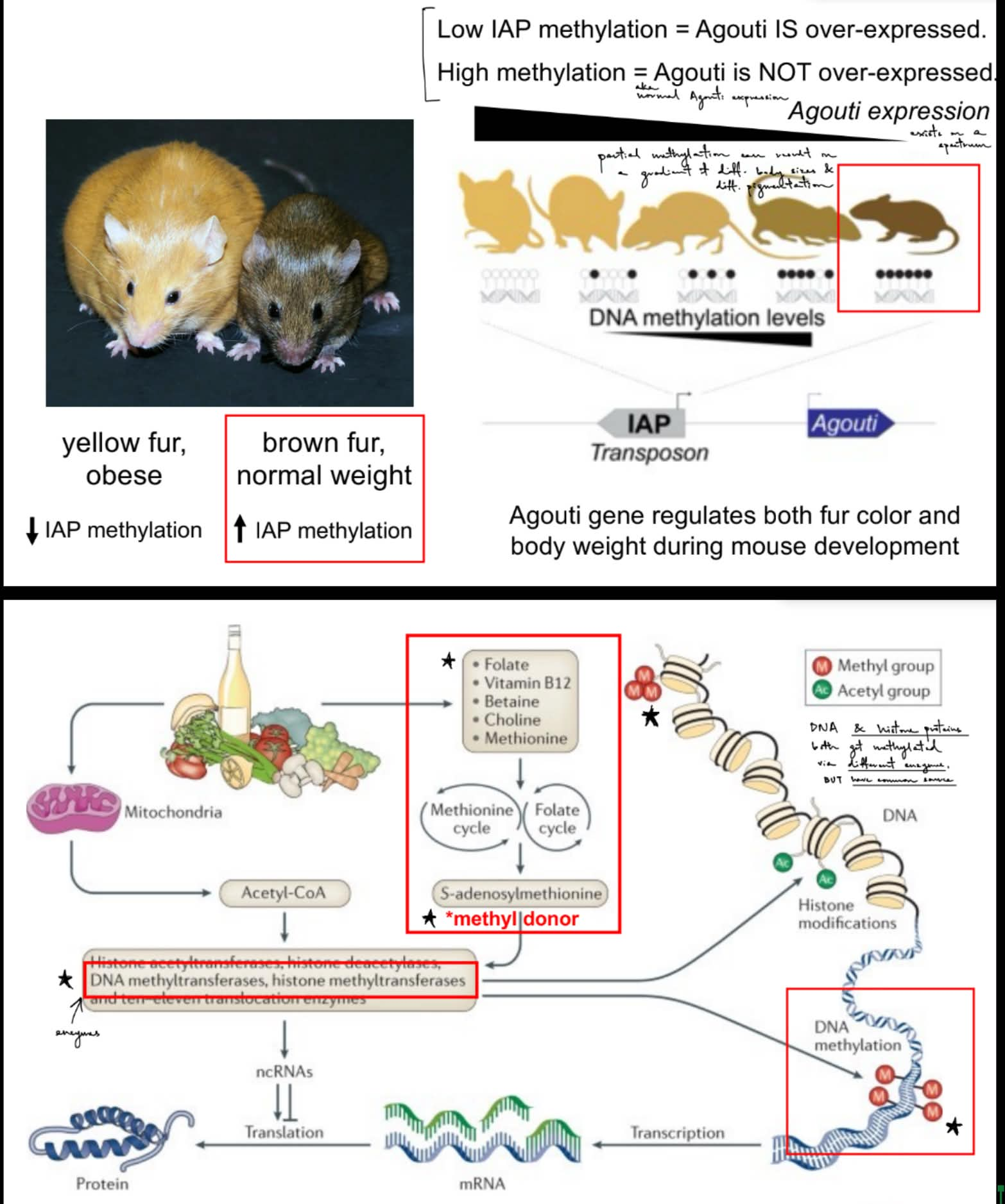
what are the 2 phenotypes of mice?
def. Agouti gene
a __ called __ is inserted upstream of the Agouti gene & present in __ mice
____
review:
3 differences b/w retrotransposon & DNA transposon
what does upstream mean?
yellow fur, obese & brown fur, normal weight
Agouti gene — produces a protein that regulates fur color & body weight
a retrotransposon called IAP is inserted upstream of the Agouti gene & is present in some mice
____
review:
3 differences b/w retrotransposon & DNA transposon:
retro- do copy+paste, DNA- does cut+paste
retro- are transcribed into an RNA inter. via RNA pol.
the RNA inter. in retro- is then converted into DNA via RT (reverse transcriptase)
upstream — sequence that’s 5’ of the promoter
cont. Agouti gene
insertion of IAP causes Agouti to be __, resulting in what pheno?
what happens when the inserted IAP is methylated? why? what pheno?
^ reworded as high IAP methylation vs. low IAP methylation causes what?
__
why does IAP methylation vary to get a spectrum of DNA methylation?
aka cytosine methylation patterns are affected by (3)
describe 1 of them
note: DNA methylation is enzyme-mediated by __ that gets __ __ from our __, so it is not spontaneous
insertion of IAP causes Agouti to be overexpressed, resulting in yellow fur & obese
inserted IAP is methylated / methylation of IAP → Agouti is NOT overexpressed aka get normal Agouti expression (b/c methylation of IAP suppresses IAP from increasing Agouti expression) → brown fur & normal weight
^ high IAP methylation → Agouti is NOT overexpressed — brown fur, normal weight
^ low IAP methylation → Agouti is overexpressed — yellow fur, obese
__
cytosine methylation patterns are affected by experience, chemicals, and diet
folate & folic acid in food affects the amount of methyl groups consumed, which then affects DNA methylation (for epigenetics)
note: DNA methylation is enzyme-mediated by DNMT that gets methyl groups from our diet, so it is not spontaneous
cont. Agouti gene
if a pregnant mouse has IAP inserted upstream of Agouti & is fed food low in methyl groups, what happens to the retrotransposon IAP? pheno?
pheno of offspring?
but if fed food supplemented in methyl group donors, then what happens to IAP? Agouti expression? pheno?
__
^^ overall, the foods you eat affect your __ & your __ cells, which would affect the next gen./your offspring
…
which is the wildtype pheno?
__
diet of the pregnant mouse affects __ generations (aka affects __ and __)
__
summary:
epigenetic diet provides building blocks for a healthy genome, where __ in diet provides __ __, which is used by different __ to do __ & __ __
t/f: a good example of multi-generation epigenetic effect is the Dutch Hunger Winter famine
t/f: fathers’ diets also have significant epigenetics effects on their offspring
(poor diet:)
IAP is weakly or NOT methylated → yellow fur & obese/poor health
(b/c low methyl groups, which cause overexpression of Agouti, so yellow fur and obese)
offspring mainly look like mother w/ yellow fur & obese/poor health, but can still have offspring w/ brown fur & normal weight/good health
__
(healthy diet:)
IAP is methylated → Agouti gene is not overexpressed aka normal → brown fur & normal weight/good health
offspring mainly look like mother w/ brown fur & normal weight/good health, but can still have offspring w/ yellow fur & obese/poor health
__
^^ overall, the foods you eat affect your genome & your germ cells, which would affect the next gen./your offspring
…
wt pheno. is brown fur & normal weight/in good health
__
diet of the pregnant mouse affects TWO generations (aka affects children and grandchildren)
(b/c F1 fetus growing in the pregnant mouse already has F2 germ cells)
__
summary:
epigenetic diet provides building blocks for a healthy genome, where folate in diet provides methyl groups, which is used by different enzymes to do histone &DNA methylation
_
true (multi-generation epigenetic effect is famine)
true (fathers’ diets also have significant epigenetics effects on their offspring)
overview:
want to keep transposons methylated to prevent __ the genome & __ of gene expression
_______________________________
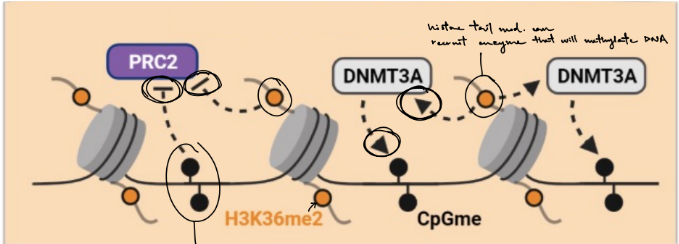
gene expression is regulated by a combination of (2) ← aka crosstalk
where some histone tail mod. are enriched/located in __ DNA, while other histone mod. are enriched/located in __ DNA
SO we can’t assume that methylation of histone proteins will correlate with __ or __ in gene expression, specifically can result in __ or __ enzymes to modify DNA + histone proteins
explain pic
____
(in this ex. w/ MeCP2:)
methylation of DNA by __ (which is a “__”) will be recognized by “__” called __ (stands for?), specifically __
describe the further process of this (3)
→ SO DNA methylation is typically ass. w/ __ gene expression
^^ t/f: methylation of genomic DNA / DNA methylation will recruit chromatin remodelers, including __
…
what’s the effect of deacetylating histones?
want to keep transposons methylated to prevent destabilizing the genome & misregulation of gene expression
_______________________________
gene expression is regulated by a combination of DNA methylation & histone tail modifications
where some histone tail mod. are enriched/located in methylated DNA, while other histone mod. are enriched/located in unmethylated DNA
SO we can’t assume that methylation of histone proteins will correlate with increase or decrease in gene expression, specifically can result in blocking or recruiting enzymes to modify DNA + histone proteins
…
(pic)
DNA methylation & histone mod. (H3K36me2) will block the enzyme (PRC2 from methylating histones)
histone mod. (H3K36me2) will recruit enzyme (DNMT3A), which methylates DNA
____
(in this ex. w/ MeCP2:)
methylation of DNA by DNMT (which is a “writer”) will be recognized by “readers” called MBD (methyl-binding proteins), specifically MeCP2
(in methylated DNA) MeCP2 recognizes & binds to methylated CpGs
MeCP2 recruits HDAC (histone deacetylase)
deacetylation of histone tail will promote chromatin formation aka chromatin compaction/condensation, which blocks gene expression
→ SO DNA methylation is typically ass. w/ reduced gene expression
^^ true: methylation of genomic DNA / DNA methylation will recruit chromatin remodelers, including HDACs
…
deacetylating histones via HDACs will restore the “+” charge on lysine, which strengthens the electrostatic attraction b/w histone & DNA → promotes wrapping DNA around histones to get chromatin compaction/condensation
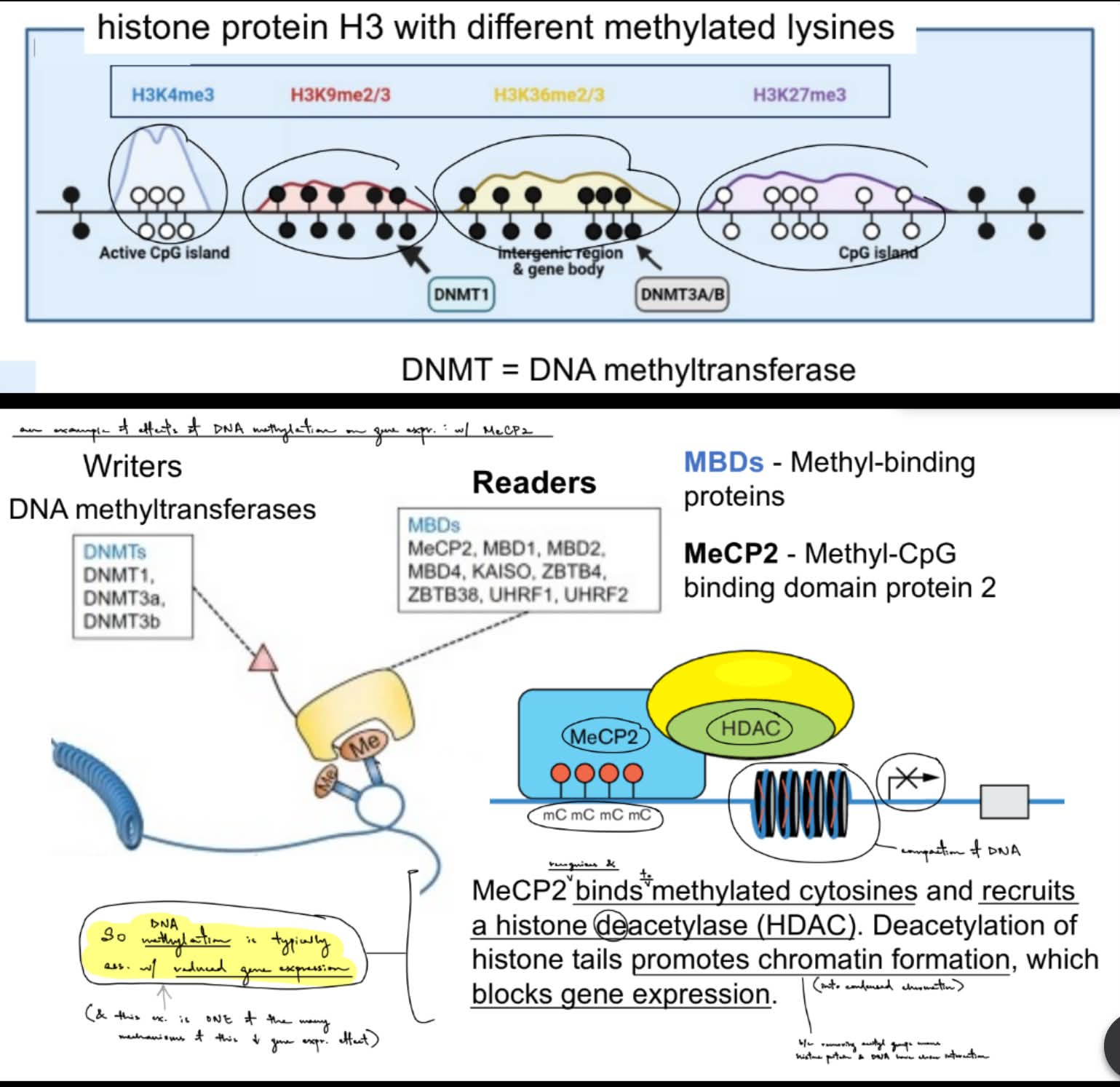
for DNA methylation
what happens to DNA methyl. as organisms age? (3, where 1 of them has 2)
__
def. epigenetic clocks (another name?)
aka is the robust correlation b/w changes in __ __ & age
what changes in DNA methylation (& resulting changed in gene expression) occur in what specific parts of a genome (comparing child & older adult)? (← 2)
generally, DNA methylation decreases w/ age in humans
decreased DNA methylation (will change how DNA is packaged)
altered histone modifications, where:
increase of activating histone mod.
decrease of repressive histone mod.
(^ both for more open chromatin, so can do gene expression ← shown in below point too)
global reduction in heterochromatin (closed chromatin)
__
epigenetic clocks / Horvath’s clock — align epigenetic age w/ chronological age (aka predict chronological age w/ DNA methylation of epigenetic age)
aka is the robust correlation b/w changes in DNA methylation & age
in older person:
decreased methylation of “non-CpG island”/methylated promoters → increased gene expression
increased methylation of “CpG island”/unmethylated promoters → decreased gene expression
decreased methylation of transposons → increased transposon expression
(remember that methylation of CpGs in promoter or transposon will decrease or silence gene or transposon expression)
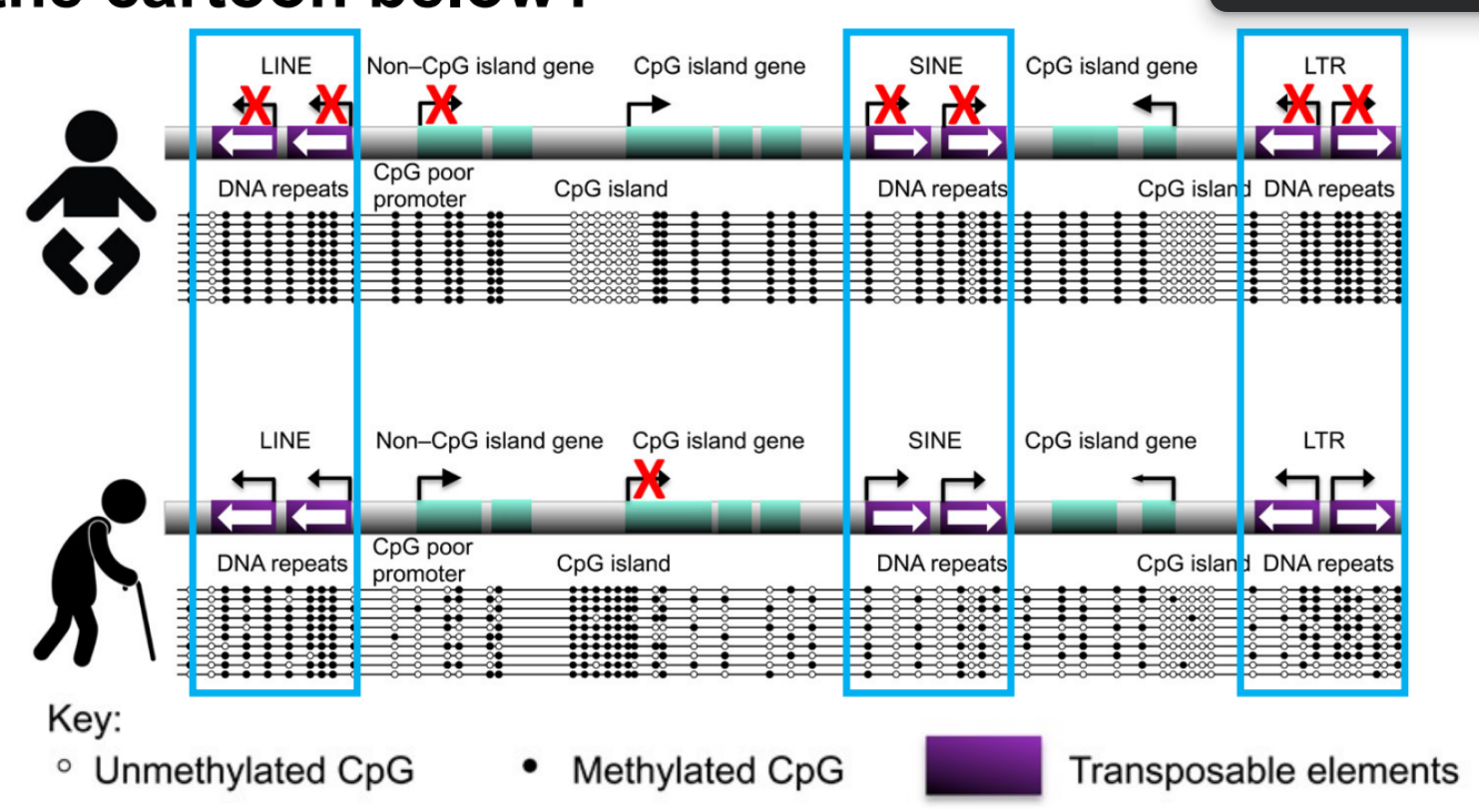
what mutation alter the methylation of a gene, which affects that gene’s expression? (1 ← incl. how & the result)
describe the gene (1)
does this mutation affect protein expression? explain
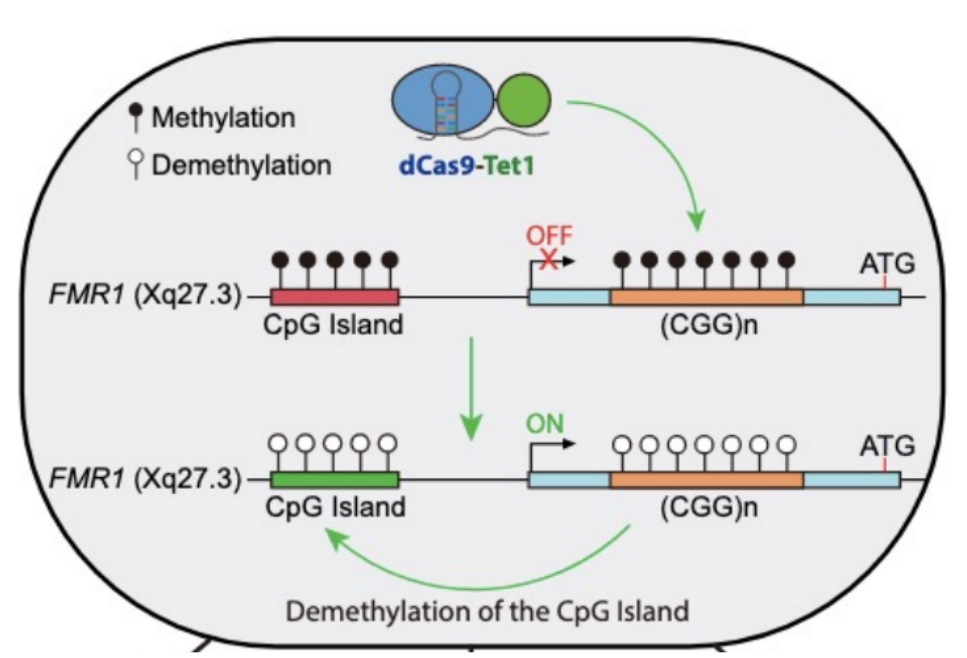
mutation: increased CGG repeat expansions that increase DNA methylation to get methylated repeats → FMR1 gene silencing
FMR1 / FRAXA gene (Fragile X gene) has a CGG repeat in the 5’ UTR of the mRNA
(^^ mutation can be called FXS)
doesn’t affect protein expression
b/c the affected CGG repeats are in the 5’ UTR, which is a non-coding region for proteins
originally, gene editing relied on __ __, such as __ __
limitation of gene editing (1 ← explain)
__
2 types of mech. to repair double-stranded break/DSB in DNA
which is precise vs. error-prone
researchers will make a researcher-designed __ __ to pair with the target site in the genome
_
briefly describe steps preceding homologous recombination (2)
the introduced homologous sequence can either come from __ __ or __ __
drawback of homologous recombination (1)
so, what is done instead?
…
—→ so now that you used an endonuclease to create a DSB / nuclease-induced DSB, it can undergo 2 different DNA repair pathways: (2)
originally, gene editing relied on homology-directed repair (HDR), such as homologous recombination
limitation of gene editing:
genomes are constantly changing b/c get mutations due to replication, aging, and the environment & b/c have many transposons that can affect the genome
__
repair double-stranded break/DSB in DNA via HDR/homology-directed repair (precise) or NHEJ/non-homologous end-joining (error-prone)
researchers will make a researcher-designed donor template to pair with the target site in the genome
__
lab-created allele containing the researcher-designed donor template in introduced into the cell → pair w/ target site in genome → homologous recombination can occur
either the donor template OR a sister chromatid can be introduced that contains homologous sequences
drawback:
frequency of homologous recomb. in many organisms is low, so can’t rely on this method to edit a genome
SO instead, use an endonuclease to create site-specific DSBs that can increase frequency of DNA repair pathway (at specific site in the genome) SO can do gene editing
…
—→ so now that you used an endonuclease to create a DSB / nuclease-induced DSB, it can undergo 2 different DNA repair pathways: HDR via homologous recombination & NHEJ
(So the purpose of inducing a DSB is not because DSBs don't exist, but because we need a DSB at the precise target site to strongly stimulate repair and editing there
Without a targeted DSB, the cell has little reason to incorporate your donor DNA at that particular locus, so homologous recombination remains very inefficient.
Think of it this way:
Natural DSBs = random potholes scattered throughout the genome.
CRISPR-induced DSB = deliberately digging a hole at the exact address you want to renovate.)
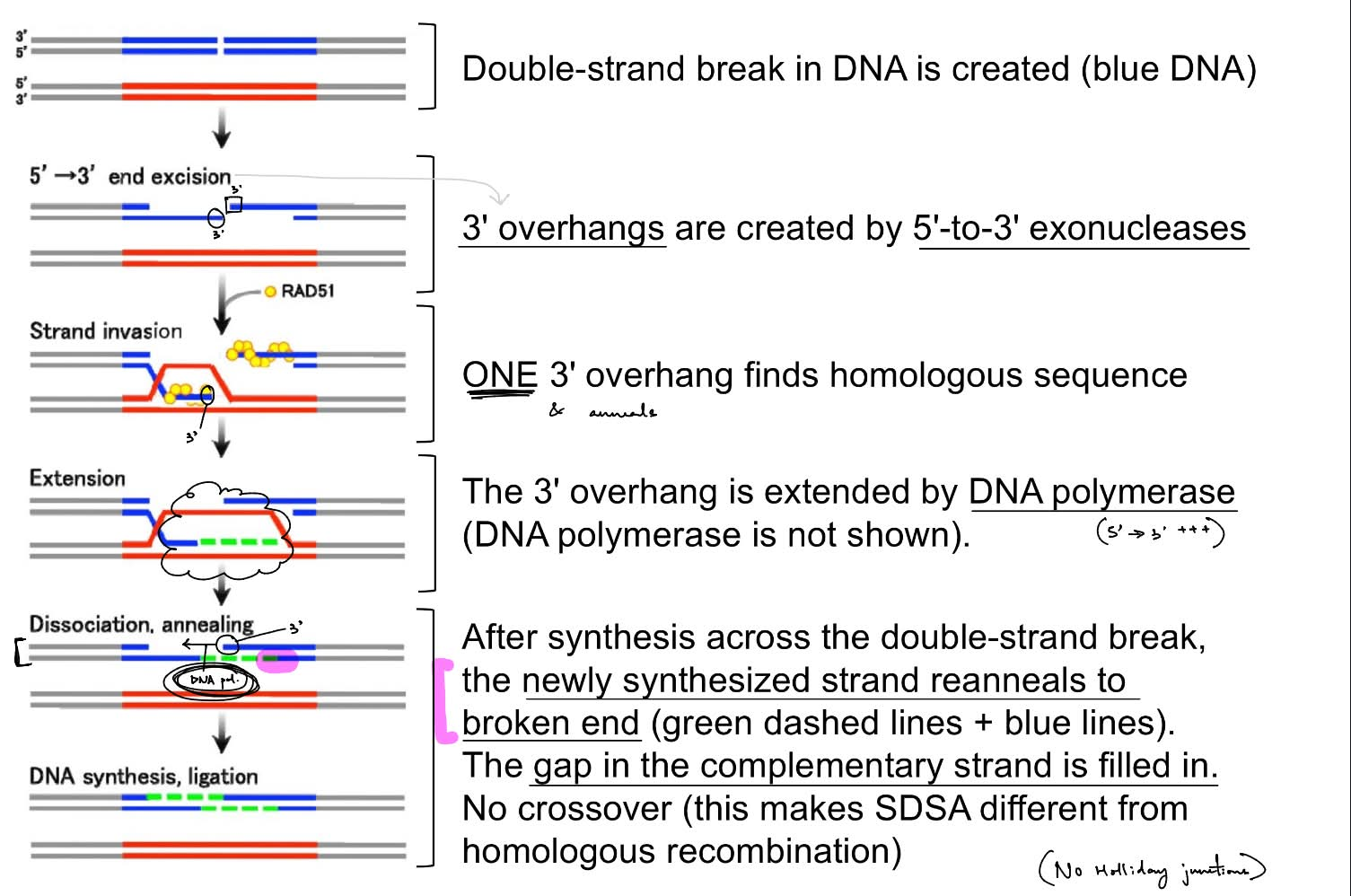
for DNA repair pathway: HDR
process of HDR: homologous recombination requires (1 ← could be 1 or 1)
what is name of a predominant HDR pathway?
steps of this pathway (6)
this HDR pathway does NOT involve __, so doesn’t form __ __
draw out this specific HDR pathway
process of HDR: homologous recombination requires homologous sequence either from homologous allele on sister chromatid OR researcher-designed donor template containing homologous sequence
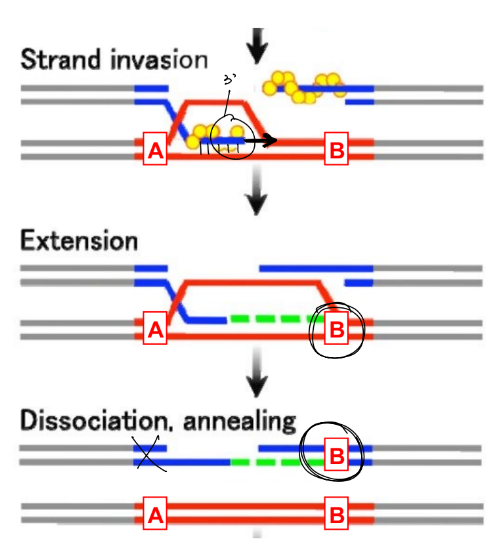
predominant HDR pathway: SDSA (synthesis-dependent strand annealing)
endonuclease creates a DSB in DNA
5’→ 3’ exonuclease will make 3’ overhangs
ONE 3’ overhang finds & anneals to the homologous sequence (in repair template)
that 3’ overhang is extended by DNA pol.
this newly synthesized strand will reanneal to the broken end
the gap in the complementary strand is filled in via DNA pol. that uses the newly synthesized strand as the template
^ the DNA in SDSA pathway does NOT crossover, so doesn’t form Holliday junctions
(the donor template is in blue, the repair template provided is in red)
for DNA pathway: HDR, spec. SDSA
SDSA is directional, what does this mean?
so if there is mutation A upstream of the 3’ end & mutation B downstream of the 3’ end when it anneals, then whifch mut. is included?
(^ when the goal is to introduce a change to the endogenous DNA sequence)
SDSA is directional, meaning that the 3’ overhang that initiates repair will determine which sequences in the repair template are included
in this “ “ case, mut. B is incorporated b/c it’s downstream (and the 3’ end that anneals will extend via DNA pol in the 5’ → 3’ direction to include that mutation B), and mut. A is not incorporated b/c is upstream of the 3’ end repairing the DNA break
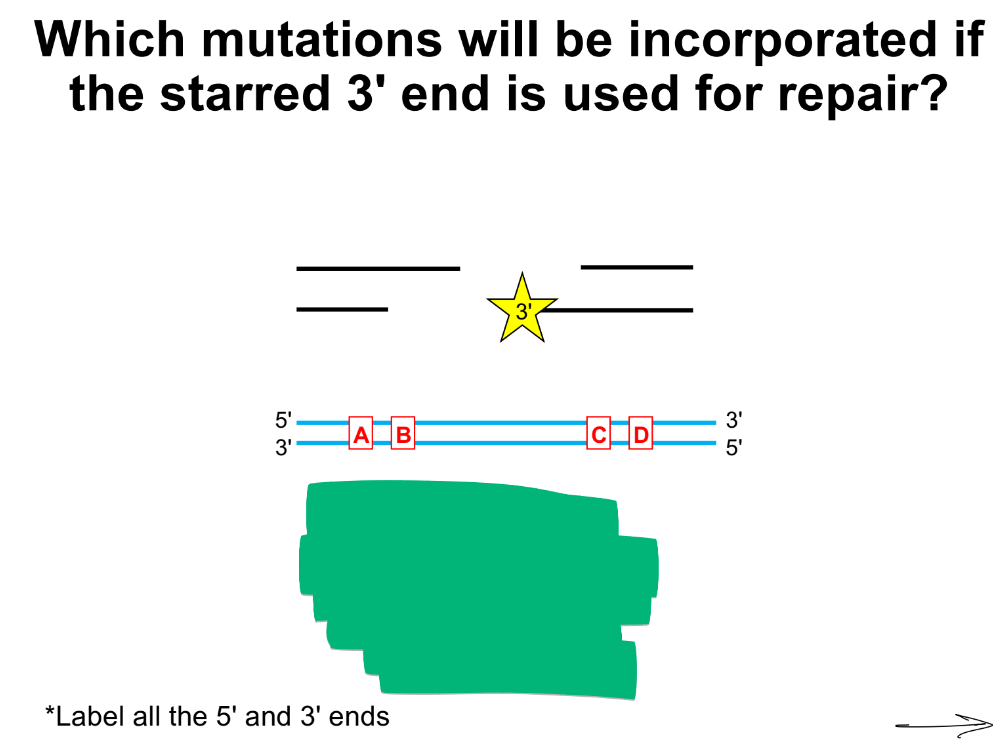
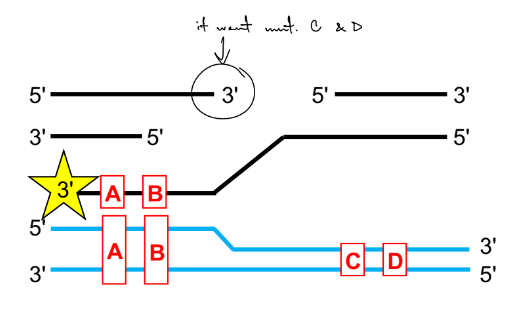
mut. A and B are incorporated, not mut. C and D
the starred 3’ end is comp. to the upper blue strand
b/c DNA synthesis is 5'-to-3' (new nucleotides added to the 3' end), and thus
DNA will be synthesized to the "left" of the double-strand break. The newly
synthesized DNA will only include mutations in the template that correspond
to the "left" of the double-strand break.
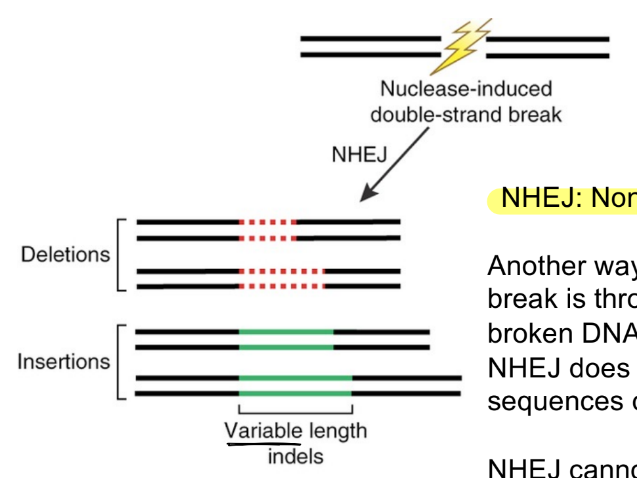
for DNA repair pathway: NHEJ
describe NHEJ
does NOT rely on (2)
result is called __, where that will be variable in length
__________
summary of both DNA repair pathways:
researchers can control (2)
while the cell ultimately deter. (1)
also, which repair pathways are done in dividing and/or non-dividing cells?
broken DNA ends are fused together
does NOT rely on homologous sequences or donor templates
result is called indels, where that will be variable in length
__________
summary of both DNA repair pathways:
researchers can control where DSB occurs & if donor template is given or not
while the cell ultimately deter. which DNA repair pathway is used (to repair the break)
HDR via SDSA in dividing cells
NHEJ in dividing & nondividing cells
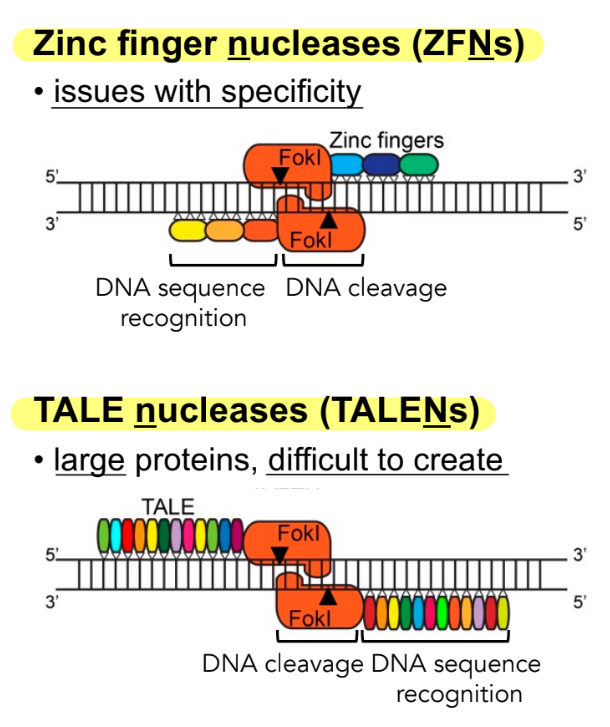
what are the 2 commonly used DNA-binding domains used in making a DSB?
problem w/ using either of these 2 DNA-binding domains
^^ how they work: these DNA-binding domains ZFNs and TALENs are made of __ __ (← name 2) that are fused to an __ (← name)
where… (aka how do they form DSBs)
t/f: both parts of the DNA-binding domain have to be in pairs to create the DSB
zinc finger nucleases (ZFNs)
TAL effector nucleases (TALENs)
problem: ZFNs and TALENs are hard to produce
^^ how they work: these DNA-binding domains ZFNs and TALENs are made of transcription factors (TALEs, ZFs) that are fused to an endonuclease (Fok1)
where ZFNs and TALENs are modified to recognize specific DNA sequences & direct the Fok1 endonuclease pairs to cleave DNA where these DNA-binding domain proteins/TFs pairs (TALE pairs, ZF pairs) bind at
_
true
aka
ZFNs and TALENs each have to be used in pairs, and Fok1 is the endonuclease used to cut the DNA when in pairs (Fok1 pairs) at specific places on the genome that are directed by ZFN pairs or TALEN pairs → create a DSB
about a decade ago, we started using the gene editing machinery called CRISPR-Cas9:
what are the 2 parts of CRISPR-Cas9? def. each
describe (3 each)
__
diff. b/w TALENs and ZFNs vs. CRISPR-Cas9 (in terms of knowing where the genome target is & how it is cleaved to make DSBs)
how do they all relate to the DNA repair mechanisms/pathways?
what are the 2 nuclease domains in Cas9?
Cas9 — an endonuclease that has 2 endonuclease domains (where each endonuc. domain is like a catalytic regions)
the 2 endonuc. domains create a/one DSB
ONE endonuclease domain can be mutated to become a nickase / Cas9 nickase
Cas9 doesn’t bind to DNA, but is instead recruited by guide RNA to attach to the DNA sequence
guide RNA (gRNA) — guides Cas9 to the target genome
guide RNA is short: 20 nucleotides
is highly specific, where guide RNA binds to the target genome via standard base-pairing w/ H-bonds
____
ZFNs and TALENs recognize where to cleave genomic DNA via sequence-specific DNA-binding domains; cleave via Fok1 endonuclease pairs → DSB
Cas9 relies on guide RNA to where the target genome is, b/c Cas9 does NOT have DNA-sequence specificity; cleave via 2 nuclease domains of Cas9 that cleave both strands simultaneously → DSB
Cas9, ZFNs, and TALENs create DSBs that are repaired by DNA repair pathways of HDR via SDSA or NHEJ
…
Cas9’s 2 nuclease domains are RuvC1 & HNH
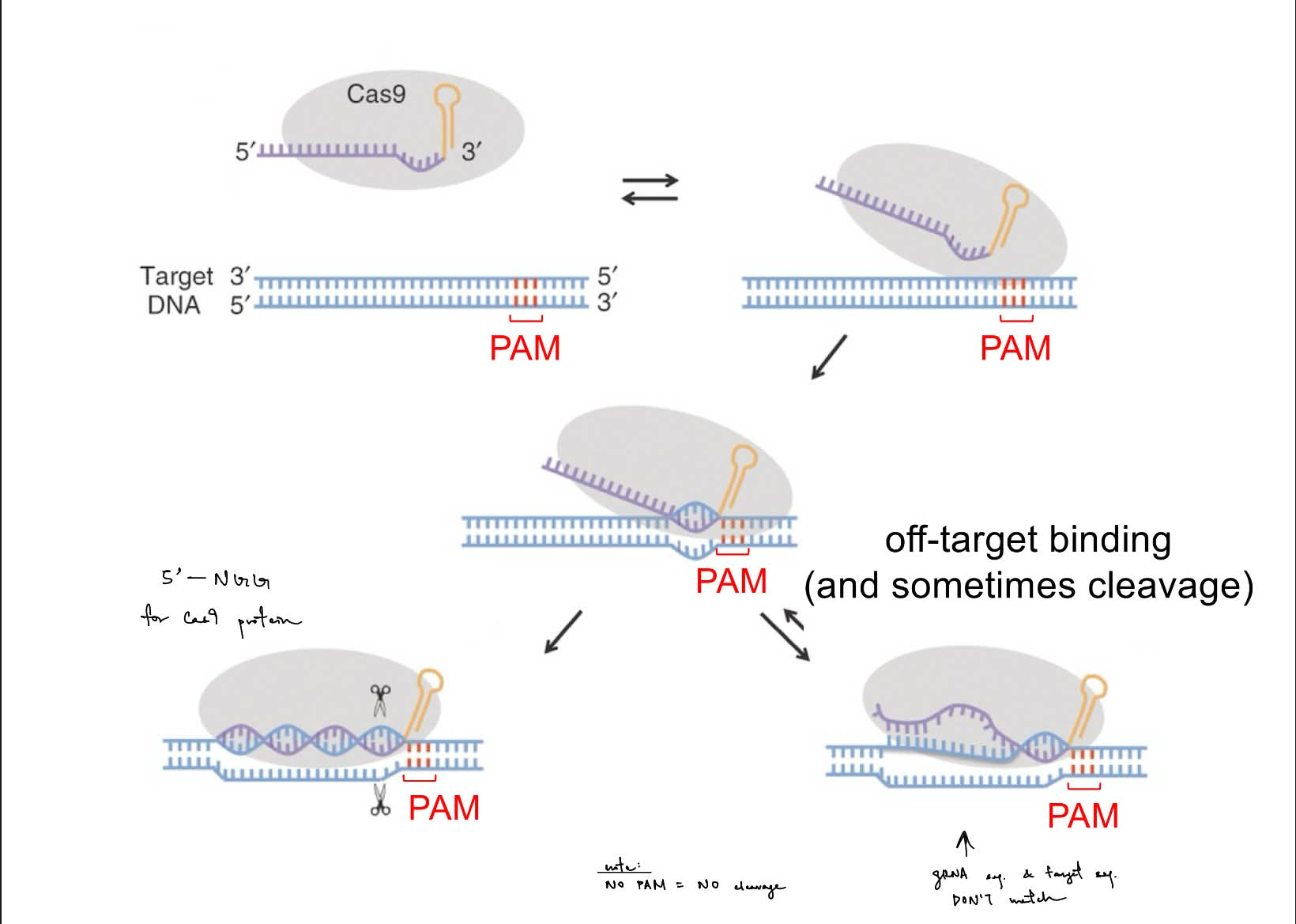
what is required for the CRISPR-Cas9 system? (~4/5)
Cas9 2 key functions (aka Cas9 has activity and activity) ← just name
describe how the CRISPR-Cas9 gene editing works (via the 2 key functions above)
Cas9
gRNA
target DNA sequence
PAM site
optional is the (researcher-designed) donor template
__
Cas9 has:
helicase activity
endonuclease activity
__
Cas9 helicase activity: Cas9 denatures the target DNA helix, so gRNA can form standard base-pairing H bonds w/ the target sequence
Cas9 endonuclease activity:
Cas9 complex scans for the PAM site (is 5’-NGG-3’) & to see if the target sequence matches/is ~same as the gRNA
Cas9 complex cleaves 3 nucleotides upstream (5’) from PAM

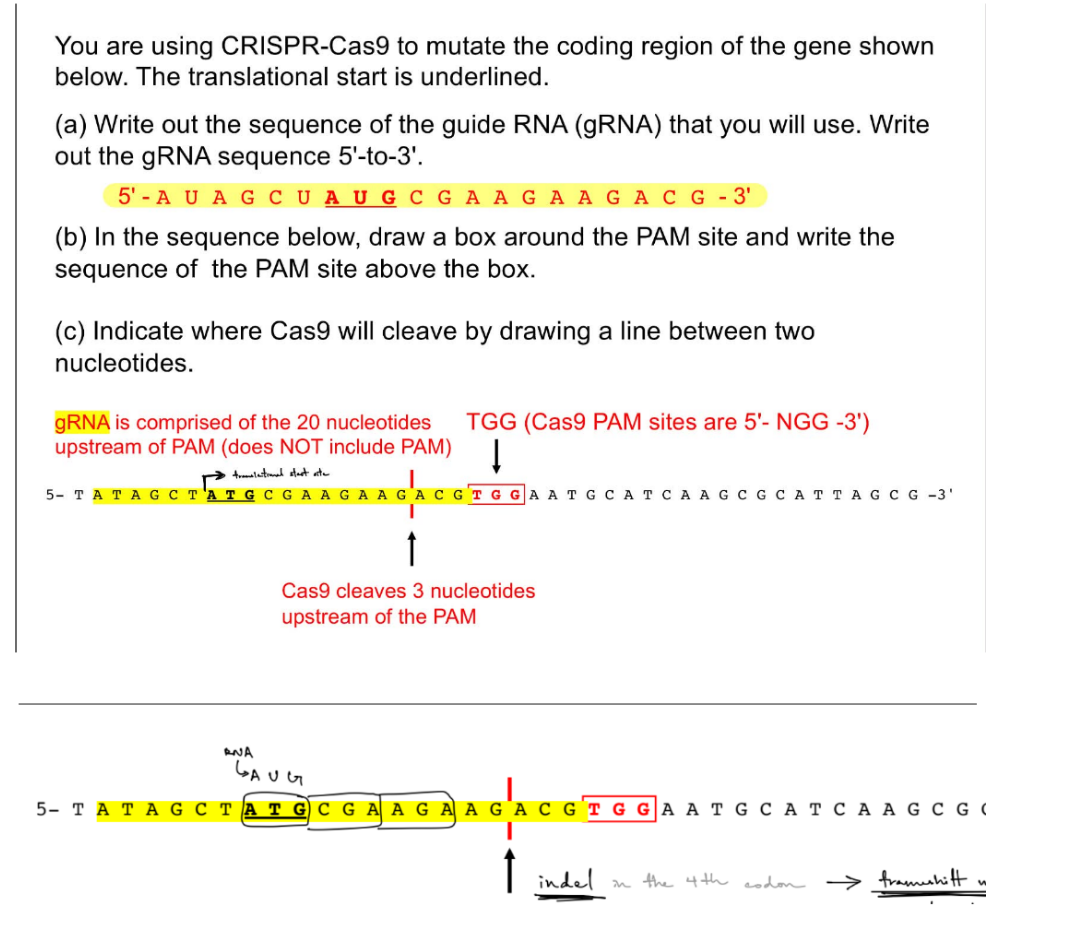
& indicate translational start site w/ an arrow
__
Which DNA repair mechanism would result in a mutation if a donor template is not used? What type of mutation could be introduced & where in the given DNA?
b/c the mutation is at this specific place in the DNA, it is a __ mutation
what is repair mech. & type of mut. introduced if has donor template?
no donor template → use NHEJ, which introduces indels WITHIN the 4th codon
is a frameshift mutation shown in the pic
donor template → use HDR, which has no mutations b/c is a homologous chromosome
where the gRNA sequence includes the 20 nucleotides upstream from PAM, not including PAM, but including the 3 nts b/w Cas9 cleave site & PAM
remember that translation start site is w/ ATG on the target DNA, while TSS (transcriptional start site) is at the promoter

(to introduce precise change to the genome via CRISPR-Cas9 & HDR)
give answer & explain the thought process
(~eh) when looking at the starting DNA in black, how would you do HDR via SDSA if using the top strand as the one that FIRST STARTS annealing to the repair template?
which part would be the donor template vs 3’ overhang? explain the thought process
b/c:
donor template is similar to the target gene, so the donor template is complementary to the strand opposite of the target gene strand
SO
change the specific DNA strand to have the mutation
draw out the complementary sequence to that strand (b/c donor template is complementary to the non-target gene/strand & similar to target gene/strand
__
SDSA:
Cas9 cleaves & forms DSB in DNA
5’→3’ exonuclease forms 3’ overhangs (only 1 is indicated in the pic)
the indicated overhang will anneal to the homologous seq. on the repair template
will be extended via DNA pol. in the 5’ → 3’ direction
that newly synthesized strand will reanneal to its broken end
(not known, but complementary stand gap is filled in via DNA pol using newly synth. strand as template)
look at bottom pic for donor template & 3’ overhang
(You use the lower strand because HDR copies from a complementary template; the ssDNA donor is designed to match that template strand (5′→3′), except at the target base where the mutation (G→A) is introduced so that repair installs the desired change.)

for SCD
you can use gene editing to treat sickle cell disease that is caused by mutations in the __ gene
mutations in this gene will do what? (1)
t/f: nearly all sickle cell disease (SCD) patients have some type of variant that is treatable w/ gene editing
state 1 very common SCD variant
____
normally, not when have SCD:
relate beta- and gamma-globin
what leads to the decrease in gamma-globin expression at birth? explain how
…
SO summary: people w/ SCD will express __ levels of beta-globin, but the expressed beta-globin is __/abnormal
you can use gene editing to treat sickle cell disease that is caused by mutations in the beta-globin gene (HBB)
mutations in beta-globin gene will disrupt red blood cells that carry oxygen
true: nearly all sickle cell disease (SCD) patients have some type of variant that is treatable w/ gene editing
A-to-G mutation in HBB (beta-globin gene/hemoglobin subunit beta gene)
____
normally, not when have SCD:
gamma-globin expression decreases after birth
beta-globin expression increases after birth
gamma-globin expression is turned off at birth by the BCL11A transcription factor b/c:
BCL11A (TF) binds to an enhancer upstream of the gamma-globin gene & inhibits gamma-globin expression
…
SO summary: people w/ SCD will express NORMAL levels of beta-globin, but the expressed beta-globin is MUTATED/abnormal
for SCD
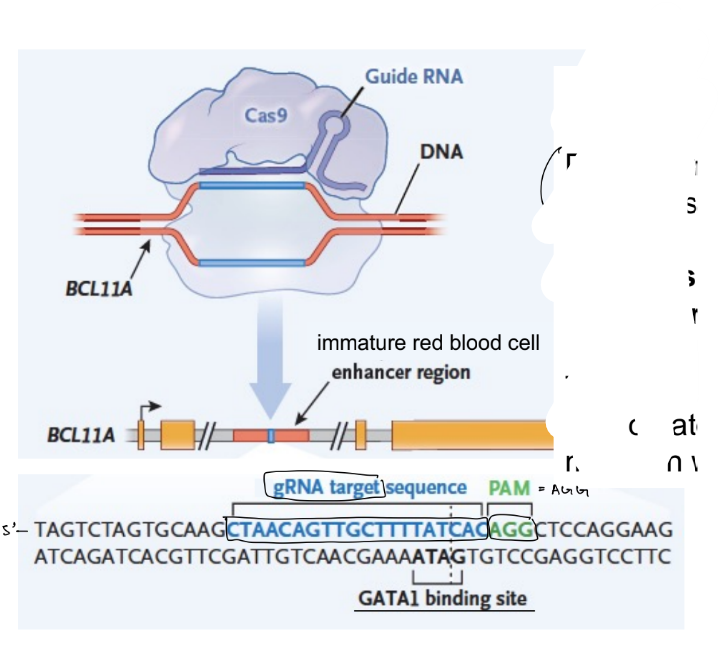
what is the COMMON gene editing treatment for SCD? (3 parts in one sentence)
specifically, you use CRISPR-Cas9 to (1 ← which usually/normally does what?) located in (1)
& within this enhancer, there is a …
why do you target the enhancer of BCL11A (gene)?
why not target the coding region of the BCL11A gene?
SCD treatment:
substitute mutant beta-globin w/ gamma-globin by prolonging gamma-globin expression via eliminating BCL11A in blood cells using CRISPR-Cas9
specifically, you use CRISPR-Cas9 to mutate an enhancer located in the intron of the BCL11A gene (where the enhancer that usually drives BCL11A expression blood cells)
& within this enhancer, there is a binding site for a TF necessary for BCL11A expression in red blood cells
target enhancer of BCL11A b/c want to selectively turn off/eliminate BCL11A expression ONLY in blood cells
if you target the coding region of BCL11A, then it can cause intellectual disability b/c BCL11A is expressed in multiple cell types & tissues
for SCD
if a donor template was NOT used. how was mutation in BCL11A enhancer created?
explain the steps/process
mutation in the BCL11A enhancer was created via NHEJ, so the indel mutation was random → which causes gamma-globin reactivation SO can treat SCD
cut out the enhancer located w/in intron of the BCL11A gene (via…)
NHEJ occurs to introduce a mutated enhancer / enhancer w/ indels
cause the TF binding site (aka GAT1 binding site) to break
SO enhancer stops working
SO BCL11A gene expression decreases
SO little to no inhibition of gamma-globin expression AKA gamma-globin reactivation, which will treat SCD
for SCD
what is the TAILORED gene editing treatment for SCD? (← incl. specific term & def.)
why is b/c normally, SCD is commonly caused by (1)
overview:
base editing involves what 2 types of base editors?
base editors create (3), rarely create (3)
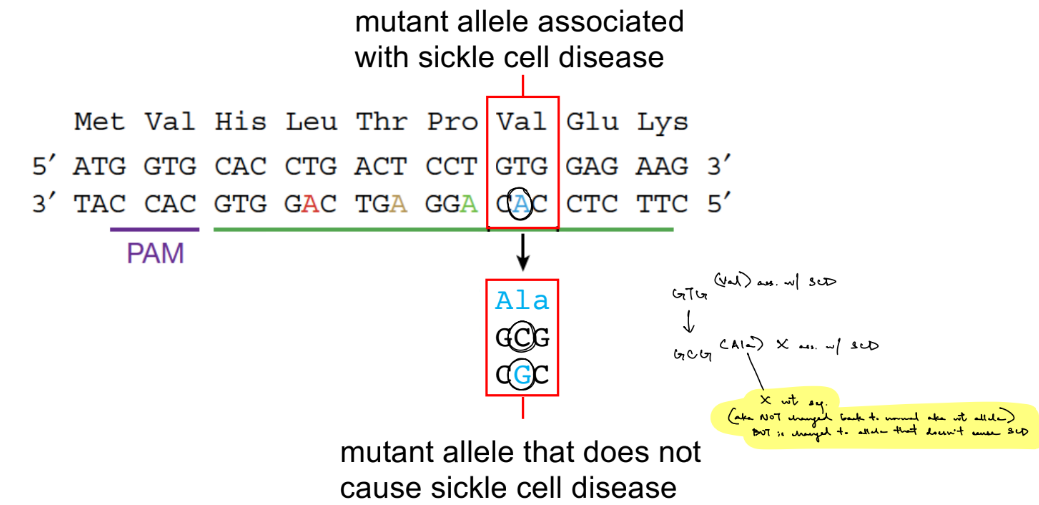
revert SCD by changing a single nucleotide via base editing ← combining CRISPR-Cas9 w/ nucleotide deaminases
b/c normally, SCD is commonly caused by missense mutation in the beta-globin gene (from wt allele w/o SCD → mutant allele ass. w/ SCD)
overview:
base editing involves what adenosine base editors & cytosine base editors
base editors create:
single nucleotide mutations
mutations of coding DNA (silent, missense, nonsense)
mutations of non-coding DNA (promoters, enhancers, mRNA splice sites, etc.)
base editors rarely create:
deletions (in general / small or large)
insertions (in general / small or large)
frameshift mutations
for SCD treatment via base editing (which is a TAILORED gene editing treatment for SCD)
purpose of adenosine base editor
steps of adenosine base editors (ABEs) (← 5)
_
why does adenine base editor (ABE) use Cas9 w/ nickase activity?
adenosine deaminase creates __ mutations
_
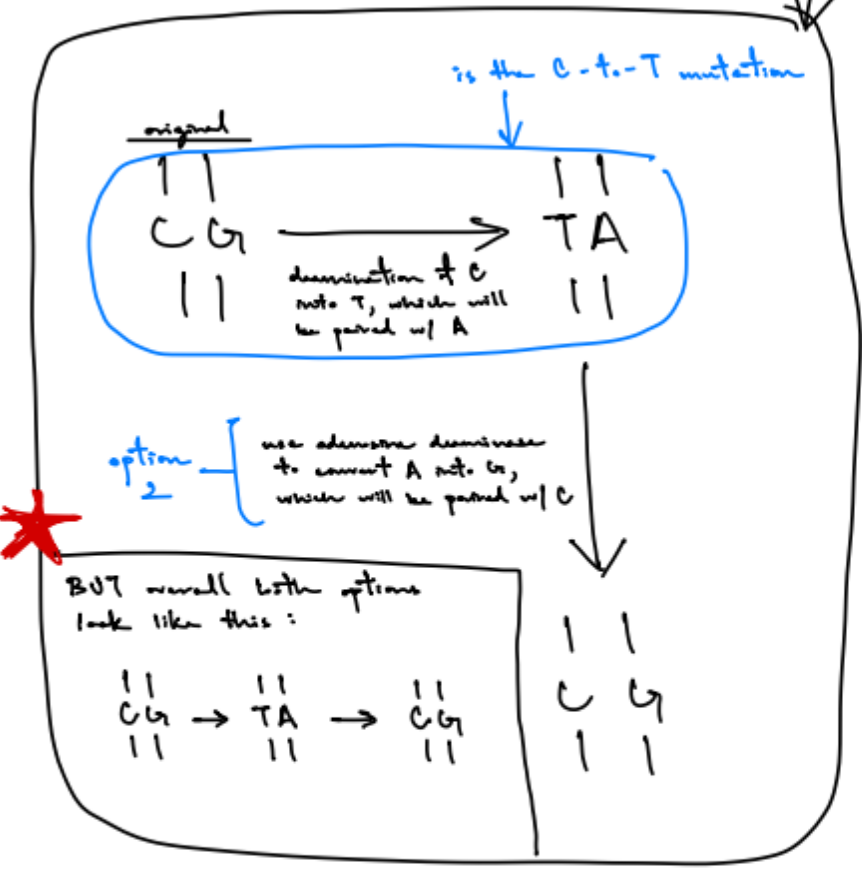
draw out adenosine base editing w/ A-to-G mutations
note: inosine does NOT pair w/ __, only pairs with (3)
targeted mutagenesis to change A into a nucleotide that does not cause SCD/disease
adenosine deaminase will attach to Cas9 nickase
adenosine deaminase use Cas9 nickase activity to deaminase A → I (inosine)
Cas9 nickase cleaves ONE strand of DNA / the unedited strand
T → C via DNA replication or BER (base excision repair), where BER uses the intact edited strand
(DNA repl. is cell divides, DNA pol pairs C to the I)
(BER is DNA mismatch repaire that adds C opposite to I)
convert I → G during DNA replication
_
adenine base editor (ABE) use Cas9 w/ nickase activity b/c it forces the cell to use I (inosine) as template
adenosine deaminase creates A-to-G mutations
note: inosine (I) does NOT pair w/ guanosine, only pairs with A, C, T
…
bottom pic shows adenosine base editing w/ A-to-G mutations
(top pic is what the cell would normally do)
for SCD treatment via base editing (which is a TAILORED gene editing treatment for SCD)
def. BER
what strand is used as template for repair
the Cas9 nickase directs the BER pathway to use the __ strand for repair, to __ the likelihood of incorporating the mutation (which neutralizes the disease allele from: mutant allele ass. w/ SCD → __ allele that __ cause SCD aka diff. from the wildtype allele)
overall, adenine base pairing __ the symptoms of SCD in a mouse model & in a patient
_
t/f: any adenosines in the gRNA could be mutated by the adenosine base editor b/c multiple codons have an A
BER — repairs changes to DNA that involve 1 or few nucleotides that don’t cause major change to DNA
use intact edited strand as template for repair
(for changing I/T → I/C, where “intact” is same I & “edited” is A → I)
the Cas9 nickase directs the BER pathway to use the intact edited strand for repair, to increase the likelihood of incorporating the mutation (which neutralizes the disease allele from: mutant allele ass. w/ SCD → mutant allele that doesn’t cause SCD aka diff. from the wildtype allele)
overall, adenine base pairing reverts/reverses the symptoms of SCD in a mouse model & in a patient
__
true
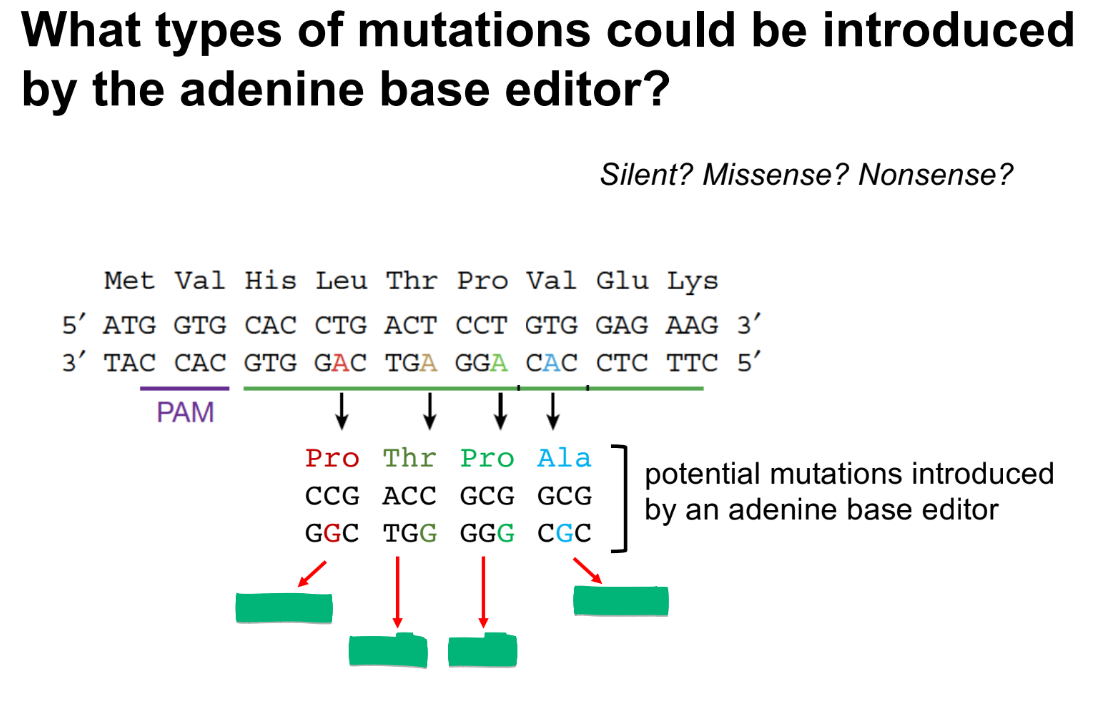
from L to R is:
missense, silent, silent, missense
for SCD treatment via base editing (which is a TAILORED gene editing treatment for SCD)
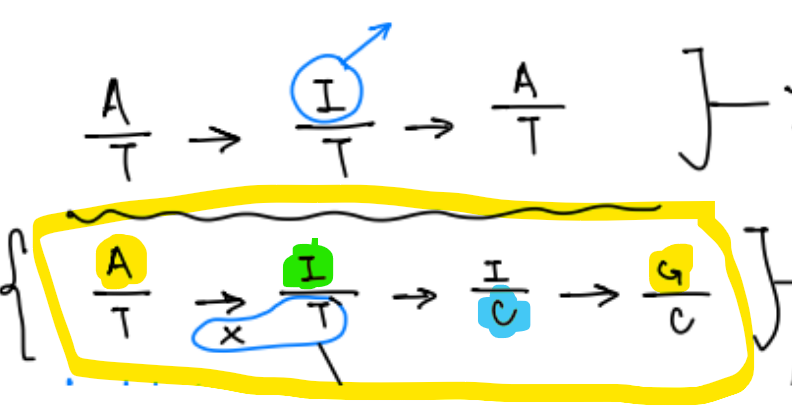
for cytokine base editing, it causes __-to-__ mutations that are highly ass. with cancers
2 ways to reverse this mutation
^ draw what this looks like
causes C-to-T mutations, highly ass. w/ cancers
reverse C-to-T mutations by changing:
T → C
changing A → G, then T → C
relate #’ ends of coding strand gene, #’ ends of mRNA, N or C-terminal or protein
5’ end of coding strand = 5’ end of mRNA = N-terminal of protein
(think N-terminal has positive amine groups that DNA moves away from b/c of “-” backbone & b/c DNA pol moves from 5’→3’, then N-terminal is at 5’ end)
3’ end of coding strand = 3’ end of mRNA = C-terminal of protein
for FXS
what are the 2 CRISPR-Cas9 approaches to reverse silencing of the FMR1 gene in FXS?
__
for FXS:
the gene that produces FMR1 is located where? it appears “fragile” b/c …
normal alleles for FMRI has about __ repeats of the CGG sequence in a 5’ UTR near the TSS, while FXS has __ repeats
CGG repeat expansion __ gene expression, so (what happens to the protein?)
__
in premutation state of FXS, FMR1 mRNA levels __, but protein levels __
in general:
mRNA levels increase due to __ transcription of the FMR1 gene
if protein levels decrease, it is b/c there is __ translation of the mRNA
use Cas9 to cleave near CGG repeats & do DNA repair to reduce the # of repeats
OR
use an enzyme-dead Cas9 to recruit a demethylase (Tet1)
__
for FXS:
the gene that produces FMR1 is located during in the “fragile” region of the X-chromosome; appear “fragile” b/c decreased chromatin compaction
normal alleles for FMRI has about 54 repeats of the CGG sequence in a 5’ UTR near the TSS, while FXS has >200 repeats
CGG repeat expansion SILENCE gene expression, so FMRP/FMR1 protein is not produced
__
in premutation state of FXS, FMR1 mRNA levels increase, but protein levels decrease
in general:
mRNA levels increase due to enhanced/increased transcription of the FMR1 gene
if protein levels decrease, it is b/c there is stalled translation of the mRNA
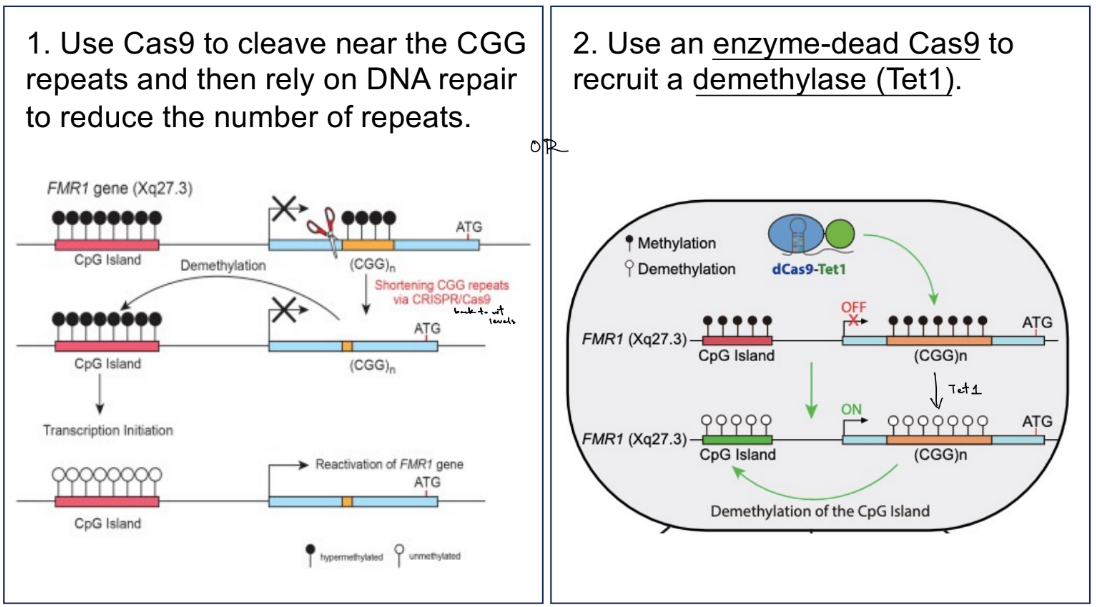
for the CRISPR-Cas9 approach to reverse silencing of FMR1 gene in FXS: w/ living Cas9
what is the method involving living Cas9?
(reminder that the CGG repeats are located in the 5’ UTR of the mRNA)
which DNA repair pathway will likely be targeted? why?
furthermore, HDR is only active in dividing cells BUT (is or is not?) active all the time in dividing cells; while NHEJ occurs in dividing & non-dividing cells, meaning that it is the most __ repair mechanism for repairing DSBs
^^^ knowing all this, state the very general steps of this CRISPR-Cas9 approach to reverse FMR1 silencing in FXS (4)
reducing the number of CGG repeats by doing Cas9 cleavage near the repeats & then DNA repair
use NHEJ
you don’t need precise deletions or changes to reduce the # of CGGs b/c the CGGs are located in the non-protein-coding region of the gene, SO can use imprecise repair method
furthermore, HDR is only active in dividing cells BUT IS NOT active all the time in dividing cells; while NHEJ occurs in dividing & non-dividing cells, meaning that it is the most ACTIVE repair mechanism for repairing DSBs
i.e. HDR is not active in post-mitotic cells, like in neurons
^^^ knowing all this, the general steps are;
identify PAM site
find gRNA target site (AKA design/form gRNA)
find Cas9 cleavage site → form DSB
then, do NHEJ repair
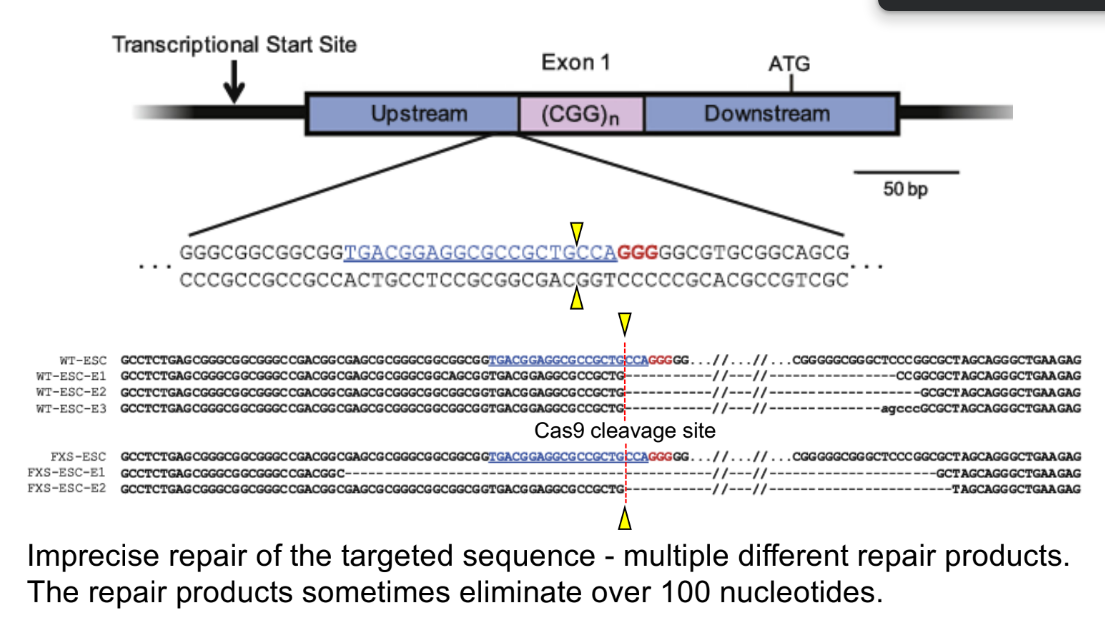
for the CRISPR-Cas9 approach to reverse silencing of FMR1 gene in FXS: w/ living Cas9
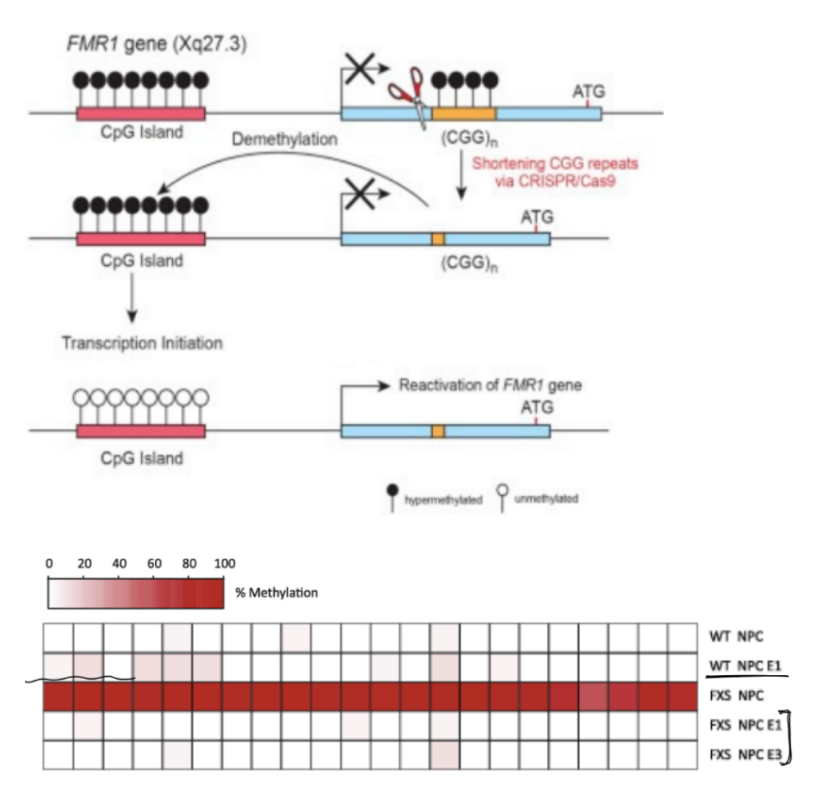
DNA is very/abnormally HIGHLY methylated in __ __ cells from __ patients, while DNA has low methylation in __/__ patients
these __ __ cells are treated w/ CRISPR-Cas9 to target the CGG repeats, to get:
(1 ← 1) cells that show NO change in DNA methylation patterns
(1 ← 2) cells show DECREASE in DNA methylation compared to FXS NPC cells, where DNA methylation is reduced to NORMAL WILDTYPE levels in these cells
__
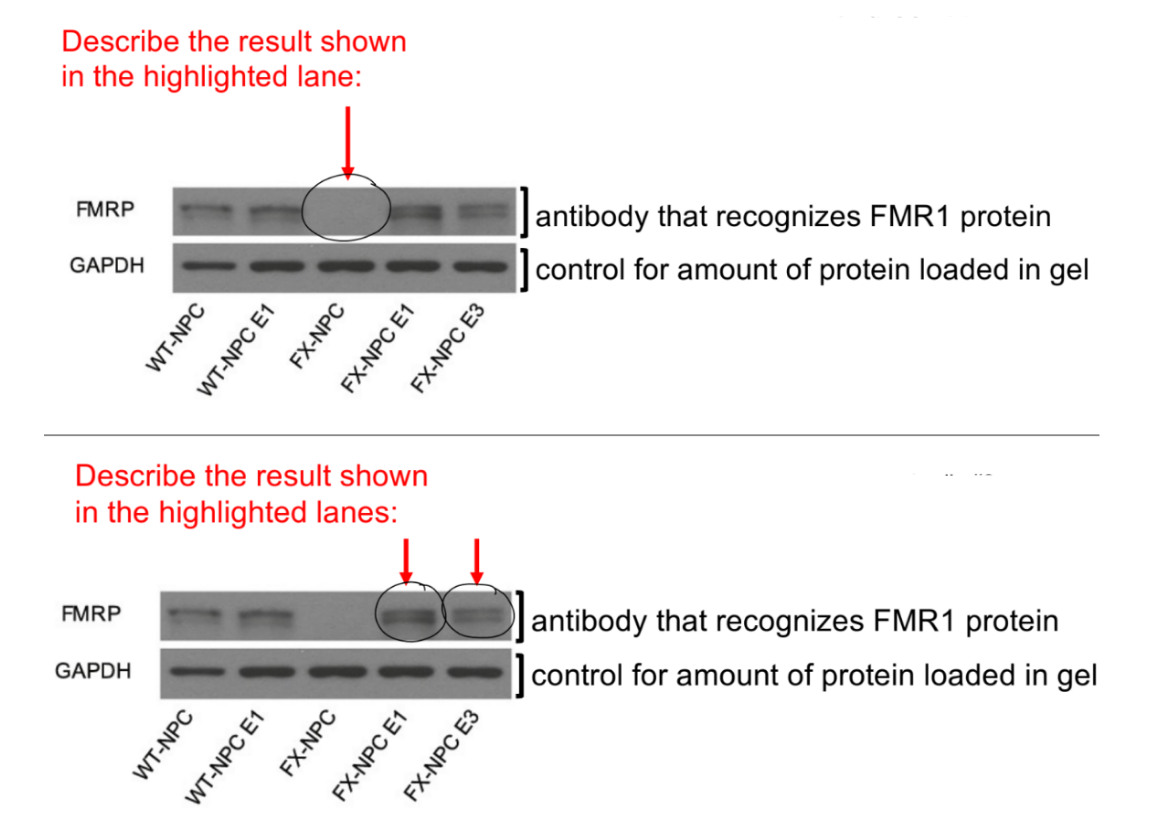
answer 2Qs in the pic, explain
conclusion (1)
DNA is very/abnormally HIGHLY methylated in neural precursor cells (NPCs) from FXS patients, while DNA has low methylation in normal/wildtype patients
these neural precursor cells (aka both wildtype & FXS NPCs) are treated w/ CRISPR-Cas9 to target the CGG repeats, to get:
edited wildtype cells (E1) cells that show NO change in DNA methylation patterns
edited Fragile X cells (E1, E3) cells show DECREASE in DNA methylation compared to FXS NPC cells, where DNA methylation is reduced to NORMAL WILDTYPE levels in these cells
(^ WT NPC, WT NPC E1 vs. FXS NPC, FXS NPC E1, FXS NPC E3)
__
(top pic) no FMR1 protein is expressed in Fragile X cells
b/c DNA methylation typically reduces gene expression, and FXS NPCs have a lot of highly methylated DNA / DNA with abnormally high amount of methylated NPCs in FXS patients
(bottom pic) there is now FMR1 protein being expressed in the Fragile X cells that were edited by CRISPR-Cas9
b/c CRISPR-Cas9 editing of FXS NPCs to get FXS NPC E1/3 will decrease DNA methylation back to normal wildtype levels
conclusion:
CRISPR-Cas9 can be used to edit FXS NCPs to restore FMR1 protein (restore FMR1 gene expression)
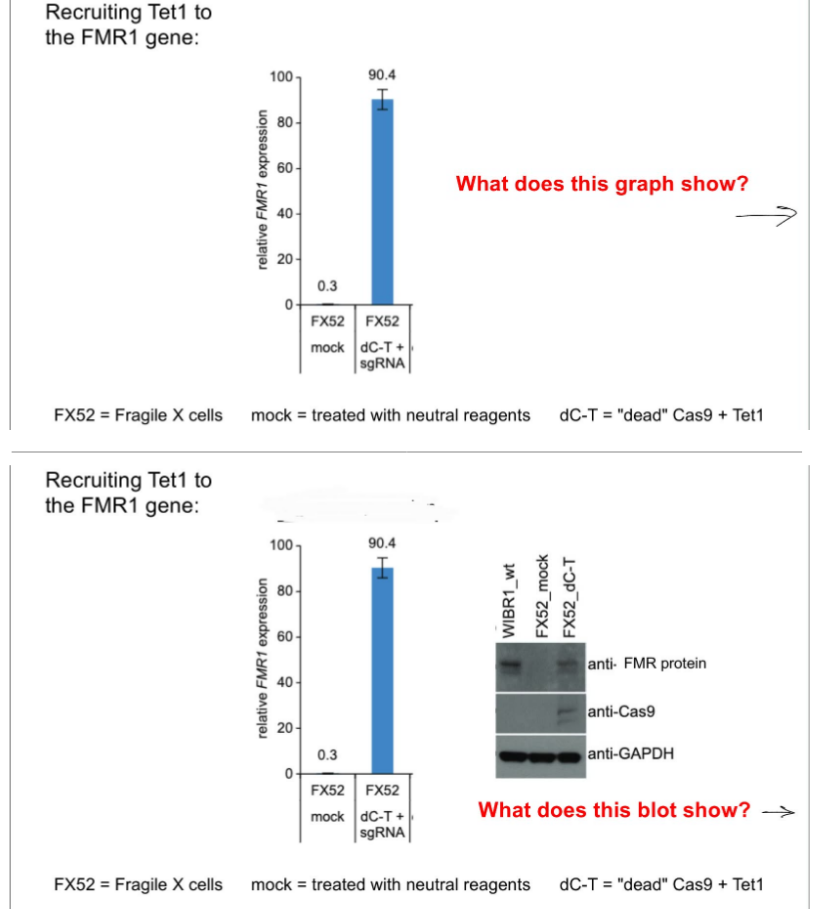
for the CRISPR-Cas9 approach to reverse silencing of FMR1 gene in FXS: w/ enzyme-dead Cas9
what is the method involving enzyme-dead Cas9?
state the steps of this CRISPR-Cas9 approach to reverse FMR1 silencing in FXS (1 step w/ 2 results)
_
answer the 2Qs in pic (don’t have to explain/self-explanatory)
use enzyme-dead Cas9 to recruit regulators for transcription and chromatin, specifically cytosine demethylase called Tet1, to reverse silencing of the FMR1 gene
(where there can be many diff. enzymes recruited b/c many functions of transcription activator/repressor, DNA demethylase, histone acetyltransferase, methyl transferase)
steps:
the enzyme-dead Cas9 recruits Tet1 to the FMR1 gene, where:
dCas9-Tet1 / dC-T will increase FMR1 gene expression
as a result, FMR1 protein levels increase in cells treated w/ dCas9-Tet1
__
(top pic) dCas9-Tet1 / dC-T recruited to the FMR1 gene will increase FMR1 gene expression
(bottom pic) FMR1 protein levels increase in cells treated w/ dCas9-Tet1
whereas, FX52_mock where there’s no dCas9 or Tet1 (is just the control) shows little to no FMR1 protein expression b/c of abnormally high DNA methylation at FXS NPCs that reduce FMR1 gene expression and resulting FMR1 protein expression
CRISPR-based treatments are already showing promise in treating patients with sickle-cell disease.
SO why are CRISPR-based treatments not being used to treat patients with neurological disease at this time? (2 similar reasons)
for 1 of the reasons, explain why it is hard (aka the process)
t/f: you can’t just inject Cas9 and gRNA into somatic cells & expect it to cause precise mutations in specific target genes (i.e. mutated enhancers in BCL11A gene in blood cells)
b/c unlike blood cells, you can’t isolate neurons from patients, edit the neurons’ genomes, and re-inject these edited genomes into the patient
these neurological diseases (i.e. FXS) are affecting neurons, which are mostly made in utero, SO would have to treat patients in utero which is unethical
&
b/c it's hard to deliver CRISPR-Cas9 gene editing parts like Cas9 and gRNA into cells
^ CRISPR-Cas9 targets somatic cells by:
isolating somatic adult stem cells from the patient & injecting these cells w/ CRISPR reagents
identify cells that have the intended/wanted gene edits & inject those specific gene-edited stem cells back into the patient
^ true: you can’t just inject Cas9 and gRNA into somatic cells & expect it to cause precise mutations in specific target genes (i.e. mutated enhancers in BCL11A gene in blood cells)
(there are many biological, chemical, and physical methods to introduce these CRISPR-Cas9 gene editing components into somatic cells)