Thermodynamics S2
1/94
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
95 Terms
first law (general form)
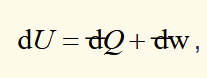
second law (general form)
inequality applies when dQ is for an irreversible process
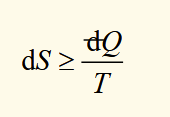
equations for a closed system where reversible PV work is involved
F
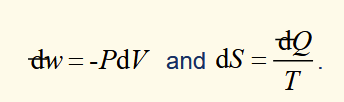
Fundamental equation for internal energy (U) for a closed system only involving PV work
the combined first and second law for a reversible PV work system (U, T, S, P and V are state functions)
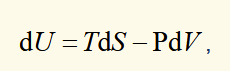
what does the fundamental eqtn for U show
it shows that the internal energy of a closed system only involving PV work is a function of S and V: U(S,V)
dU as an exact differential
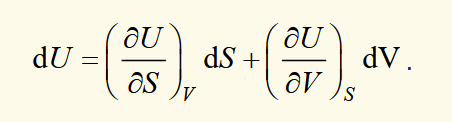
expressions for P and T by equation exact differential of dU to the fundamental equation
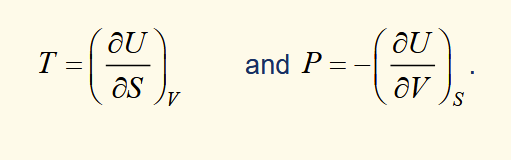
Why are S and V the natural variables of U
They are the basis for the fundamental equation of U, from which all the thermodynamic properties of the system (U, T, S, P, V) can be yielded. Considering U(T,P) or U(T,V) doesn’t let you calculate all these properties.
why consider different thermodynamic potentials
introducing new functions of state with intensive variables in them, such as P and T, allows us to explain thermodynamic questions when they are the independent variables. Also, the idea of thermodynamic potentials will show us when processes may occur.
functions of state table

thermodynamic potential
a scalar potential energy function. They don’t have an easy, insightful physical interpretation but are tools used to analyse situations depending on the variables we know.
internal energy and heat capacity
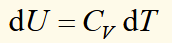
heat capacity at constant pressure
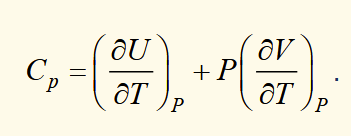
heat capacity and enthalpy
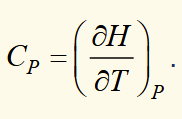
differential form of enthalpy (1)
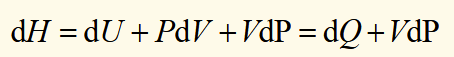
heat needed for an isobaric process
heat absorbed in a process at a constant pressure is equal to the change in enthalpy IF the only work done is reversible PV work.
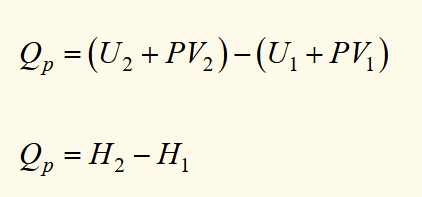
enthalpy definition
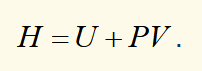
differential form of enthalpy (2)
natural variables of H are S and P
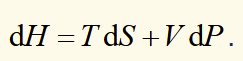
T and V using H
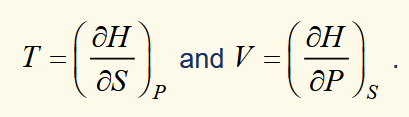
enthalpy exact differential
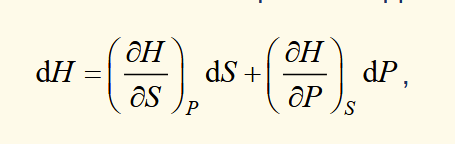
for a phase transition assuming constant pressure
change in enthalpy is the latent heat put into a system and measured for a phase transition
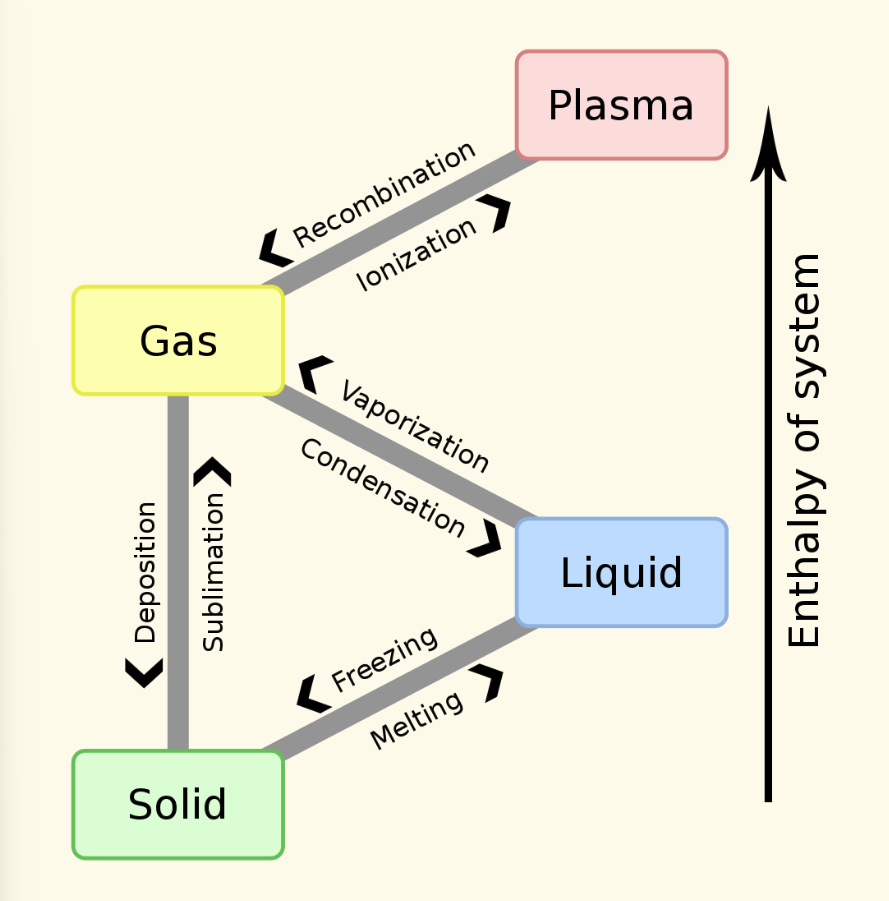
enthalpy of fusion
enthalpy change required to completely change on mole of a substance from solid to liquid
enthalpy of vaporisation
enthalpy change needed to completely change one mole of a substance from liquid to gas
enthalpy of sublimation
enthalpy change needed to change one mole of a substance completely from solid to gas
phase transitions and enthalpy of a system
condition for spontaneous change (S, V)
a change can occur spontaneously if the internal energy decreases when the change occurs at constant entropy and volume
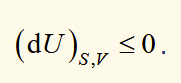
differentlial of H inequality
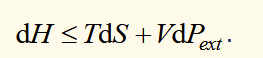
condition for spontaneous change (S,P)
if an infinitesimal change takes place in a system of constant entropy and pressure, dH is negative if the change is spontaneous and 0 if the change occurs in the system at equilibrium.
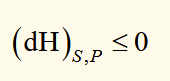
Helmholtz function
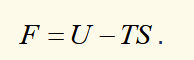
differential of helmholtz function
natural variables are T and V
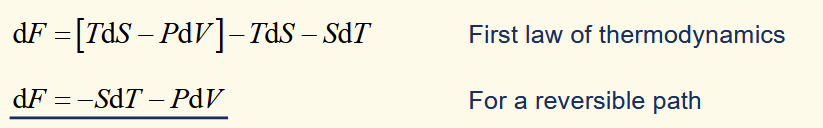
helmholtz function for an isothermal process
at constant temperature, a positive ΔF represents the reversible work done on a system by the surroundings.
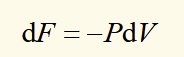
condition for spontaneous change (T,V)
for any spontaneous or irreversible process at constant temperature and volume, there is a decrease in F, and for a reversible process it is constant. When this system experiences a spontaneous change, the helmholtz function decreases until equilibrium is reached.
derivatives of the helmholtz function
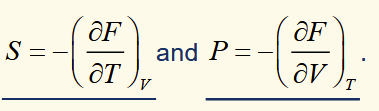
Gibbs Function
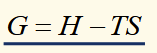
differential of gibbs function
(for a reversible process) natural variables are T and P
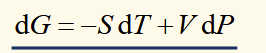
gibbs function for isothermal, isobaric process
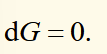
derivatives of gibbs function
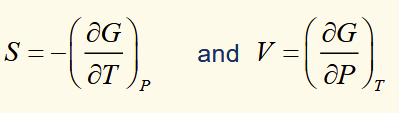
spontaneous change (T,P)
for any irreversible process in which only PV work is done, G decreases and for a reversible process it is constant. when a system undergoes spontaneous change at constant T and P, G will decrease until equilibrium is reached.
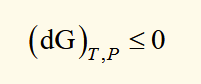
alternate expression for G
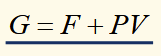
significance of natural variables
if the function of state can be determined as a function of its natural variables, then the function will yield all the thermodynamic properties of the system.
Gibbs-Helmholtz equation
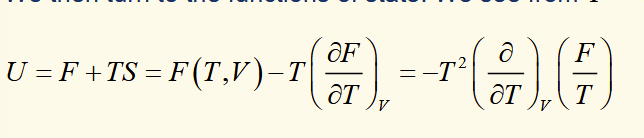
equation of state with H
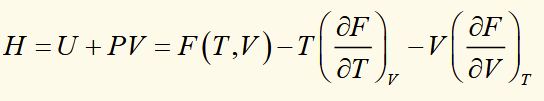
equation of state with G
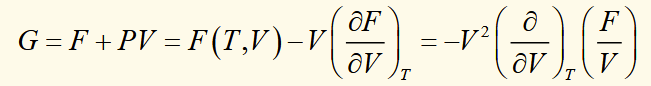
equations of state using G(T,P)
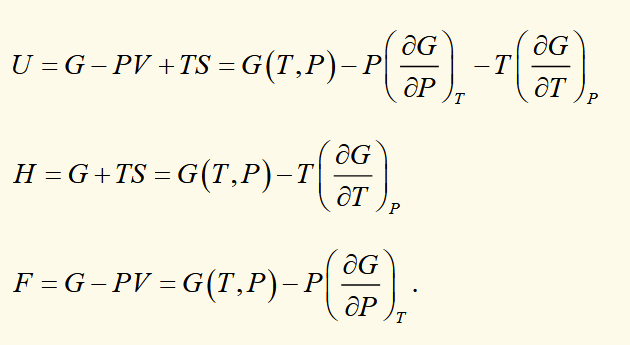
maxwell relations
[the variables that are kept constant are the natural variables for the function each relation comes from]
![<p>[the variables that are kept constant are the natural variables for the function each relation comes from]</p>](https://assets.knowt.com/user-attachments/239cbba2-bc0f-4b7f-9826-e28e947084c7.png)
internal energy of an ideal gas is not a function of volume
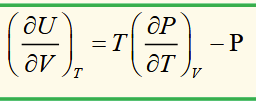
internal energy of an ideal gas is not a function of pressure
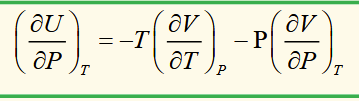
isobaric cubic expansivity (volumetric coefficient of thermal expansion)
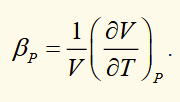
linear expansion coefficient
the fractional change in length per degree of temperature change
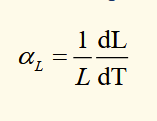
isothermal compressibility
the (-) sign because materials reduce in volume when pressure increases
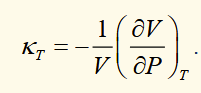
heat from a compressed object
general result for heat flow from a material that is being compressed
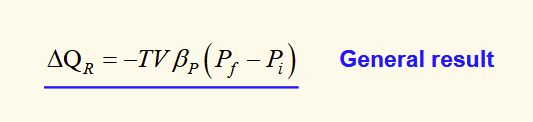
heat capacity at constant pressure (directly observables)
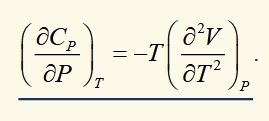
heat capacity at constant volume (directly observables)
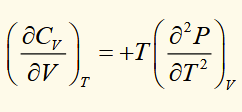
difference between heat capacities
This is a general relationship.
because κT is positive for all substances, it’s always true that CP > CV
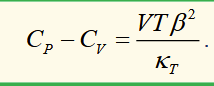
difference in heat capacities for ideal gas
for n moles of ideal gas
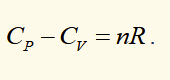
molar entropy of an ideal gal (P,T)
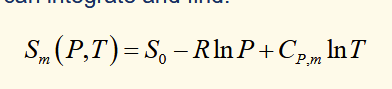
molar entropy of an ideal gas (P,V)
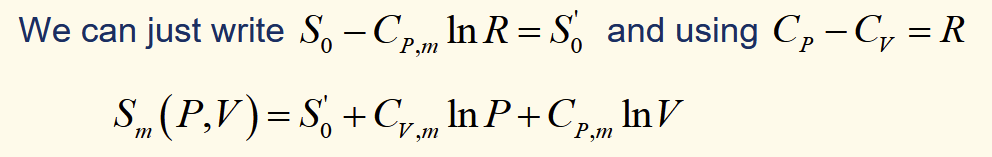
alternate expression for molar entropy of ideal gas (P,T)
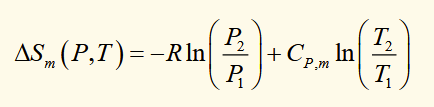
internal energy of a gas
general relation
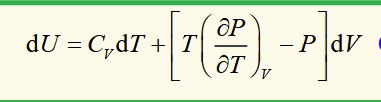
internal energy of a van der waals gas
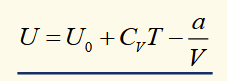
joule expansion
no work is done and no heat enters, so U is constant
joule coefficient (for expansion under constant internal energy)
rate of change of temperature with a change in volume when no work is done.
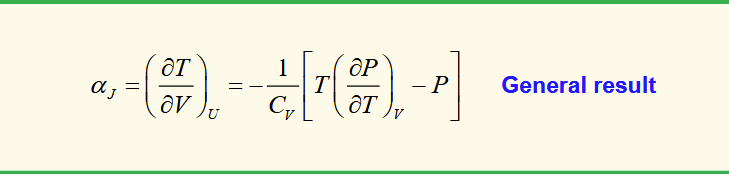
joule coefficient for an ideal gas
from PV = nRT
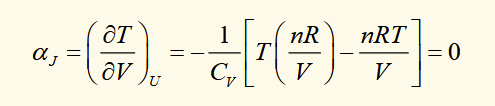
joule coefficient for a van der waals gas
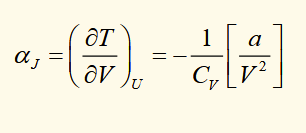
free expansion of a real gas…
always results in cooling.
chemical potential
N is the number of particles
this is an intensive variable
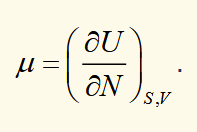
modified first law for a change dN
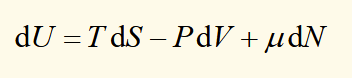
conjugate pairs of extensive/intensive variables
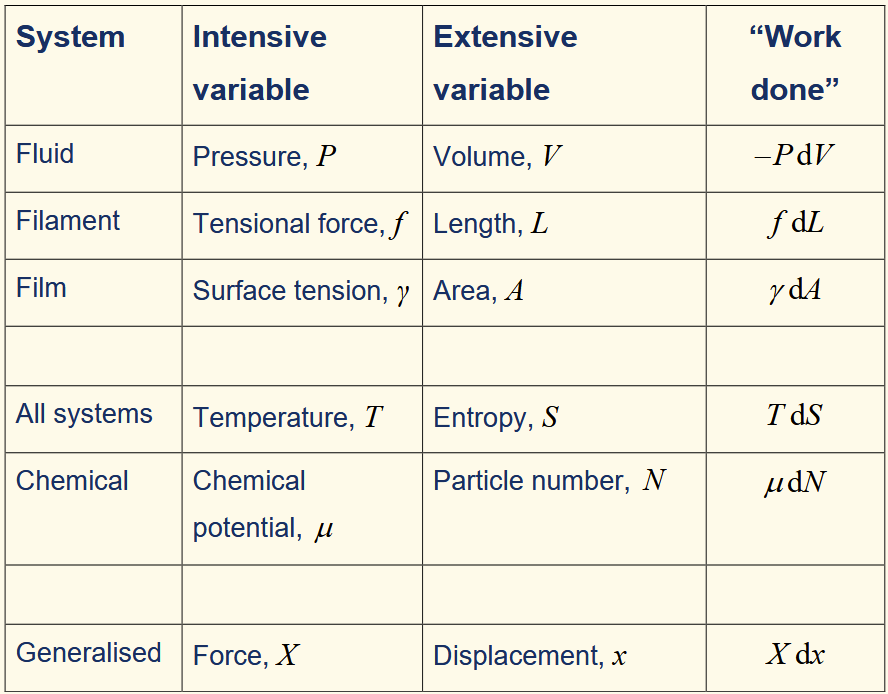
expression for dS
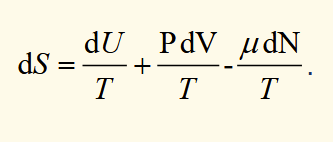
derivatives of S
particle exchange to gain equilibrium
equilibrium is found for T1 = T2 when μ1 = μ2.
for dN > 0, particles flow from system 1 to 2 when μ1 > μ2
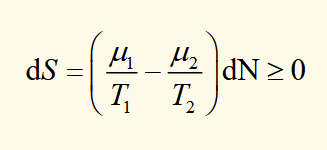
chemical potential (2)
it’s the gibbs function per particle
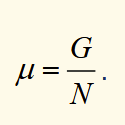
gibbs function for many types of particle in a system
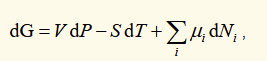
helmholtz function for many types of particle in a system
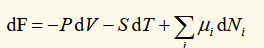
general case for heat capacities
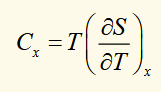
latent heat (L)
For two phases in thermodynamic equilibrium at a critical trainsition temperature (Tc), to change an amount of material between these phases some extra heat must be supplied while the system stays at constant temperature
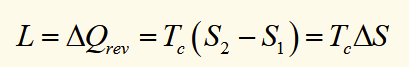
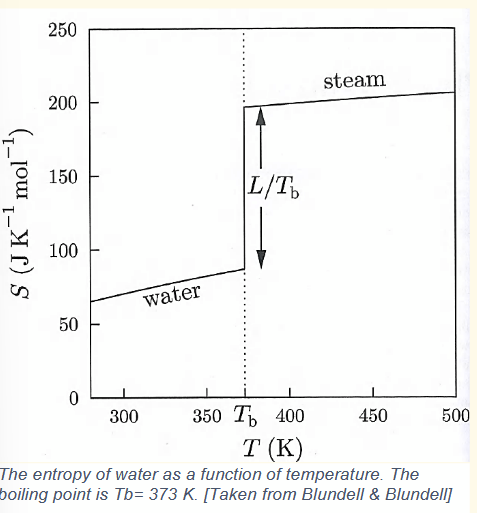
liquid-gas transition of water
[change in entropy at boiling point]
![<p>[change in entropy at boiling point]</p>](https://assets.knowt.com/user-attachments/8d112e16-63a6-4bac-a29c-93b26f87ac54.png)
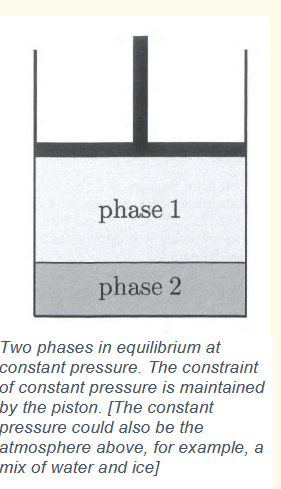
chemical potential and phase changes
for a system in equilibrium, gibbs free energy is constant, and also equal to ΣNiμi. If the number of particles in phase 1 increases, then the number of particles in phase 2 decreases by the same amount. This require μ1 = μ2 (along a line of coexistence). The phase with the lowest chemical potential is the stable phase.
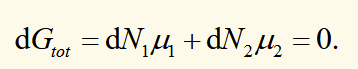
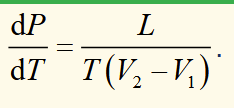
Clausius-Clapeyron equation
two phases in the P-T plane can coexist at the phase boundary. The equation describing this boundary line is the Clausius-Clapeyron equation. It shows that the gradient of the phase boundary is determined by the latent heat, and the volume difference between the phases.
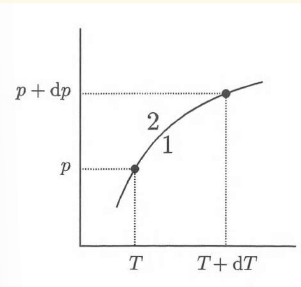
Clausius-Clapeyron for an ideal gas
using the substitutions R = NAk and l = L / NA, it can be shown that this exponential is a boltzmann factor for the energy step for a particle to move from liquid to gas
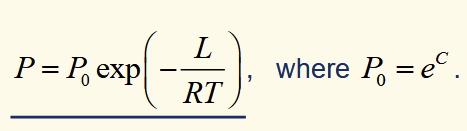
P-T boundary between a liquid and solid phase
rearranging the CC equation and ingoring any temperature dependence for L and ΔV, then integrating to get this expression.
T0 and P0 are known points on the phase boundary.
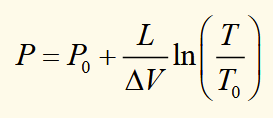
triple point
the point where solid, liquid and gas phases exist simultaneously.
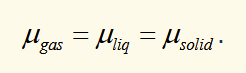
critical point
when the phase boundary between a liquid and gas terminates (end of vapour-pressure curve). At this point, liquid-gas coexistence is reached
termination of solid-liquid boundary
does not exist. after triple point, the boundary is very steep but does not terminate
phase diagram for a hypothetical pure substance
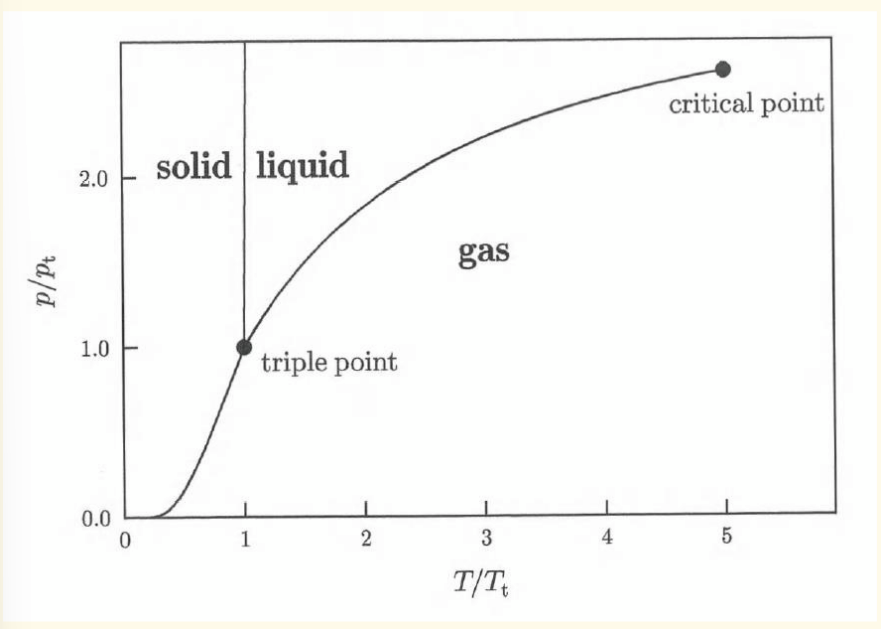
what happens exactly at a phase boundary
change in gibbs free energy is 0 and the chemical potentials of the two phases are identical
what happens upon approaching a phase boundary
the specific volumes (inverse of density) before and after a phase boundary are different. The slope of the Gibbs function must change at the boundary - the system will try to minimise it energy and travel along the lower of the two curves
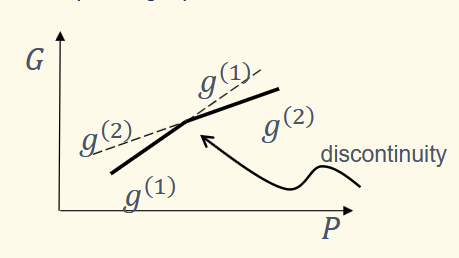
what drives phase transitions
when a system undergoes spontaneous change at constant T and P, G decreases until equilibrium is reached
for any irreversible process where only P-V work is done, G decreases
for a reversible process, it is constant
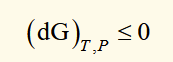
ehrenfest classification for order of phase changes
the order of a phase transition is defined as the the order of the lowest differential of the Gibbs function which shows a discontinuity at the transition
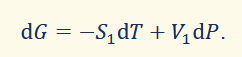
table of gibbs free energy and its first/second derivatives w.r.t. pressure
first derivative - volume
second derivative - isothermal compressibility

alternative gibbs free energy curves
by finding the derivatives w.r.t. temperature, similar curves can be found such that:
1st derivative - entropy
2nd derivative - heat capacity at constant pressure
examples of first order phase changes
solid-liquid
solid-vapour
liquid-vapour
superconducting transition in mag. field
some allotropic transitions in solids (e.g. iron)
examples of second order phase changes
superconducting transition in 0 field
superfluid transition in liquid helium
order-disorder transition in 𝛽-brass
examples of third order phase changes
curie point of many ferro-magnets (e.g. iron)
modern approach to classifying phase transitions
show a latent heat (which are still called ‘first-order phase transitions’)
vs
do not have a latent heat (continuous phase transition)