L16- Stem cells and cellular plasticity in cancer
1/50
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
51 Terms
what is a stem cell
An undifferentiated cell which is capable of indefinite self-renewal and has the capacity to give rise to other cell types via differentiation.
how are stem cells produced
Cell division produces two daughter cells:
- One maintains stem properties (self- renewal)
- One differentiates to give rise to committed progenitors
what are the types of stem cell
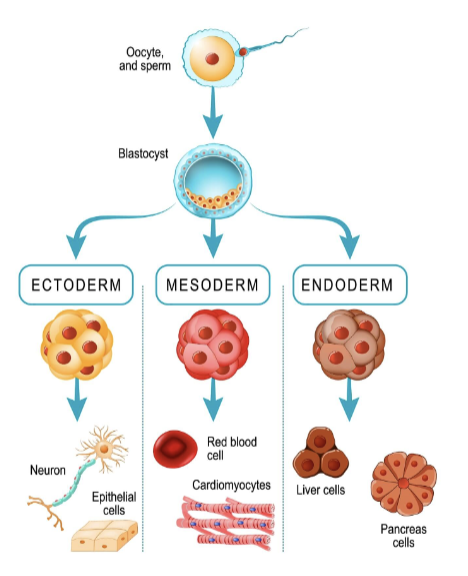
Totipotent (e.g. zygotes)- Differentiate into any cell type
Pluripotent (e.g. embryonic stem cells)- Differentiate into cells from any of the three germ layers
Multipotent/Oligopotent (e.g. tissue- specific stem cells)- Differentiate into a limited number of cell types
Unipotent- Differentiate into a single cell type
what are the 3 germ layers
ectoderm
mesoderm
endoderm
what is the history of stem cell research
• 1968 first bone marrow transplant to treat severe combined immunodeficiency
• 1978 Stem cells were discovered in human cord blood
• 1981 First stem cell line developed from mice
• 1995 First embryonic stem cell line derived from a primate
• 1996 Mammal cloned from a hybrid stem cell (nucleus from a mammary gland cell injected into an empty egg cell)
• 1994 First cancer stem cell identified in human leukaemia
How were cancer stem cells first identified in acute myeloid leukaemia (AML) and what are their key features?
First identified in 1994 (John Dick’s team) in AML
Only a small subset of cells could repopulate leukaemia in immunocompromised mice
These cells have a stem-like phenotype (CD34⁺ CD38⁻)
Resemble normal haematopoietic stem cells (HSCs)
Suggests leukaemia may originate from or mimic normal stem cell populations
wha are the 2 models of carcinogenesis
classical stochastic model
cancer stem cell (CSC) model
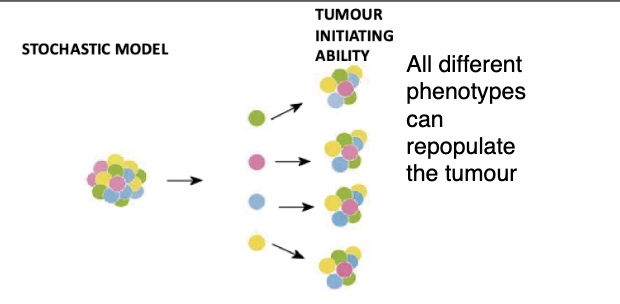
what is the classical stochastic model of carcinogenesis
Cancer can arise in any cell and can be propagated by any cell in the population (or most cells at least)
Cancer develops through a process of continual mutation and clonal selection of the fittest clones (Darwinian selection).
Allows it to populate and continue tumour growth
what is the cancer stem cell (CSC) model of carcinogenesis, what does ot say about incidence
Cancer arises in a cell that has or acquires stem cell properties of self-renewal
Tumours display some level of hierarchical organisation with the CSC at the apex of that hierarchy.
Incidence of cancer is directly proportional to the number of stem cell divisions in that tissue (Vogelstein model).
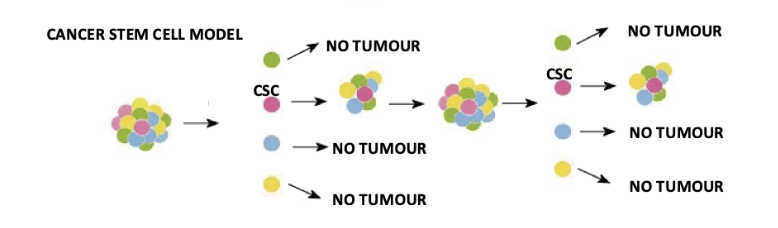
what is the hierarchical structure of the cancer stem cell model
Cancer stem cells (CSCs) sit at the top and can self-renew
Only CSCs can repopulate and sustain tumour growth
Other tumour cells are more differentiated and have limited proliferative capacity
Evidence requires serial transplantation showing tumour regrowth from CSCs
non malignant tissues also have a hierarchical structure
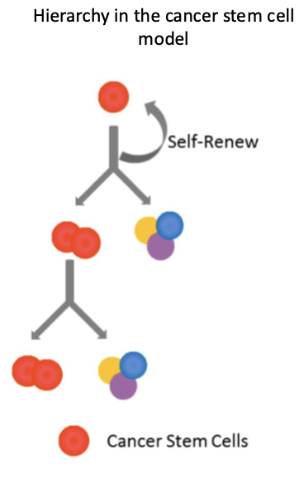
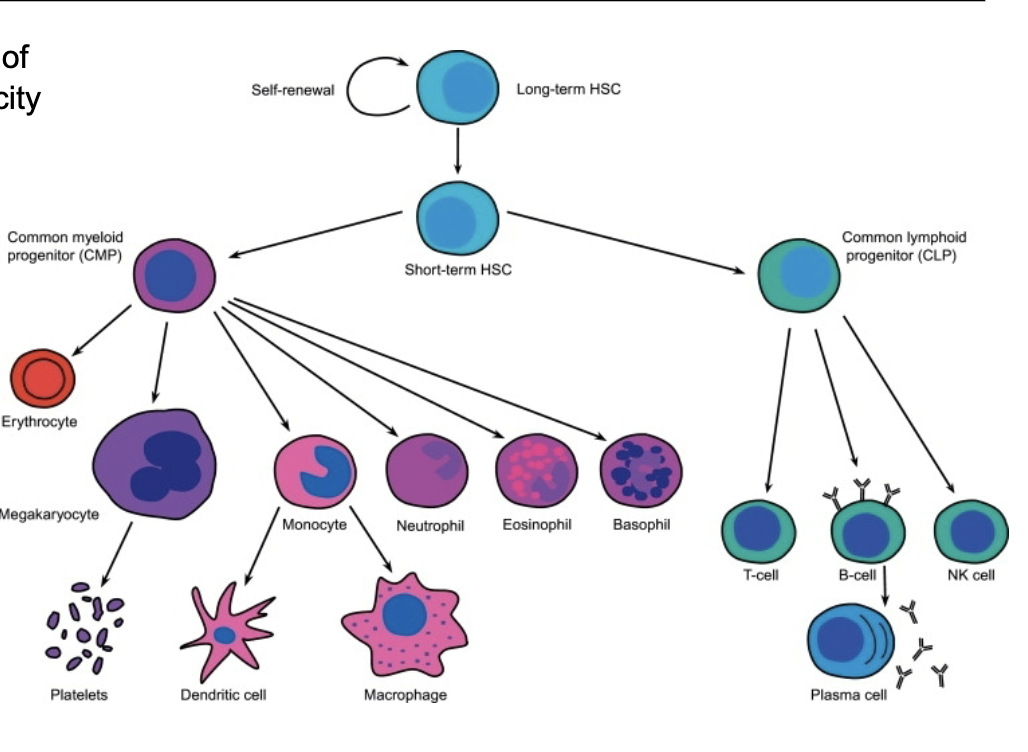
What characterises the normal haematopoietic stem cell hierarchy
HSCs at the top with self-renewal ability
Give rise to lymphoid and myeloid progenitors
Cells undergo controlled proliferation and differentiation
Leads to fully differentiated mature blood cells
Clear hierarchy based on increasing differentiation
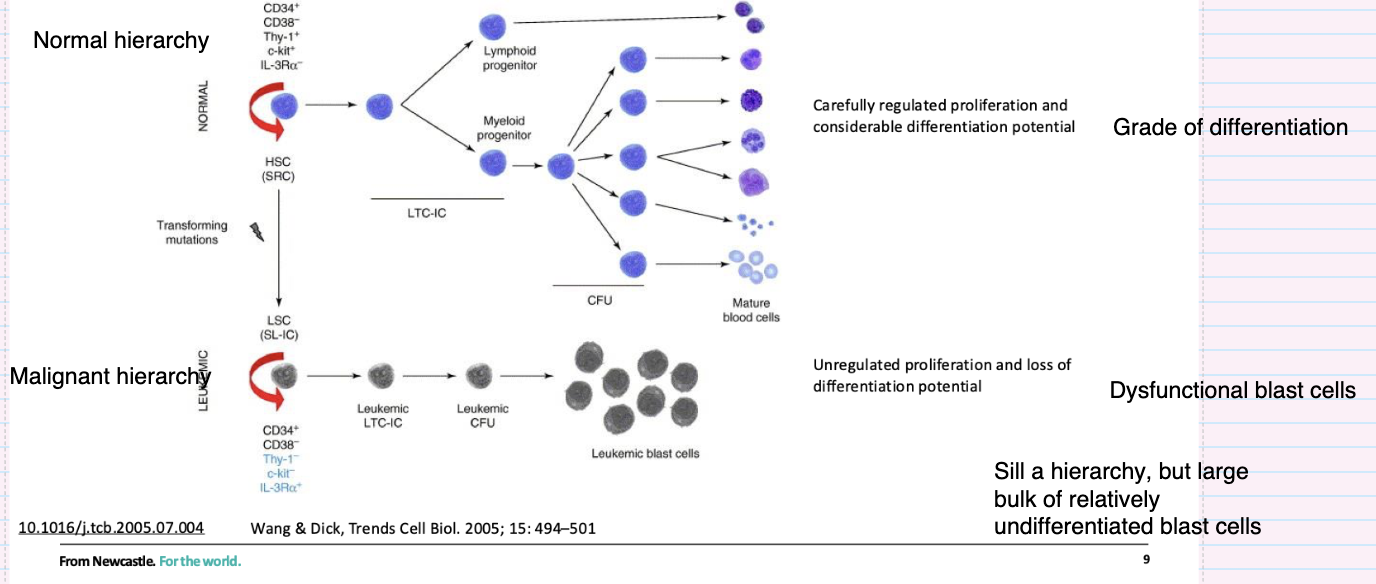
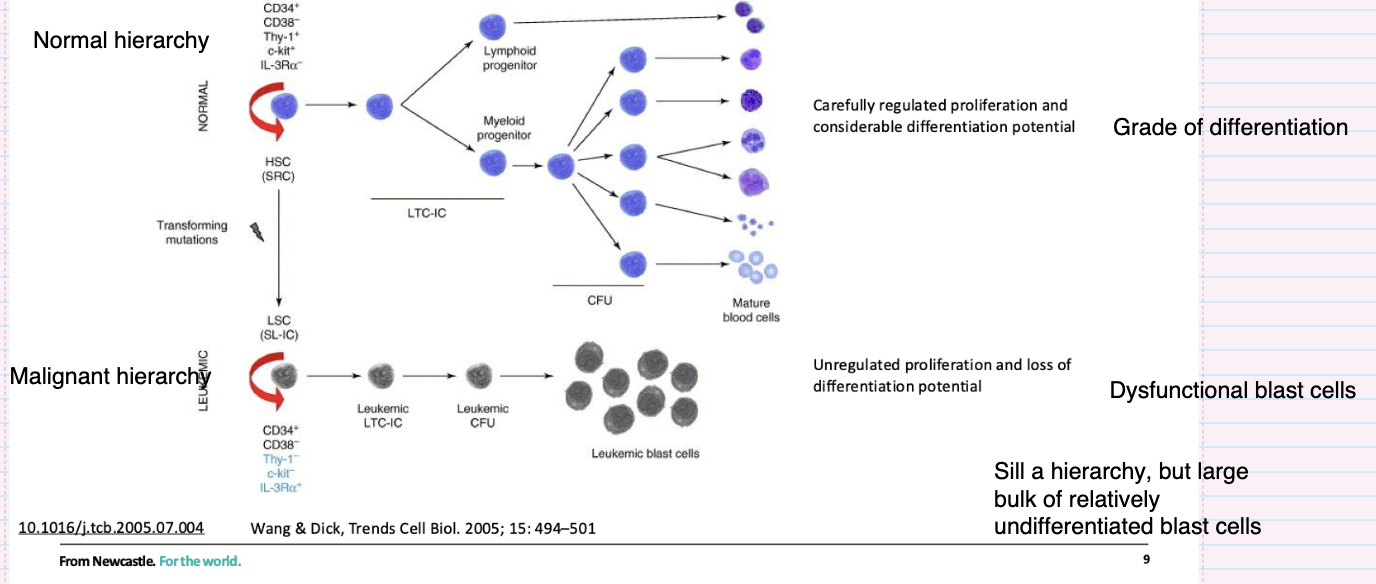
How does the leukemic stem cell hierarchy differ from the normal hierarchy?
Originates from transforming mutations in stem/progenitor cells
Leukaemic stem cells (LSCs) retain self-renewal
Produces a hierarchy but with impaired differentiation
Results in accumulation of immature blast cells
Characterised by uncontrolled proliferation and dysfunctional cells
How have cancer stem cells been identified in solid tumours?
Identified by specific surface markers (phenotype) (e.g. CD44⁺, CD133⁺)
Defined by ability to initiate tumours in recipient mice (functional test)
Demonstrated in prostate and colon cancers
prostate- CD44+, Ɑ2β1high, CD133+
colon- CD133+
Supports idea that only a subset of tumour cells drive tumour growth
give examples of different cancers and their marker types
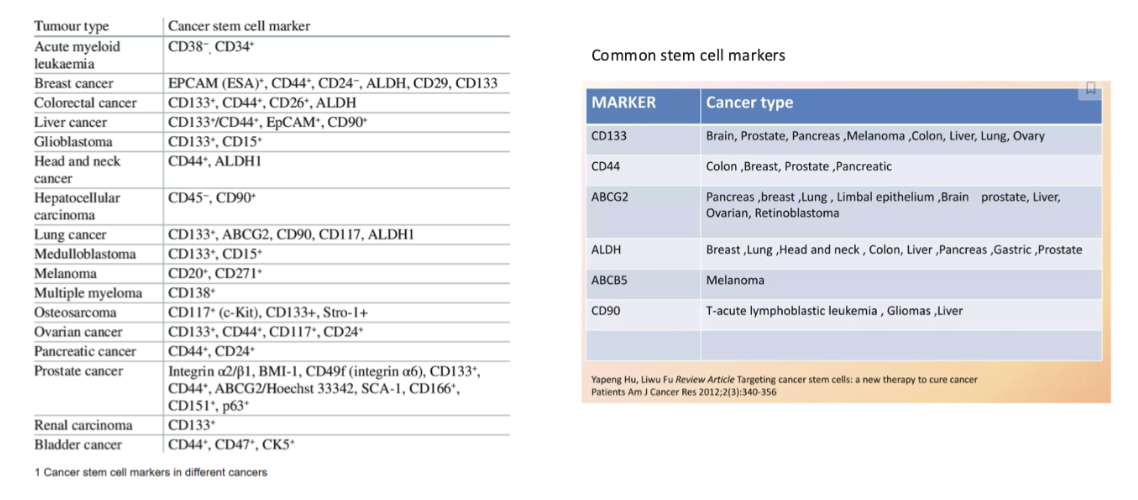
what is CD133
aka Prominin-1 is a transmembrane glycoprotein.
Precise function/activity is unknown
“Regulates” numerous pathways associated with stem cell characteristics
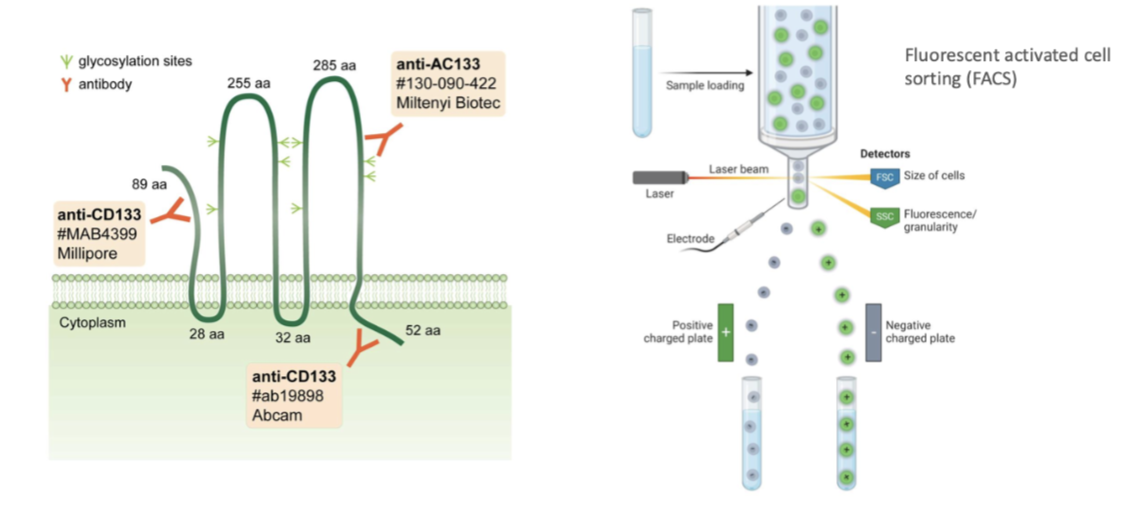
how can canser stem cells be isolated
Use markers to isolate stem cells
Commercial organisations released Abs that bind to CD133, bind to cells and isolaye the cells
Eg through fluorescence activated cell sorting dependent on if they're positive for CD-133 or not
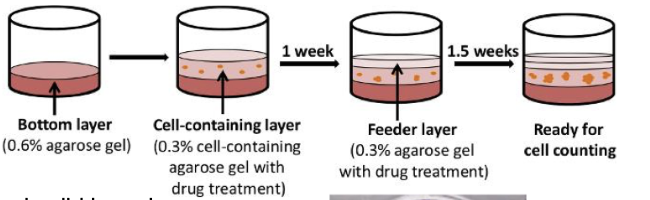
describe the functional identification of cancer stem cells via colony formation in vitro
Colony formation in vitro
• Ability to form colonies when cultured from single cells (property shared by both normal and cancer stem cells)
• Serial passage of colonies and retention of colony formation ability
key is if they can form colonies in vitro- serially
what are the limitations of the functional identification of cancer stem cells (colony formation in vitro)
clonogenicity influenced by the tumour micro-environment
Expensive cytokines or feeder layers are often required
describe the functional identification of cancer stem cells via serial transplantation in vivo
Another major mechanism to test for cancer stem cells
forms cancer in transplanted animal
serial transfer of cancer into subsequent animals
Transplant into mice
Leukaemia- normally In bone marrow in femur, see whether cells can form leukaemia in that setting
Called an orthotopic model
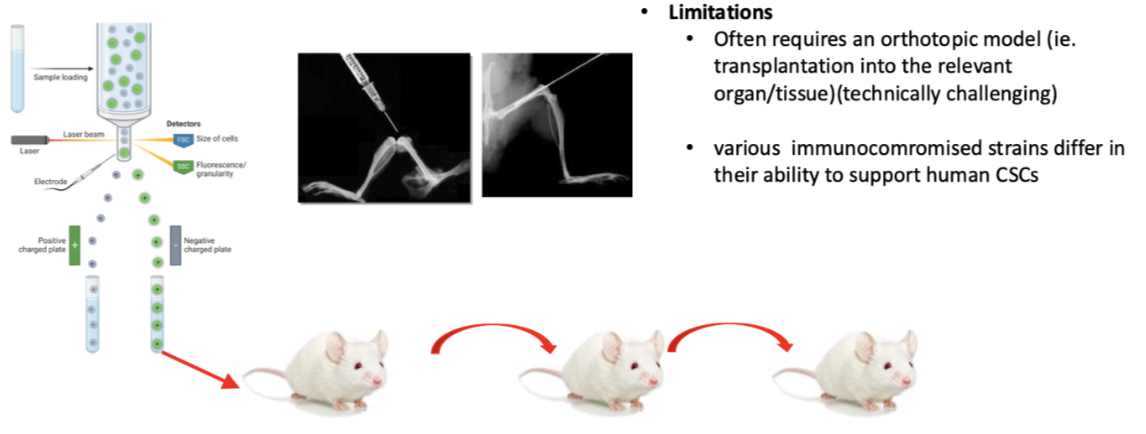
what are the limitations of the functional identification of cancer stem cells ( serial transplantation in vitro)
Hard to set up systems
Easy to get to femur but hard in breast to inject into mammary fat pad and prostate as very small
Often requires an orthotopic model (ie. transplantation into the relevant organ/tissue)(technically challenging)
Also need to be in immune compromised mice so foreign human cells aren't recognised- different immunocompromised strains of animals differ in ability to support human CSCs so results don’t necessarily mean there is specific cancer types in sample
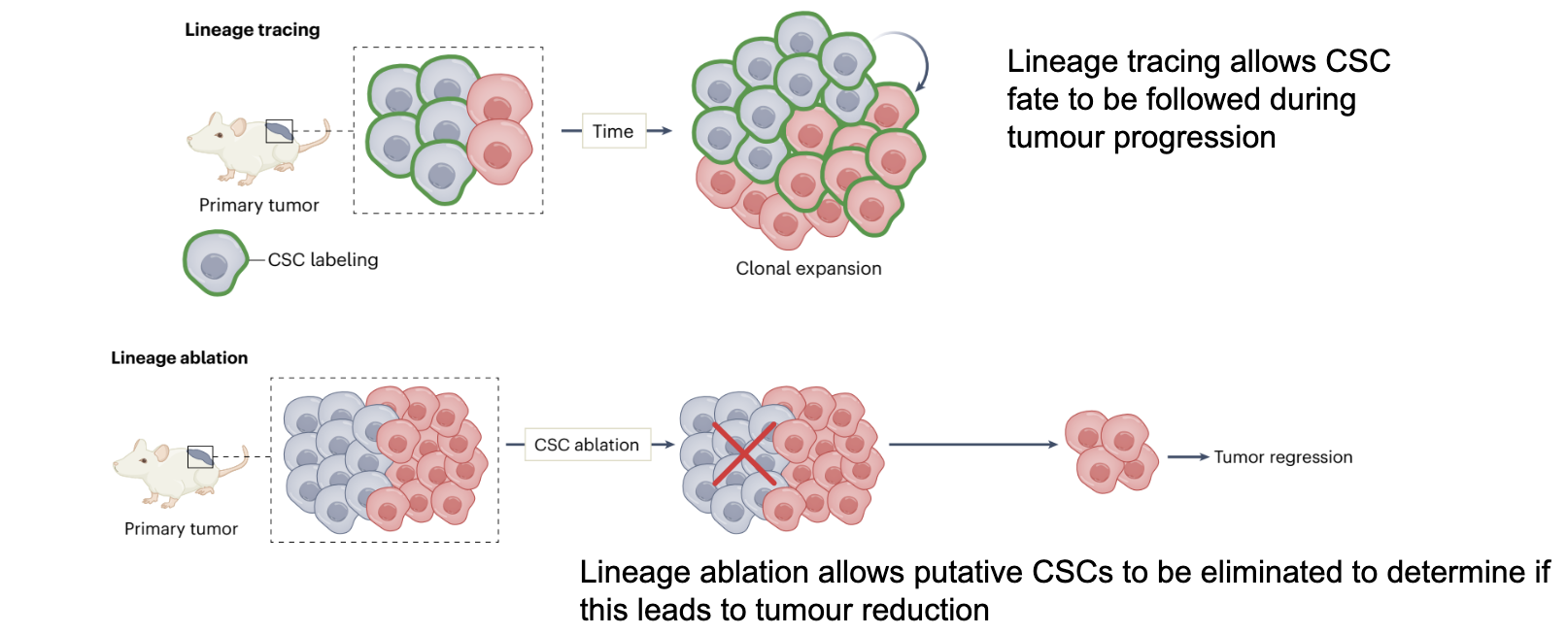
what is Lineage tracing and lineage ablation
Stem cells can also be functionally identified by lineage tracing/ablation experiments
Lineage tracing allows CSC fate to be followed during tumour progression
Lineage ablation allows putative CSCs to be eliminated to determine if this leads to tumour reduction
What happens when cancer stem cells (CSCs) are specifically targeted in therapy?
CSC-targeted therapy removes tumour-initiating cells
Tumour cannot sustain itself
Leads to gradual tumour degeneration
Prevents long-term regrowth
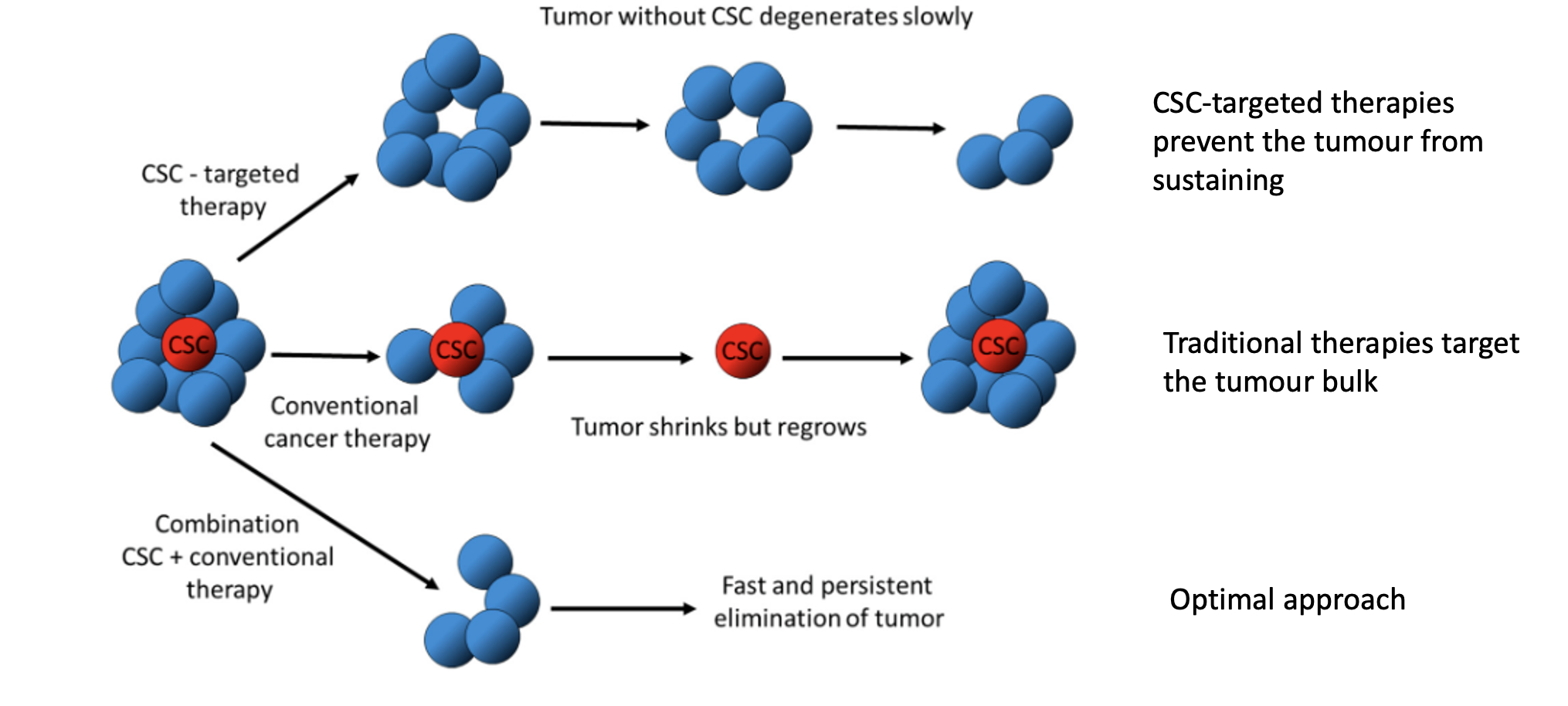
Why can conventional cancer therapies lead to tumour relapse?
Target mainly the bulk tumour cells, not CSCs
CSCs survive treatment
Tumour may initially shrink but regrows from CSCs
Explains relapse and treatment resistance
What is the optimal therapeutic strategy for treating cancers with CSCs?
Combine CSC-targeted therapy with conventional therapy
Eliminates both tumour bulk and stem cells
Leads to more complete and sustained tumour eradication
Results in faster and more durable response
what is another possible function of CSC markers
they are also attractive therapeutic targets
eg CD44 and CD133
how can CD44 be used as a therapeutic targets
CD44 is a major surface hyaluronic (HA) receptor
Both HA and CD44 are involved in chemotherapeutic resistance
CD44 can interact with P-gp (/P-glycoprotein, a major drug efflux pump) to promote cell migration and invasion of tumor cells
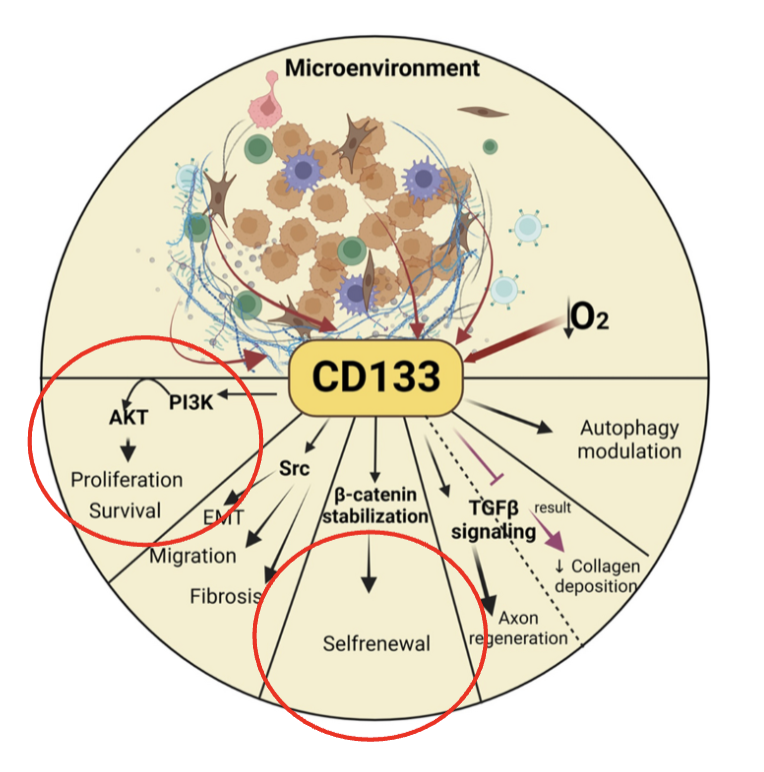
how can CD133 be used as a therapeutic targets
CD133, a member of the prominin family, consists of five transmembrane single-chain glycoproteins
CD133 can play a dominant role in drug resistance
CD133 can enhance the activity of histone deacetylase 6 (HDAC6) via the Wnt/β-catenin signaling pathway
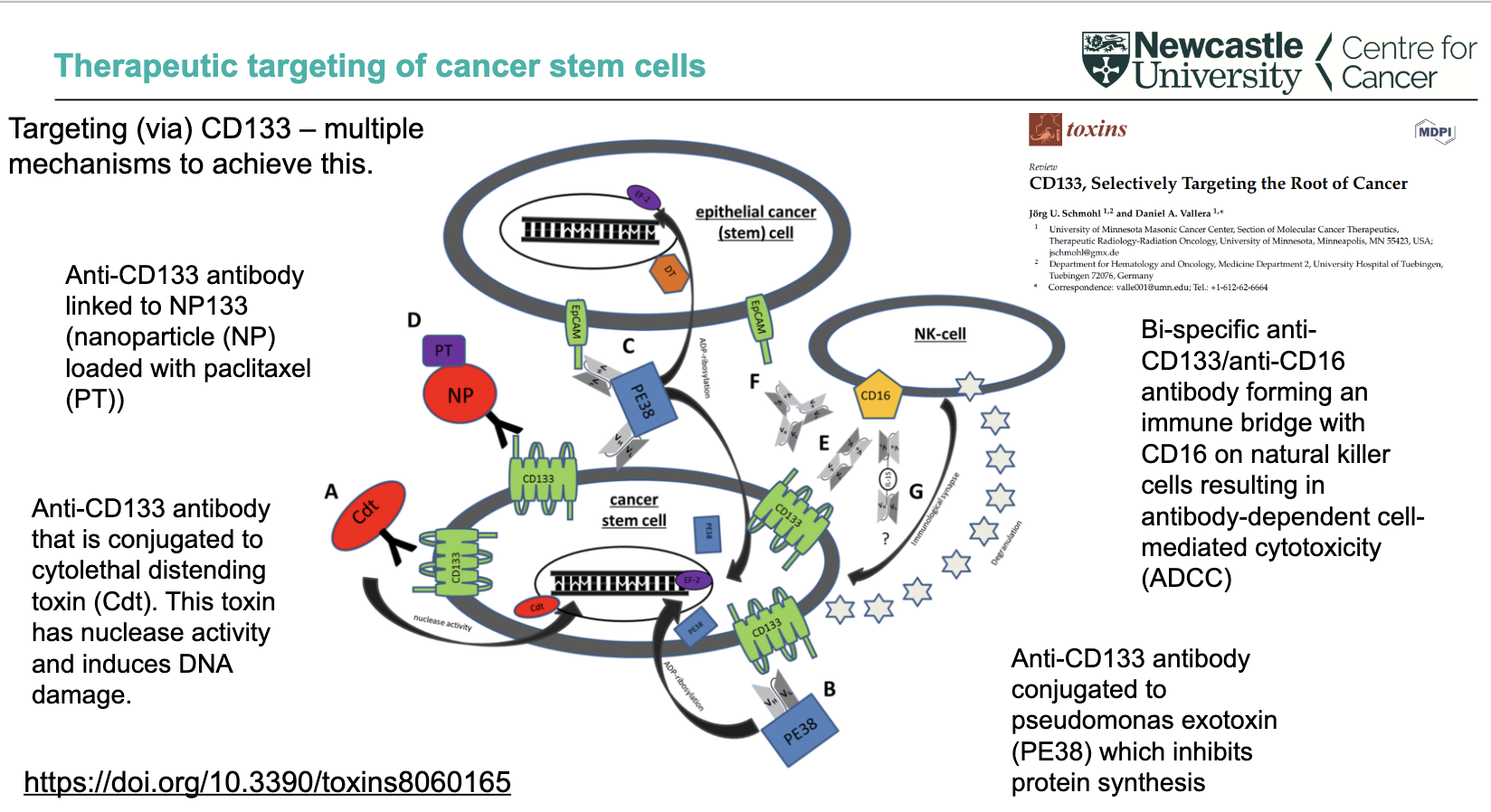
What are direct targeting strategies using CD133 in cancer stem cells?
Anti-CD133 antibody linked to nanoparticles (NP) loaded with paclitaxel (PT)
Anti-CD133 antibody conjugated to cytolethal distending toxin (Cdt) → nuclease activity → DNA damage
Anti-CD133 antibody conjugated to Pseudomonas exotoxin (PE38) → inhibits protein synthesis
These approaches deliver cytotoxic agents directly to CD133⁺ cancer stem cells
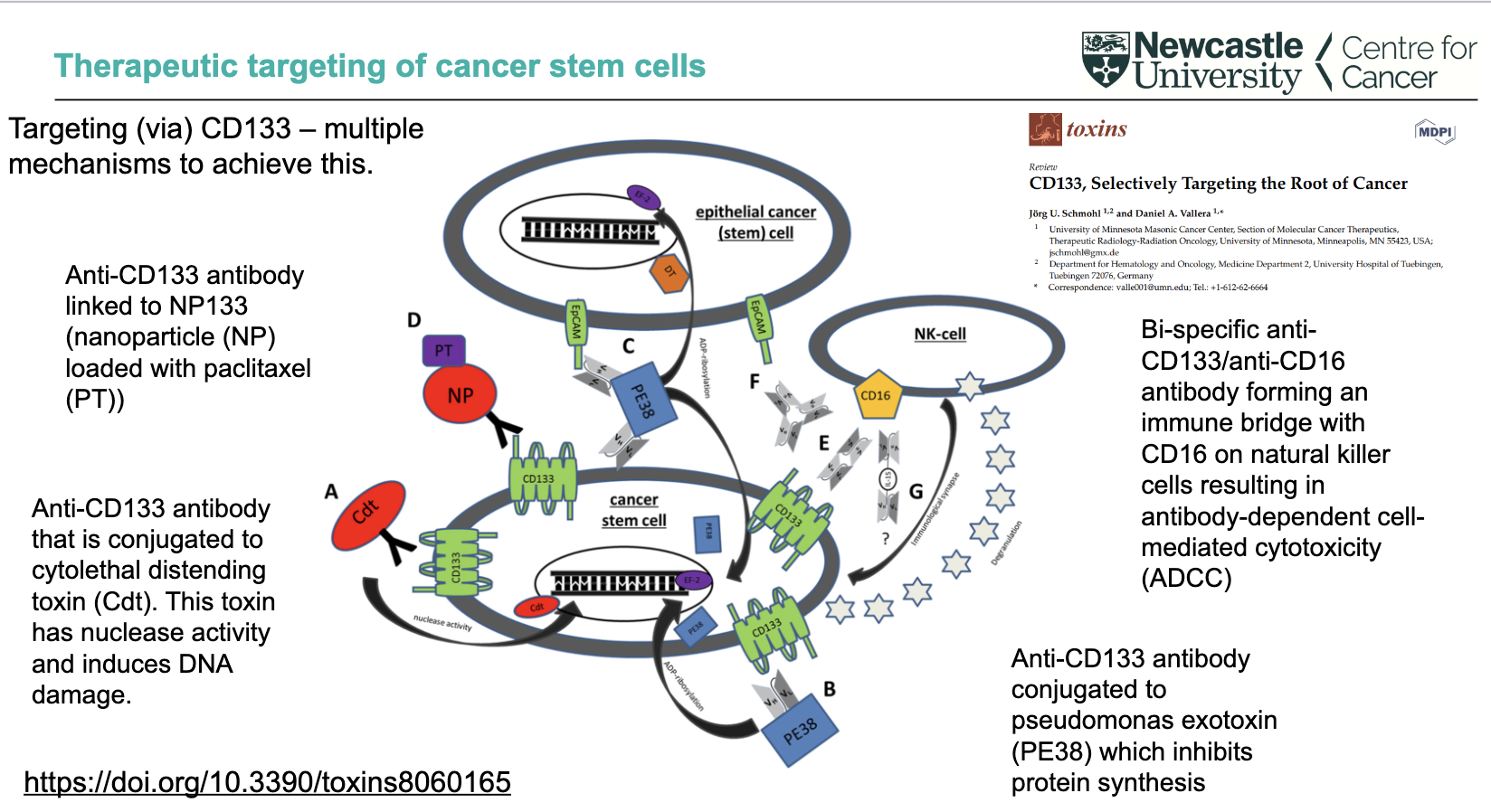
How can CD133 be targeted via immune-mediated and multi-mechanism approaches?
Bi-specific anti-CD133/anti-CD16 antibody
Links tumour cells to NK cells (via CD16)
Triggers antibody-dependent cell-mediated cytotoxicity (ADCC)
CD133 targeting can involve multiple mechanisms simultaneously
Overall aim: selectively target and eliminate cancer stem cells (“root of cancer”)
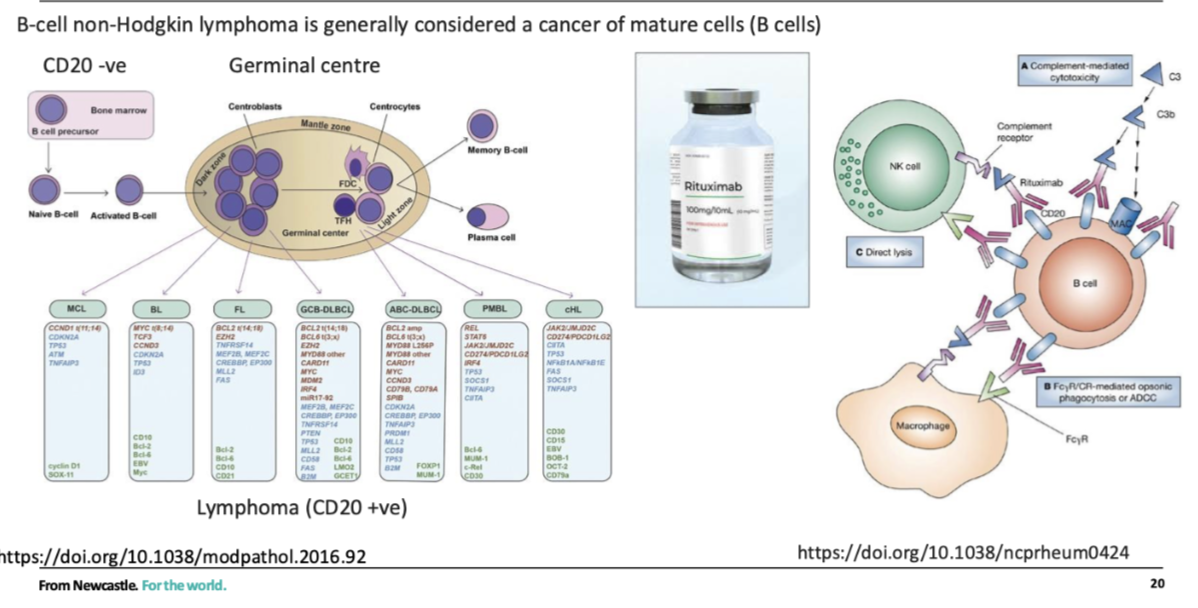
how can normal stem cells be protected during cncer treatment
Protect normal stem cells
Eg need HSCs to repopulate normal haematopoiesis in bone marrow
Lymphomas- positive for CD20, HSCs In bone marrow are CD20-ve, so can target CD20 +ve
Makes the B cells more visible to NK cells
what is cancer cell plasticity
the ability of cancer cells to change their phenotype to affect their identity and fate
Often (but not always) this is achieved via non-mutational mechanisms (epigenetic, transcriptomic, proteomic changes)
what can cancer cell plasticity do
• drive transformation to a cancer phenotype
• confer resistance to treatments
• be exploited as a therapeutic option
What is epithelial-to-mesenchymal transition (EMT) and where is it seen?
EMT = cells switch from epithelial to mesenchymal phenotype (with reverse process = MET)
Occurs naturally during embryogenesis
Demonstrates that cell phenotype switching is a normal biological process
Important concept for understanding cancer cell plasticity
what is EMT (epithelial-mesenchymal transition)
a process whereby polarized epithelial cells (which normally interact with basement membrane) assume a mesenchymal cell phenotype, thereby gaining migratory and invasive properties.
And becomes unpolarised, allowing it to migrate
what does EMT state correlate with
in carcinomas, which arise from epithelial cells, the EMT state correlated with tumour stage, disease aggression and patient outcome.
High grade disease – poorly differentiated
Low grade disease – well differentiated
What is EMT regulated by
EMT (& MET) regulated primarily by non- genetic mechanisms, including transcriptional and post-transcriptional mechanisms
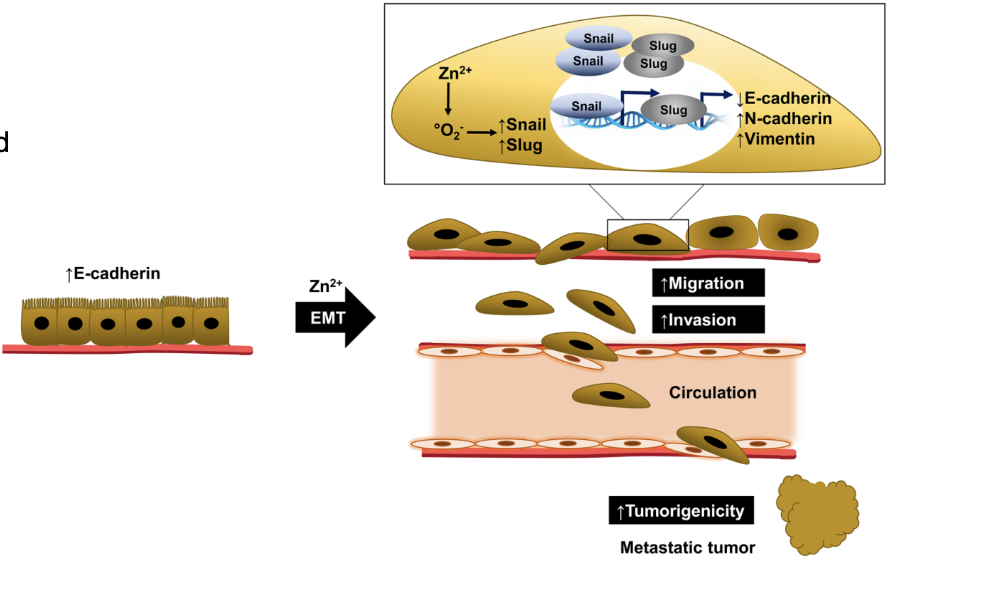
How can environmental exposures drive cancer through cell plasticity?
Cell plasticity = ability of cells to change phenotype
Often triggered by environmental exposures (e.g. inflammation, infection, smoking)
Involves processes like exposure-driven EMT
Leads to metaplasia/transdifferentiation
Can initiate pathways toward cancer development
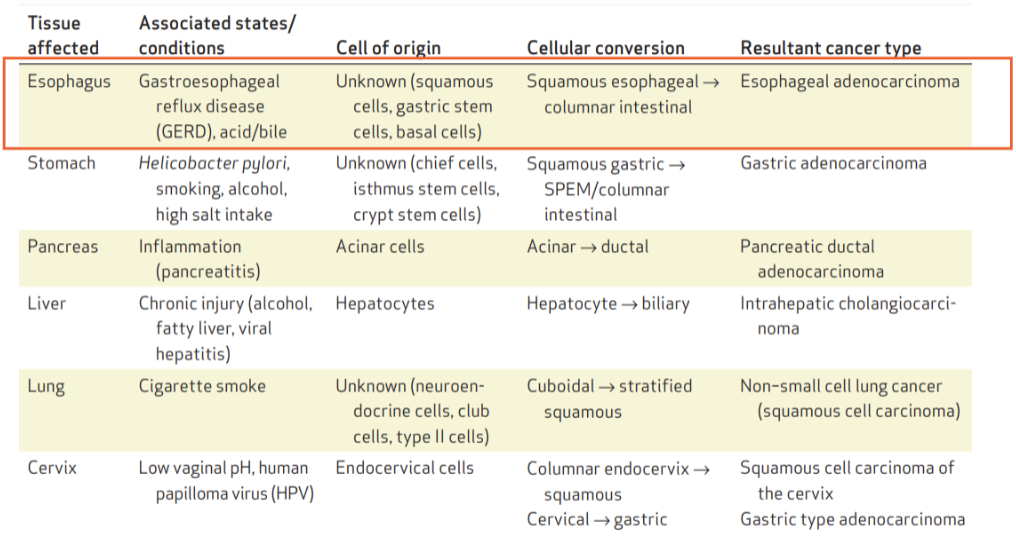
What are examples of cell plasticity leading to cancer in different tissues?
Oesophagus (GERD/acid reflux): squamous → columnar → oesophageal adenocarcinoma
Stomach (H. pylori, smoking, alcohol, salt): gastric → intestinal → gastric adenocarcinoma
Pancreas (inflammation): acinar → ductal → pancreatic cancer
Liver (chronic injury): hepatocyte → biliary → cholangiocarcinoma
Lung (smoking): cuboidal → squamous → squamous cell carcinoma
Cervix (HPV): columnar ↔ squamous → cervical cancer
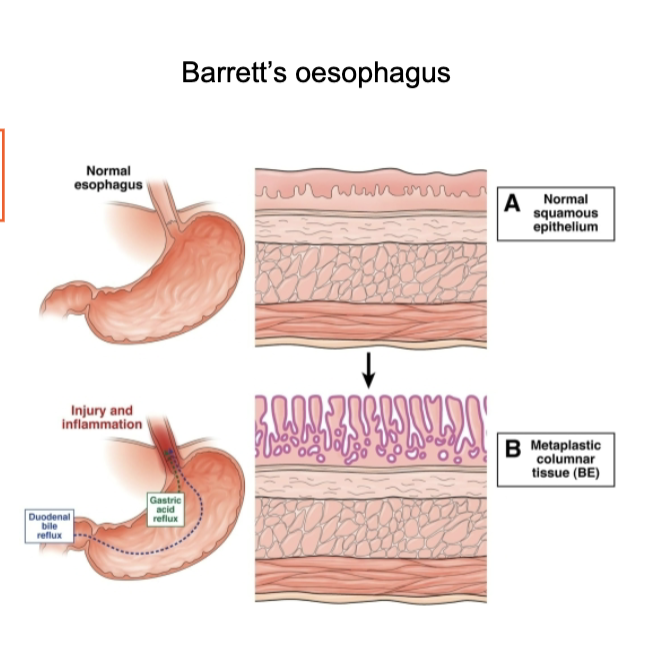
What is Barrett’s oesophagus and why is it clinically important?
Caused by chronic acid reflux (GERD) → injury and inflammation
Normal squamous epithelium transforms to columnar epithelium (metaplasia)
Represents exposure-driven EMT / phenotype switching
Acts as a precursor to oesophageal adenocarcinoma
Key feature: transition from squamous → columnar epithelium → important for diagnosis and risk monitoring
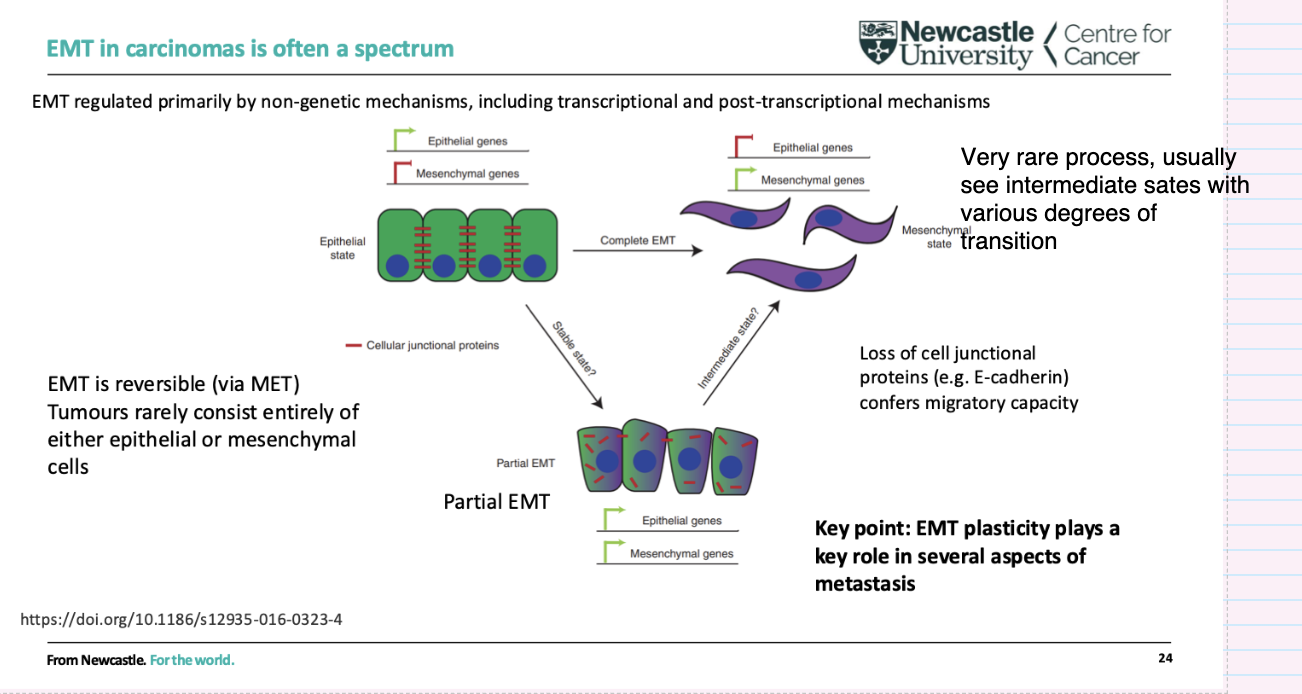
What are the key features of EMT in carcinomas?
EMT exists as a spectrum, with many intermediate (partial EMT) states
Rarely complete—tumours usually contain a mix of epithelial and mesenchymal features
Reversible process (via MET)
Driven mainly by non-genetic mechanisms (transcriptional/post-transcriptional)
Loss of cell junction proteins (e.g. E-cadherin) → increased migration
EMT plasticity is important for metastasis
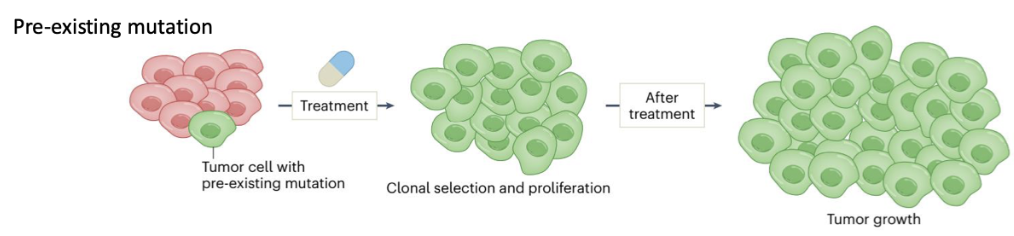
How do pre-existing mutations cause cancer therapy resistance?
Some tumour cells already have a pre-existing resistance mutation before treatment
Treatment kills sensitive cells but selects resistant clones
These cells undergo clonal selection and proliferation → tumour regrowth
Key idea: mutation is already present prior to therapy
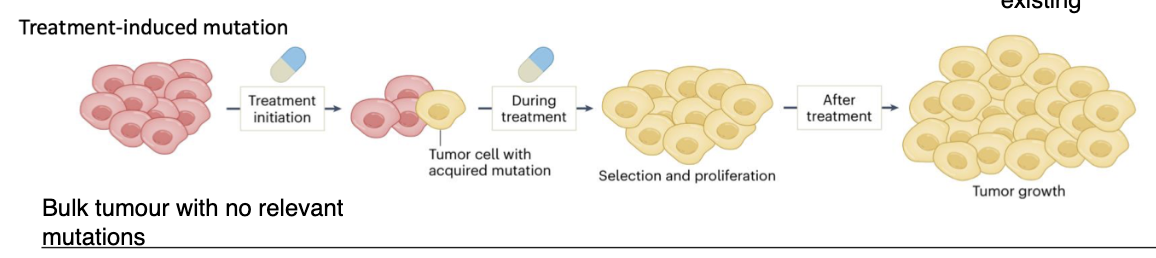
How do treatment-induced mutations lead to cancer therapy resistance?
Tumour initially has no relevant resistance mutations
Exposure to treatment (agents) induces mutations in one or more cells
Mutations occur in key genes → resistant phenotype
These cells are then selected and proliferate during treatment
Leads to tumour regrowth after therapy
How does cellular plasticity contribute to therapy resistance (non-genetic)?
Resistance often associated with a mesenchymal state
Driven by non-genetic mechanisms (transcriptional changes)
Multiple resistance mechanisms (e.g. ↓ drug influx transporters)
EMT plasticity linked to immunotherapy resistance
Resistance to dendritic and cytotoxic T-cell killing
Creation of an immunosuppressive microenvironment
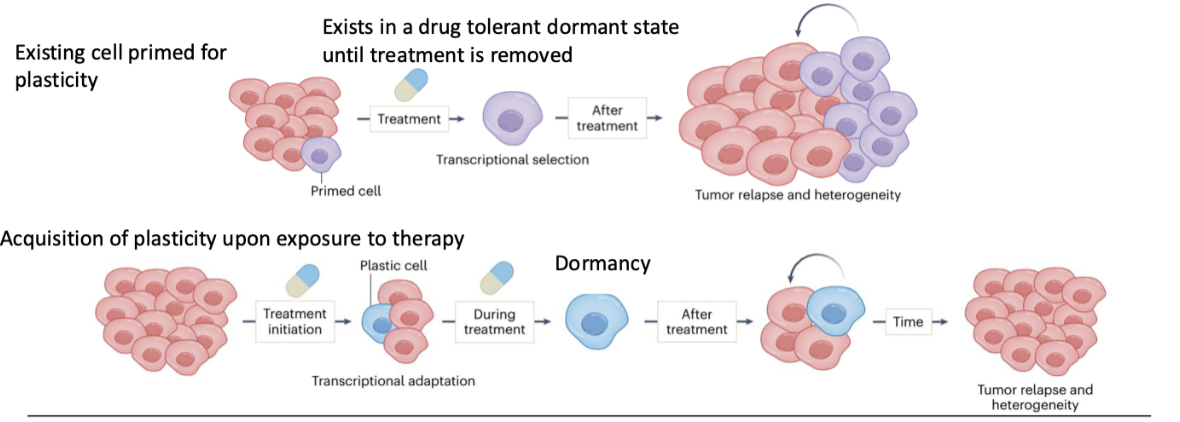
How can pre-existing plasticity lead to therapy resistance?
Some cells are already primed for plasticity
Under treatment → transcriptional selection
Cells enter a drug-tolerant dormant state
Survive until treatment stops
Then re-expand → tumour relapse and heterogeneity
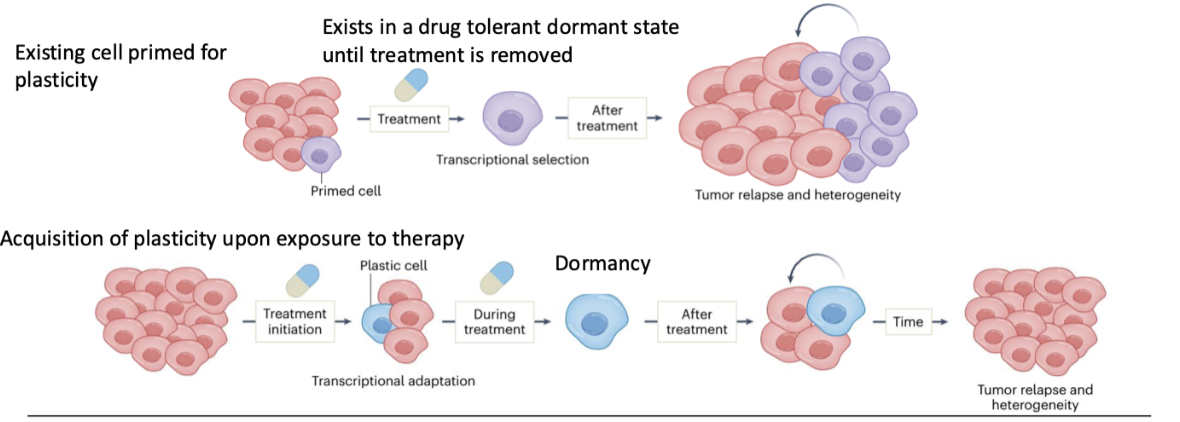
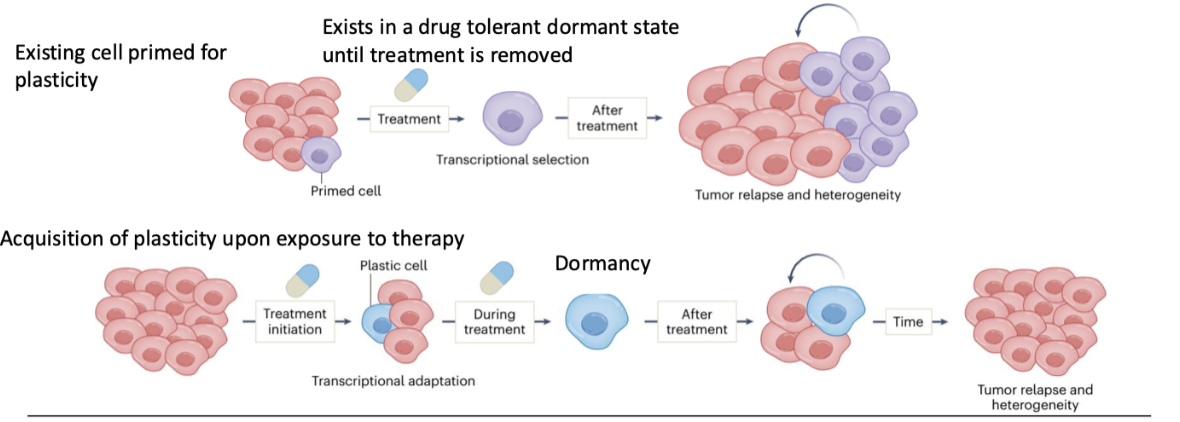
How can plasticity be acquired during therapy and lead to relapse?
Cells adapt during treatment (transcriptional adaptation)
Gain plastic phenotype in response to therapy
Enter dormancy during treatment
After treatment → cells reactivate and proliferate
Leads to tumour relapse and increased heterogeneity
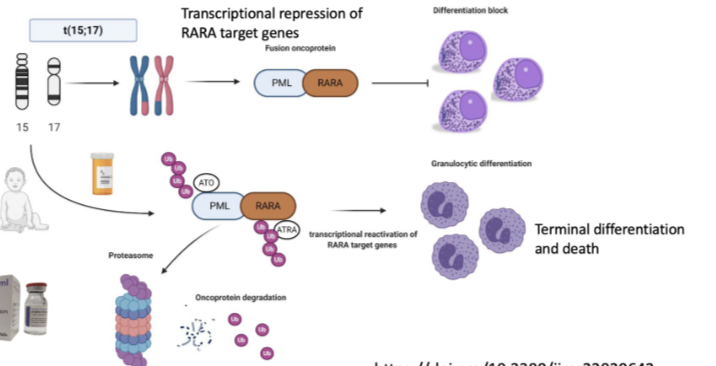
How does acute promyelocytic leukaemia (APL) arise and why is it dangerous?
Caused by t(15;17) translocation → fusion of PML + RARA genes
Forms PML-RARA fusion oncoprotein
Leads to transcriptional repression of RARA target genes
Causes a block in differentiation (promyelocytes cannot mature)
Results in accumulation of immature cells
Associated with extremely high risk of fatal bleeding → requires immediate treatment
How does APL demonstrate transcriptional reprogramming in cancer?
Fusion event alters transcriptional control (not just DNA mutation effect)
Target protein is transcriptionally changed → reprogrammed state
Causes differentiation block in myeloid cells
Shows cancer can be driven by changes in cell state (plasticity), not just mutations
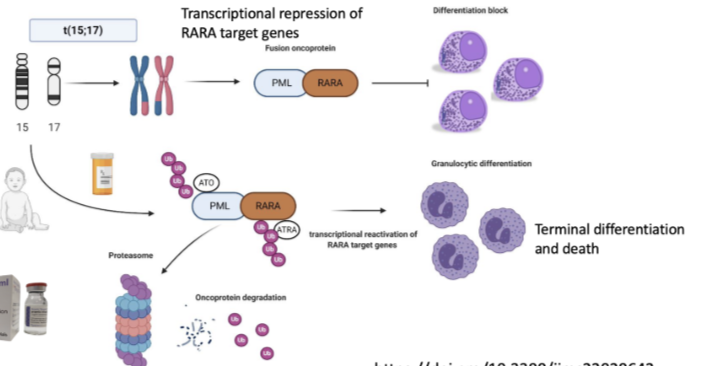
How is APL treated and what does this show about targeting cellular plasticity?
Treated with all-trans retinoic acid (ATRA) and arsenic trioxide (ATO)
These therapies degrade the PML-RARA fusion oncoprotein (via proteasome)
Leads to reactivation of RARA target genes
Cells undergo granulocytic differentiation → terminal differentiation and death
Cell returns to a more normal transcriptional state
Key point: therapy does not reverse the genetic mutation, but changes the transcriptional state to treat cancer
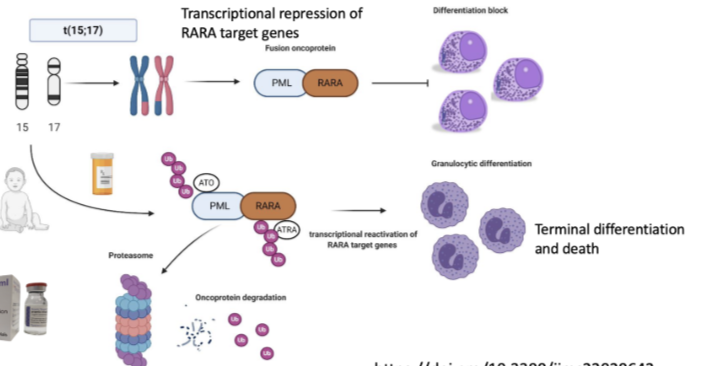
How does acute promyelocytic leukaemia (APL) relate to normal haematopoietic differentiation?
Occurs within the myeloid lineage (granulocyte pathway)
Normal pathway:
HSC → CMP → GMP → CFU-GM → granulocytes (neutrophils, eosinophils, basophils)
APL causes a block at the promyelocyte stage (failure of differentiation)
Demonstrates that cancer can arise from disruption of normal differentiation hierarchy
Key concept: cellular plasticity + differentiation block → disease
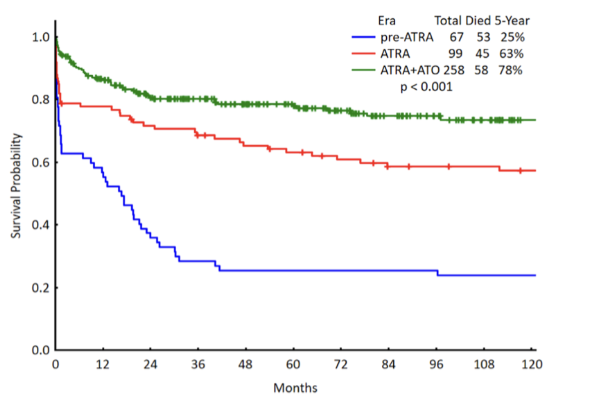
How has treatment of APL improved survival outcomes?
Historical (pre-ATRA): very poor survival
ATRA alone improves outcomes
ATRA + arsenic trioxide (ATO) gives best outcomes
Dramatic increase in long-term survival probability
Current complete cure rate ≈ 94%
Demonstrates success of targeting cellular plasticity (differentiation therapy)
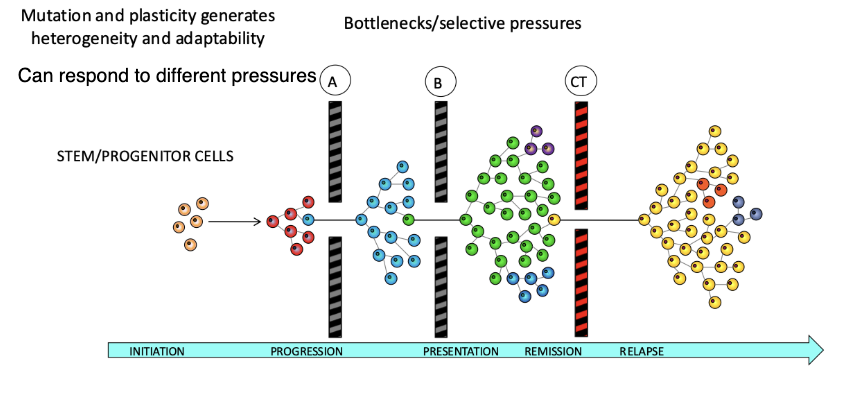
How does tumour heterogeneity contribute to cancer adaptability and progression?
Mutation + plasticity → tumour heterogeneity
Tumours contain diverse subclones that can respond to different selective pressures
Disease progression involves bottlenecks/selective pressures (A, B, CT)
Only certain clones survive and expand through each stage
Leads to stages: initiation → progression → presentation → remission → relapse
Key idea:
Heterogeneity allows cells to adapt, survive, and repopulate tumours
Enables cells to pass through bottlenecks and drive aggressive cancer phenotypes