Modern Proteomics Methods
1/26
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
27 Terms
Why is proximity labeling used in proteomics?
traditional MS struggles w/ weak/transient protein interactions and membrane proteins
proximity labeling solves this by tagging proteins near a protein of interest inside living cells
captures native, endogenous interactions that can be identified by MS
we attach an enzyme to a protein of interest (via ligand/antibody) → enzyme generates a highly reactive species (often free radical) which labels nearby proteins within a few nm
labeled proteins are then purified and identified by MS
How is the GLP-1 receptor targeted for proximity labeling in the GLP1 paper
Ligand-based targeting:
GLP-1 (ligand) is conjugated to an enzyme → GLP1 binds GLP-1R → enzyme sits near receptor → initiates labeling reaction
Recombinant tagging approach (transgene):
GLP-1 or receptor is genetically fused with labelling enzyme
Notes: GLP1 attached to APEX → GLP1 binds to receptor → biotin phenol is deposited onto protein of interest (biotin phenol is what is deposited and activated)
APEC uses H2O2 to oxidize biotin phenol to produce radicals
can later be isolated w/ streptavidin cuz it binds biotin with high affinity
ensures controlled, specific labeling
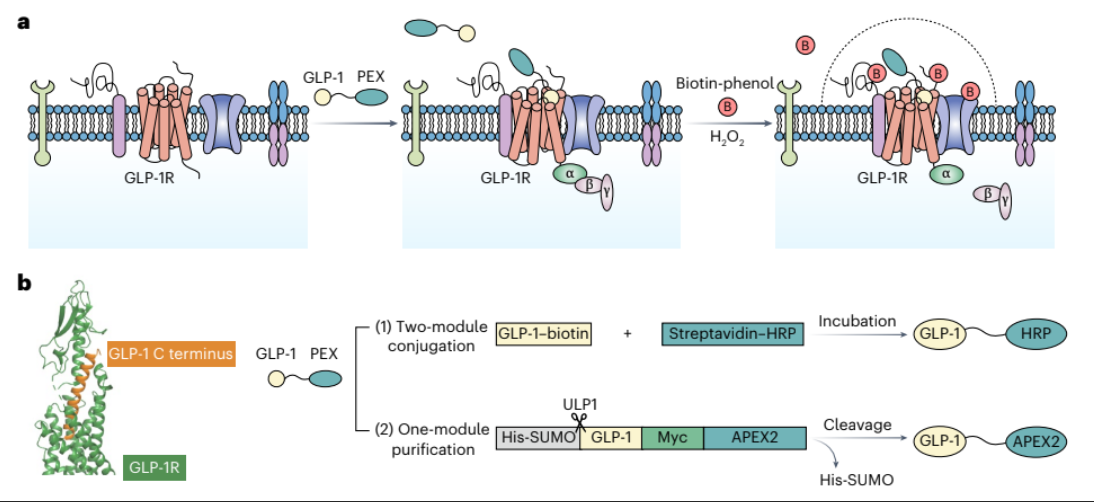
Results of proximity labeling
measuring cAMP production (downstream effect of GLP1 binding)
right = transgenically fused approach
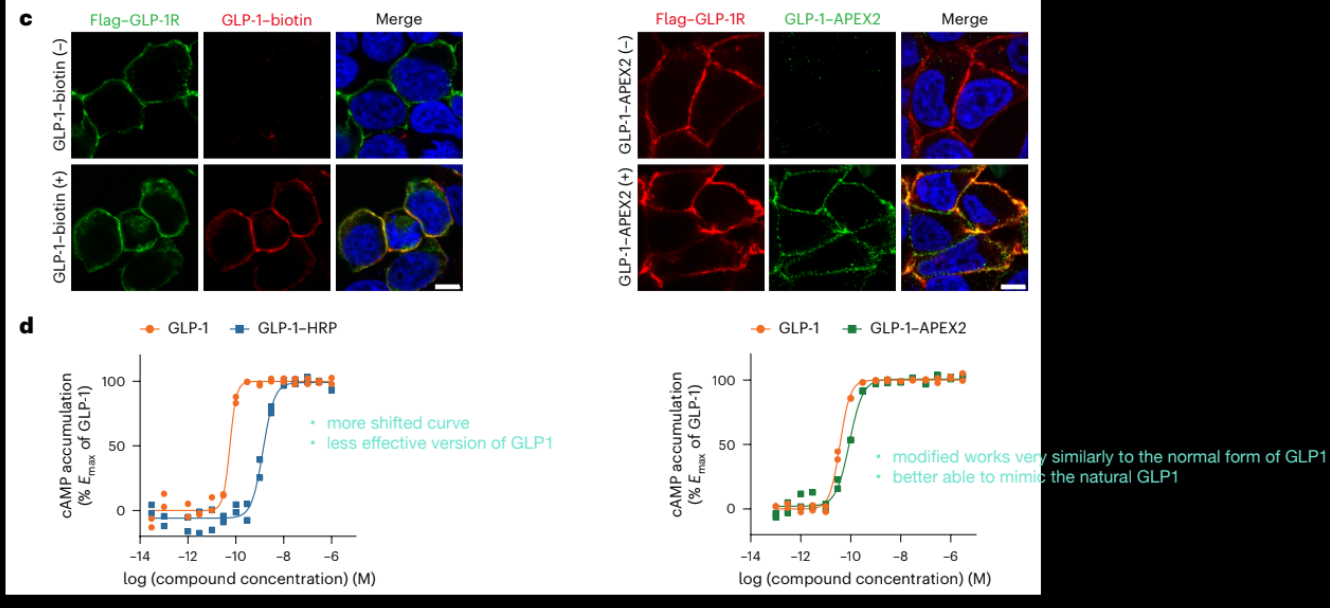
Why is observing membrane labeling important in the GLP-1 proximity labeling experiment
GLP-1R is a cell membrane protein
if the system is working correctly, labeling should occur at the membrane (ie ligand is binding correctly and enzyme is positioned at the receptor)
we see labeling when all components (GLP-1-enzyme, biotin-phenol, H2O2) are in
if labeling occurs only when everything is present, it confirms specific enzymatic labeling
Membrane-localized staining of GLP-1 construct
membrane localized staining tells us that GLP-1 is successfully binding to GLP-1R and the conjugated enzyme is correctly localized at the membrane
therefore the GLP-1 construct is functional
proper localization is essential b/c proximity labelling tags everything nearby
if localization is wrong, you label the wrong proteins and get meaningless data
correct localization ensures the labeling reflects true biological neighbors
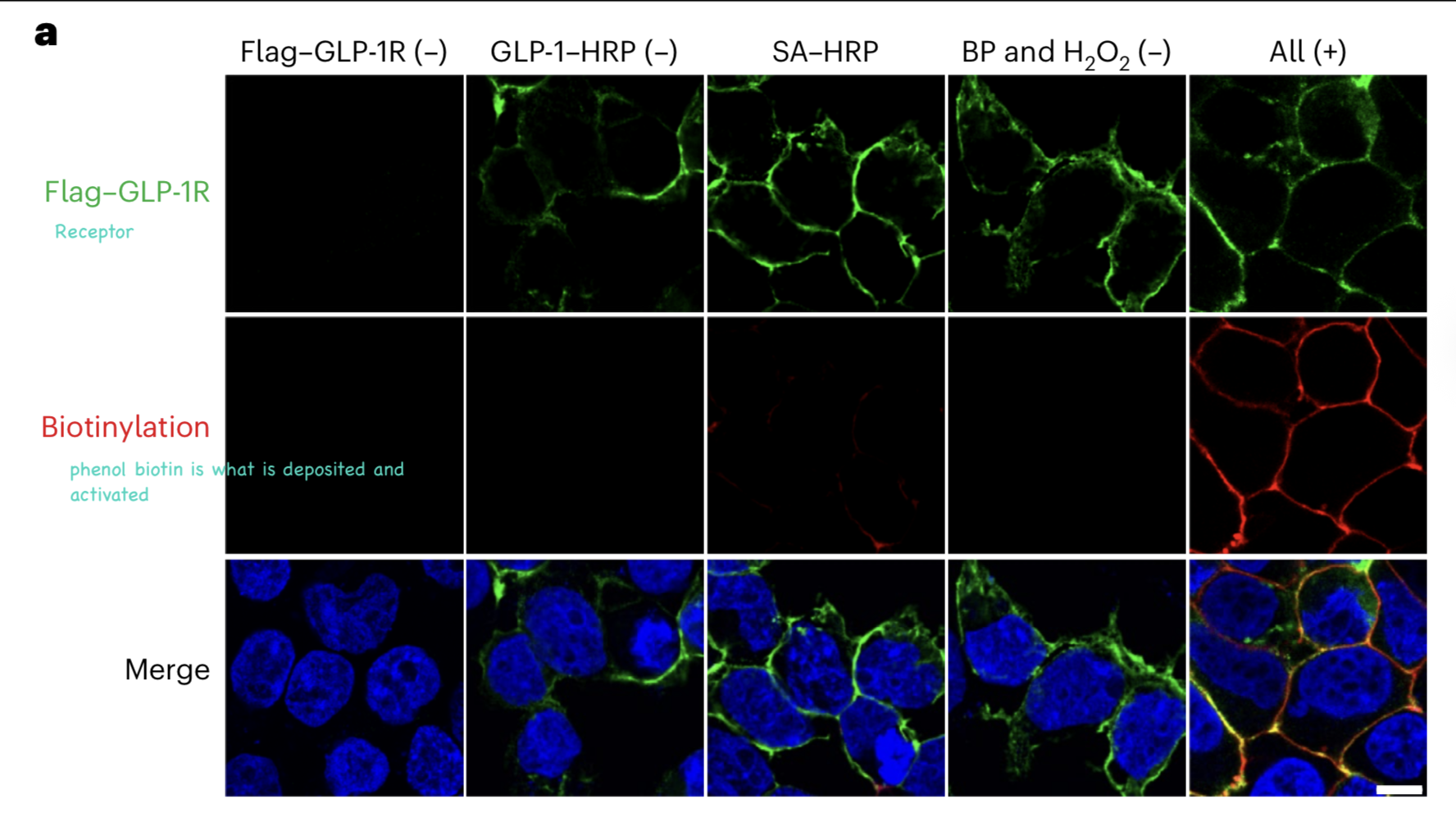
Biotin-Enrichment Approach
previously, immunofluorescence shows localization and the proteomics workflow identifies which proteins are present
biotin enrichment allows isolation of all proteins near GLP-1R
MS then identifies and quantifies these proteins
limitation: cannot confirm direct physical binding to GLP-1R b/c labeling is based on distance, not binding
Biotin-Enrichment Approach: Steps
perform labeling (biotin-phenol + H2O2)
lyse cells
add streptavidin-coated beads
biotinylated proteins will bind strongly to streptavidin
wash away non-biotinylated proteins
analyze bound proteins by MS
Roepstorff-Fohlmann-Biemann Nomenclature: What determines where the charge goes?
when a peptide bond breaks, one fragment keeps the charge → detectable in MS
charge on N-term → b-ion
charge on C-term → y-ion
if a peptide has more than one charge, we can produce both b and y ions
when fragmenting peptides, we put it enough energy to break one bond per peptide (no t too much)
Roepstorff-Fohlmann-Biemann Nomenclature: Energy
when fragmenting peptides, we put it enough energy to break one bond per peptide (not too much)
we want clean, interpretable fragment ions
if too much energy → multiple bonds break
each peptide molecule may break at a different position, and across many molecules, we could get a series of fragments covering the sequence
Residue mass vs amino acid mass
Amino acid mass:
free amino acid (includes full structure)
Residue mass:
amino acid within a peptide minus H2O (lost during peptide bond formation)
MS calculations use residue masses, not free amino acid masses
Read off the sequence: How do you determine a peptide sequence from a MS/MS spectrum?
use b-ion or y-ion series
look at mass difference between adjacent peaks, each difference = one amino acid residue mass
build sequence step-bystep
Why is a y₁₀ ion impossible for a 10 amino acid peptide?
fragmentation breaks peptide bonds between residues
therefore the max y-ion is y9
for n amino acids → max fragment = n-1
Parent Ion: What does [M+2H]2+ represent?
the intact peptide (original ion, without any fragments missing)
M= m/z + 2 protons → we have the whole peptide + 2 protons (2+ charge state)
seeing it means fragmentation was incomplete, but it’s useful because it confirms expected peptide mass
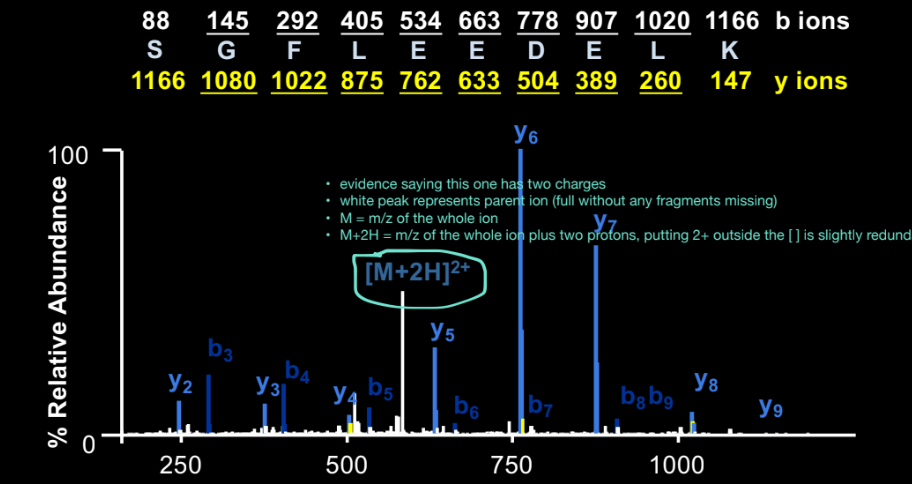
Why are singly charged fragments easier to interpret?
for z = 1, m/z ≈ actual mass + 1 proton
easy to calculate: subtract proton mass to get fragment mass
no need to divide by charge
the spectra is showing a full series of ions that are all singly charged
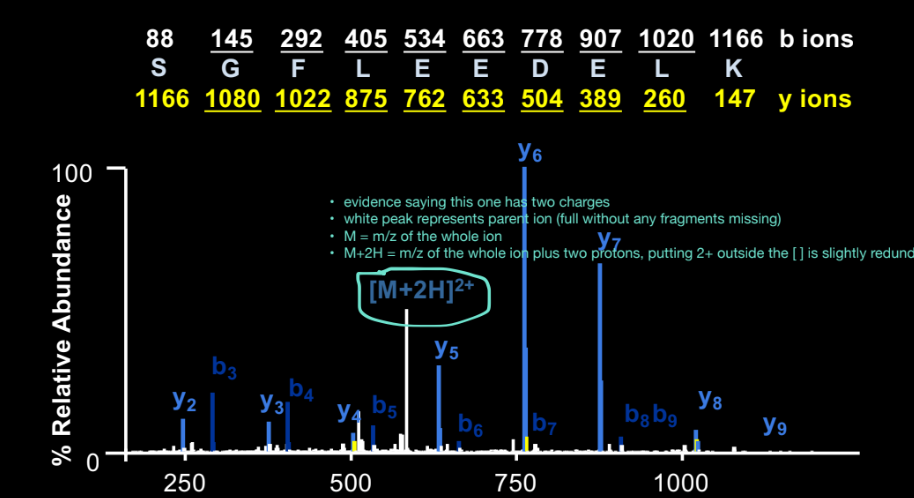
How do you use y-ion differences?
take 2 adjacent y-ions (e.g. y7 - y6), calculate mass difference and match the difference to the residue mass of an amino acid
Why are middle fragments more abundant?
bonds in the middle of the peptide break more easily and the gragments generated from cutting in the middle are more stable than very big or very small fragments
exception: Proline, remember that the bond before proline is more labile, resulting in strong fragment peaks at that position
Why don’t we always see b- and y-ions
fragmentation is probabilistic (not every bond breaks equally)
some fragments are unstable or have low abundance
you can still determine the sequence if some ions are missing by using nearby ions and known peptide mass
Why does a C-terminal K suggest a tryptic peptide
trypsin cleaves after Lys or Arg, so peptides often end in K or R
De novo sequencing
determining peptide sequence directly from MS/MS spectra
uses mass difference between fragment ions and doesn’t rely on protein databases
advantage: can recover full amino acid sequence directly, useful when protein is not in database or detecting mutations or novel peptides
disadvantages: high error rate, slow, computationally intensive, often the full sequence information is not present
use as backup option and to find ‘stretches’ of correct sequences (infer sequence directly from fragment ions)
primary method is to match spectra to known protein sequences (database search)