genet 270 pt 1
1/19
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
20 Terms
Investigators of microbes
1. Luria and Delbruck: variation is pre-existing. Darwin’s theory of evolution. Undertsanding foundation of mutation.
2. Griffiths, Hershey, and Chase: DNA is genetic material
3. Lederberg and Tatum: Genetic exchange & recombination
4. Watson and Crick: DNA structure.
5. Benzer: Gene structure. No Nobel Prize.
6. Meselson and Stahl: DNA replication. No Nobel Prize
7. Crick: Genetic code
8. Jacob and Monod: Gene regulation
Why bacteria?
- Have several features that make them excellent models for the study of fundamental biological problems
1. Physical Organization – Prokaryotes as Model organisms
o They are a single cell; 1 cell = 1 organism (doesn’t rely on any other cells)
o Two types; Gram positive and Gram negative
o Bound by single phospholipid bilayer with a cell wall
o Are prokaryotic
No nuclear membrane
mRNA transcription and translation occurs simultaneously in the cytoplasm
Chromosome is condensed in a region called the nucleoid (highly organized by non-histone proteins)
Single circular chrosomse. Genome tends to be streamlined (little “wasted” space; most of DNA is coding).
o Haploid
Single circular chromosome. One copy of every gene.
Advantage; mutations have immediate effects. Disadvantage; hard to study lethal alleles.
Chromosome is dsDNA and supercoiled; tightly wound to have a relatively large chromosome fit into a small cell
2. Bacteria are easy to grow
o Grow in simple, easy to make and defined media
o Culture initiated by placing a small amount of bacteria (inoculum) into sterile medium in a flask or tube
o Medium may be complex or minimal: diff species/strains have diff recepies
- Bacteria are often classified based on their different growth requirements:
Prototrophs: can grow on minimal media. Auxotrophs: require media supplemented w/ an organic molecule to grow.
- Or solid media
o Agar is added to media and poured into plates
o Plate; petri dish containing solid media. Plating; spread a culture across an agar surface (often require serial dilutions to get isolated colonies → mass of genetically identical cells)
3. Bacteria have short generation times
- Generation time: time an organism takes to reach maturity and give rise to offspring. Ex. E. coli → 20 minutes
- Rapid growth facilitate genetic analysis. Can observe billions of organisms on one plate.
- Progeny are clones.
- Many strains used in the lab are non-pathogenic. eg K12 common E.Coli lab strain
4. Bacteria are amenable to genetics
- Can look at 1000’s of bacteria at a time
Sheer number of organisms help identify rare mutants
- Can easily store mutants for future study – ie. freezing
- Capacity for genetic exchange – there are many mechanisms that facilitate mutant analysis (transformation, conjugation, transduction)
- Carry plasmids; small (<5 kb), circular, carry “extra” genes ie genes non-essential for normal growth.
Common plasmids like F factor (fertility factor) → used in matings/conjugation
Plasmids = free floating in cytoplasm. Can be lost during divison.
What are bacteriophage/Lytic lifecycle
- Bacteria “eaters”.
- Obligate bacterial parasites; viruses that only grow in actively metabolizing bacterial cells
- Considered non-living (not “organisms” but “particles”)
- Nucleic acids – DNA, RNA, single or double stranded, linear or circular
- Protein coat or capsid – two types
Viruses that infect euks contain a lipid envelope around a proteinaceous capsid
Bacteriophages consist of just protein and DNA
- No internal metabolism
- Lytic: phage infects bacterium, hijacks host cell resources, reproduces quickly & lysis the cell to release phage progeny (see pic)
- Virulent phage; phage that can only undergo lytic growth
1) Adsorption: phage attached to specific receptors on the bacterial surface
host cell can mutate so that they are no longer recognizd by phage tail fibers
2) Passage of DNA from phage through bacterial cell wall; not fully understood how this happens
neck/sheath of phage cell wall & memb to inject DNa into host cell cytoplasm'
3) Bacterium is turned into a ‘phage factory’…how?
Ex of temporal gene regulation
Early genes: activated by bacterial gene expression machinery. Gene expression is initiated upon entry into cytoplasm. Include genes for viral DNA replication. Turn on late genes
Late genes: degrade bacterial DNA. Encodes structural genes for phage capsid.
4) Phage particle assembly (aka morphogenesis)
Phage capsid will spontaneously self assemble in bacterial cytoplasm. Based on protein-protein interactions.
dsDNA linear phage genome is stuffed into capsid by “headful packaging”
5) Release of newly synthesized phage; requires host enzyme lysis
Holin: late gene. Viral protein that disrupts host cytoplasmic memb
Lysozyme: degrades cell wall
6) Forms plaques on bacterial lawn
plaque: region of clearing on bacterial lawn due to lytic phage growth
Start w/ a thick lawn of densly growing host cell in order to culture phage.
add diluted phage lysate
each plaque represents a clonal expansion of a single phage thus can isolate a pure phage stock by selecting a single plaque
# of plaques represents # of phage in inital lysate. Diff phage strains have diff plaque morphologies.
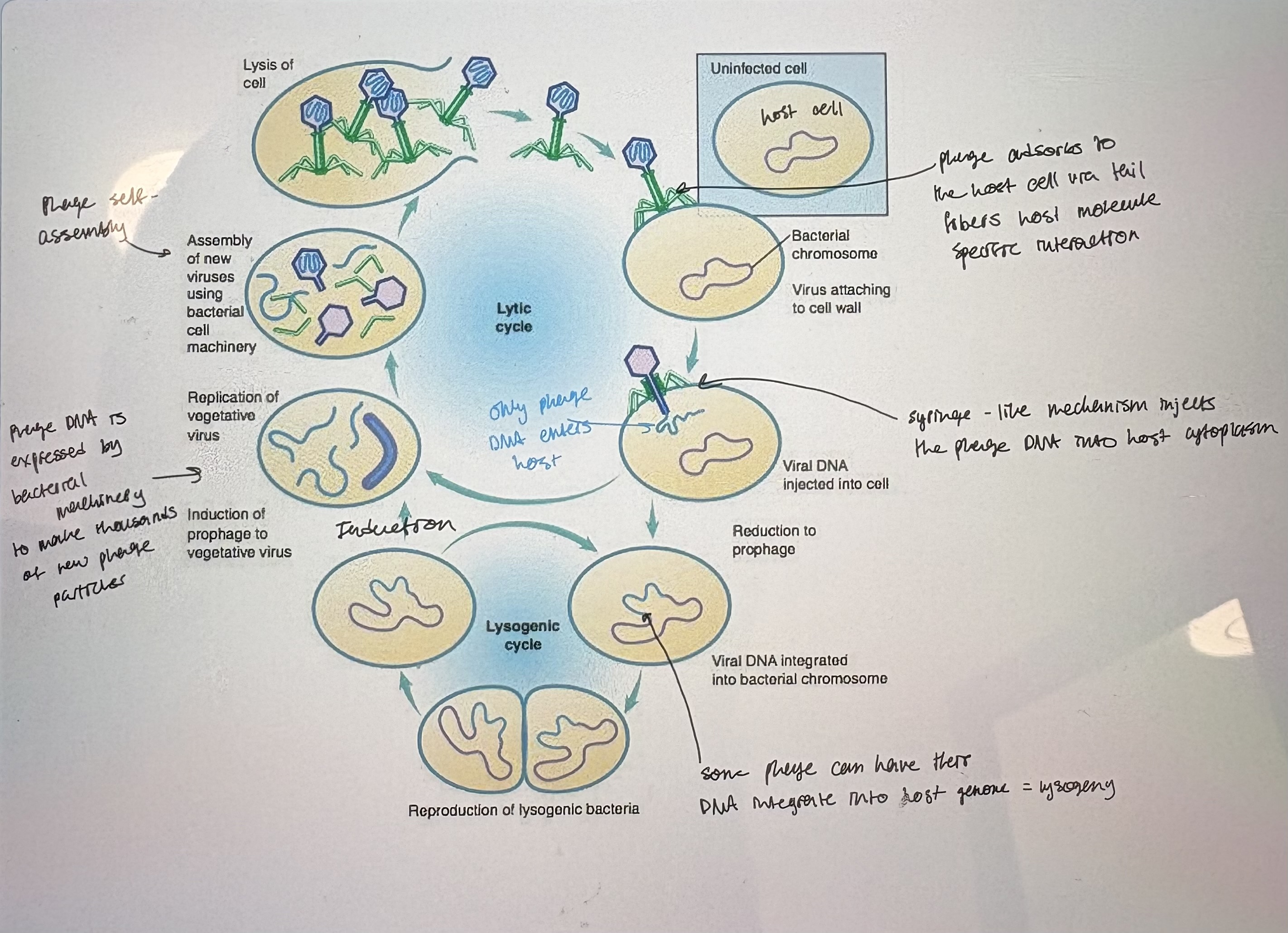
Lysogenic lifecycle/Concept for studying phage
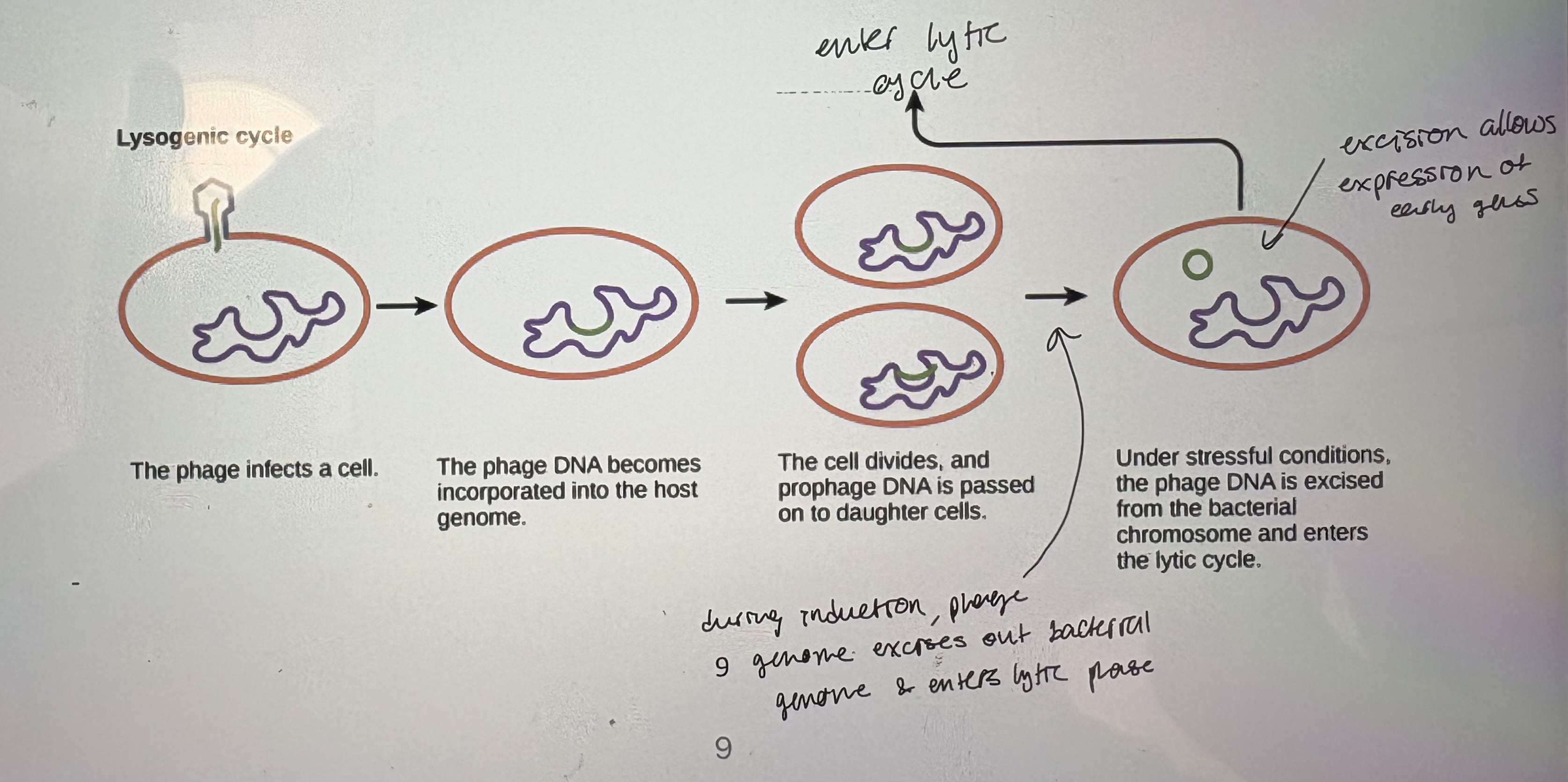
- Lysogenic: phage DNA becomes integrated into the bacterial chromosome and is silent, aka a prophage
- Lysogen: bacteria carrying a phage
- Lysogeny: process of integrating phage DNA into bacterial chromosome. A
Advantage: virus replicated along w/ bacterial host
- Temperate bacteriophage: a phage strain capable of lysogeny
1) Phage adsorption and injection of phage DNA into host cell
2) Brief period of transcription
expression of i. repressor phage protein = shut down lytic cycle, ii. site specific recombinase = facilitate host genome integration
3) Phage DNA molecule recombines into the bacterial chromosome
often into integration hot spots
iv. Phage DNA is now replicated with the bacterial chromosome as the bacterium grows
does not express phage structural proteins
v. Lytic growth may be triggered again after many cell generations by a process known as induction (occurs if host cell experinces stress)
- Efficiency of plating (EOP): fraction of phage particles that can form a phage. Most phage EOP = 1
- Multiplicity of infection (MOI): # of phage adsorbed onto bacteria. Dtermines how many bactetia will be infected
- Isolation of Phage stocks
Phage produce more rapidly than bacteria (100 phage vs 2 bacteria per generation time)
- Host range
Phage infection shows host specificity. Eg. Phage P22 infects Salmonella typhimurium but not Escherichia coli
Host range is determined by many factors; cell wall components, potential phage receptors, restriction enzymes of host
Mutants in host range are easy to identify
- Some mutant bacteria may be resistant to phage infection. No plaques/bacterial lawn. Mutant ohage can now adhere to mutant bacteria
- h mutants: mutations that extended the host range. Easy to find because of sheer # of phage allows isolation of rare events
- Phages are good genetic models because:
1) Bacteriophage are haploid; mutations manifest immediately
2) Short generation times: >100 phages in ~20 min/bacterium (allows us to identify rare events)
3) Multiply clonally: can isolate & maintain pure stocks. Easy to manipulate.
4) Easy to cross phage strains
Infect same bacteria with different mutant phage stocks at high MOI
Some bacteria will be infected by both phage
Bacterium will contain genomes from 2 diff phage strains. Genomes can inetract in this common cytoplasm; can complement, can recombine.
5) Mutant Selection: Millions of phage can be mixed with bacteria and grown under selective conditions
Ex rll mutant: creates mottled plaques. Rapid lysis.
Genetic analysis/Types of mutations
- Science of Heredity – process by which living organisms generate offspring like themselves
- In the lab – the manipulation of DNA to study cellular and organismal function
- Classic genetics: isolate mutants, locate mutations in genome (mapping), group mutations into complementation groups, determine gene function by measuring effects of muations
- Reverse genetics: clone genes, alter gene in vitro, return the altered gene to organism, asses function by effects BUT prioor knowledge of a gene is required. Limitation → can’t test what you don’t know.
- Nonconditional: displays mutant pheno under all conditions. Ex null mutant = complete loss of function
- Conditional: The mutant phenotype is dependent upon environmental conditions or other mutations. Only way to genetically analyze essential genes (in haploid → can’t keep essential gene mutations in a stock cuz 1 only one copy, without it they die)
Temperature Sensitive (TS)
Ex. Temp sensitive; displays wt phenotype at one temp (permissive temp). Displays mutant phenotype at another temp (restrictive temp).
Ex. Temp sensitive conditional lethal; eg Lac Z18 allele. 37C = wt, 42C = lethal if grown on lactose.
- Mutational screening: how do we find mutations in particular pathways? (classical approach)
Induce mutations by exposure to mutagens (UV, EMS, etc)
all mutations will be random in genome
some cells survive the mutagens & carry novel mutations
grow on permissive conditions eg complete media (let bacteria recover in best possible conditions to see what is now wrong)
If intrested in novel alleles of lac operon = challenge the mutagenized strains to grow on lactose (to check if lac operon works).
Colonies have X-gal indicator which turns bacteria blue if Lac opeon is functional.
Permissive temp = all colonies can grow on lactose (thus Lac operon works in wt temp)
At 42C = if not coloured blue, cant use lactose → has a temp sens mutant allele of Lac operon
Suppressor sensitive
mutant pheno can be observed in phage using diff bacterial host background
gene products made by bacterial host can conditionally suppress phage mutants
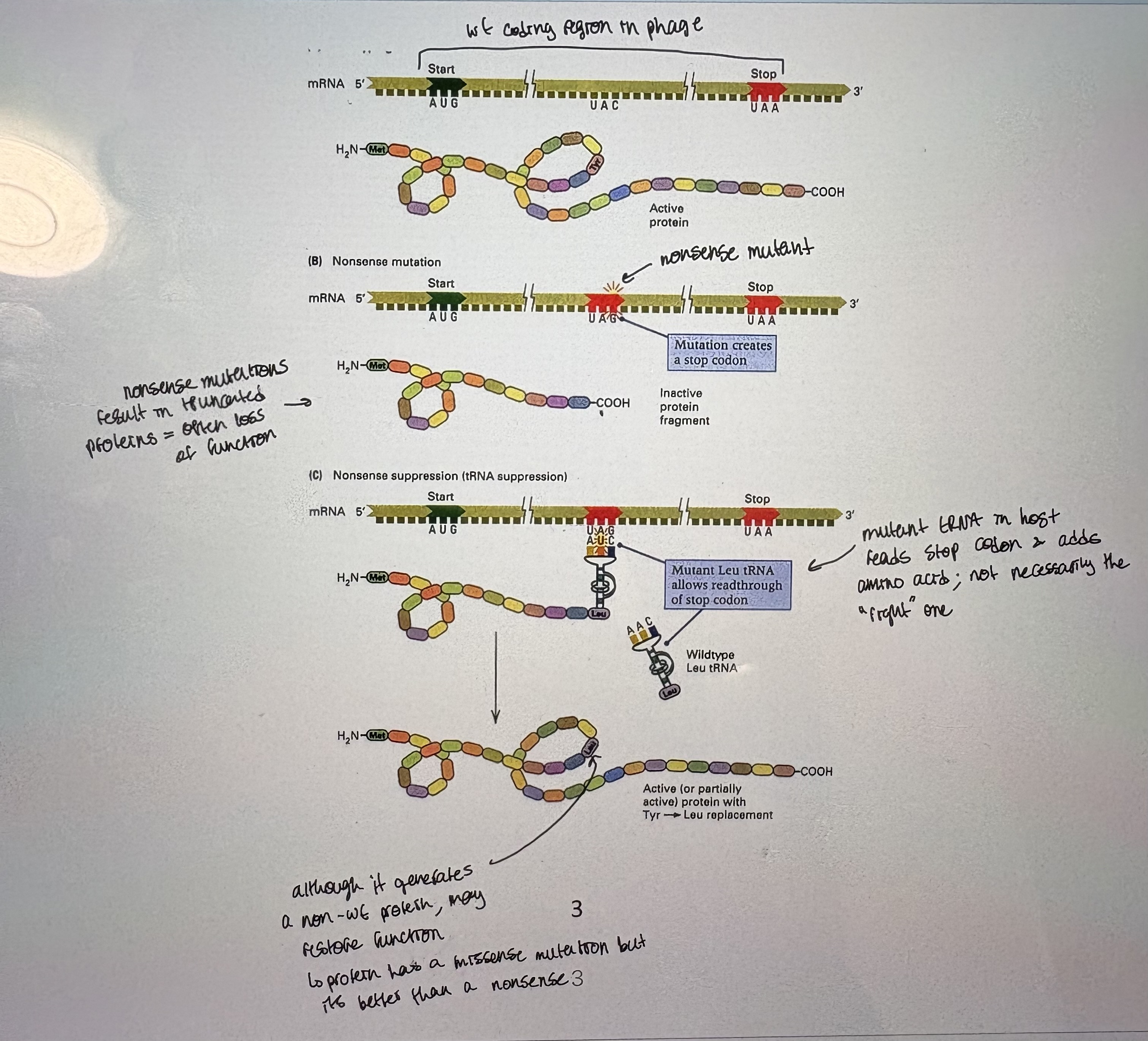
Eg. Nonsense suppressors: Nonsense mutations → introduce a premature stop codon in coding region. If a phage carries a nonsense mutation, it can be rescued/suppressed by a bacterial host that has mutant tRNA that reads nonsense codon.
Strain lacking suppressor: if phage w/ mutation is grown on wt host = get mutant pheno
Strain carrying suppressor: if phage w/mutation is grown a suppressor host = get wt pheno
Nonsense suppressor strains: host that carriers tRNAmut that reads the stop codon as amino acid encoding. (see pic)
Suppressor sensitive mutations are designated amber (Am), ochre (Oc) or opal (Op) depending on the type of nonsense codon suppressed. UAG → Am, UAA → Oc, UGA → Op (will read each codon as an amino acid)
Can use host strains to maintain lethal nonsense phage mutants.
What effect does tRNA mutation have on host? Host will have larger proteins for the proteins that use that stop codon (cuz it can’t control which stop codons get fixed and which stay)
Auxotrophic mutants: nutritional mutants are conditional mutant. Bacterial mutants may lack ability to grow on diff media.
To isolate conditional nutritional mutants by first growing on complete media
CM without biotin isolates biotin auxotrophs (the bacteria that can’t grow without biotin)
histidine auxotroph (has mutation that prevents synthesis of an essential nutrient)
Isolation of mutants
- We often need to identify rare mutants that are affected in specific processes from large numbers of phage or bacteria – we use plating or plaquing to observe large numbers of organisms
- Screen: mutants are identified by phenotypic differences compared to wt (look for mutant that doesn’t grow- hard). Much more work than selection.
- Selection: mutants are identified due to their survival ability in a selctive environment. The best screen? Where only mutants grow.
- Replica Plating: must be used to screen for conditional mutants (see pic)
Bacteria or phage first plated or plaqued under permissive conditions
A copy of plate is made and grown under non-permissive conditions
- Ex. Host dependant phage mutants (ie Conditional lethal)
T4 rll+ (wt) → small plaques on B permissive → small plaques on K restrictive
T4 rll (mutant) → large plaques (faster growing) on B permissive → no plaques (can’t grow) on K restrictive
- Selections: plate out under conditions which only wanted mutants will grow
Negative selection = assesing loss of ability to grow
Positive selection (easier) = assesing gain of ability to grow
Start w/ His- auxotrophs (can only grow on supplemented media) → allow mutagenesis recovery on CM → challenge on CM-His- thus only mutants will grow (mutation is revertant)
- Reversion: mutation that regains wt function
- Revertant: mutant containing reversion
- Reversion frequency: frequency of cells in a popl’n that required wt phenotype. Reflects type of original mutation.
Relative reversion frequency = single point mutations (common, easily revertible) > multiple bp mutations > inversions >>> deletions (non-revertible)
Genetic analysis of mutants (both phage and bacteria)
- Complementation and recombination: very often mixed up but methods and conclusions are very diff
Both concepts require 2 copies of genes in question in the same cell; merodiploid (pseudodiploid in bacteria) or coinfection (phage)
- Complementation: the products synthesized from 2 diff genomes interact in a single cell to produce wt pheno
Genetic test to determine if mutations are in sane or different genes
- Recombination/crossing over: physical exchange of DNA between 2 diff DNA molecules = progeny have diff DNA combos compared to the parents
Used to determine relative position of loci
- Complementation: in phage = using coinfection (2 diff phage strains infect same bacterial cell)
Diff mutations lead to the same loss of function. Mutation in gene b = no plaques, mutation in gene a = no plaques.
Put them in the same space (cell). If phage coinfect same bacterial cell, can share gene products IF mutations are in same pathway AND mutations are in diff genes. Plaques form if mutations complement/non-allelic
- Complementation in bacteria: don’t cross like yeast do, instead intorduce second copy via a plasmid
move genomic region of one mutant into another via a plasmid
If genes complement → grow → mutations are in diff genes thus non-allelic
If genes DON’T complement → no growth → mutations are in same gene thus allelic.
OR one mutation affects expression of another gene. OR gene product inhibits the other (to distinguish; map)
- Recombination: physical exchange of DNA between chromosomes of diff haploid strains
a crossover can form anywhere along a DNA sequence that shares homology
creates novel genetic combos in progeny
leads to the production of recombinant progeny with genotypes diff from both parents
If a bacterial cell is coinfected w/ 2 phage strains, then recombination may occur
Non-parental allelic combos. Recombinants are rare in progeny.
- Recombination frequency: # of recombination types/total # of progeny (# parental + # recombinants) x 100%
- Genetic mapping:
RF is proportional to the genetic distance (not nessecarily physical distance) between mutations/genes
RF is used to map genes relative to one another
a+ b- c- Phage 1
a- b+ c+ Phage 2
Coinfection allows crossover
There will be 2x as many a+c+ recombinants as a+ b+ recombinants because they are double the distance apart
In order to select for recombinants, choose one locus as the “anchor” and map other loci relative to the “anchor”.
Types of T4 mutants used in genetic analysis
1. Plaque morphology mutants
- wt = small plaques → wt shows lysis inhibition at high MOI
- other T4 mutations affecting plaque formation (T4 mutant phenotypes):
rll mutants; mutants in lysis inhibition → show large plaques
r = rapid lysis. tu = turpid plaques (lawn not fully cleared w/in plaque). sm = small plaques. ac = allow phage to grow on acridine.
2. Host range mutants
- host range mutants (h mutants) → h = host
- eg. mutants fail to absorb to one strain of host vs the other
- T4 → E.coli B = clear → E.coli B/4 (normally resistant to T4 phage) = none
- T4h → E.coli B = clear → E.coli B/4 = clear (mutant has expanded host range compared to wt)
3. Conditional lethal mutants
- permit the isolation of mutations in many genes (essential)
- eg. #1 - Nonsense mutants
wt phage → sup- host = plaques → sup0/sup+ host = plaques
Nonsense mutant → sup- host = no plaques → sup0/sup+ host = plaques
- “Brute-force” screening → plaque on sup- bacteria → replica plate to sup+ lawn to screen for nonsense mutants (shouldn’t grow)
- Eg. ts mutants.
rll- → replica plate? yes → screen for plaque morphology or select for host range
ts → replica plate? yes → negative selection for loss of plaques @ restrictive temp
Nonsense → replica plate? yes → positive selection on nonsense suppress host
tu → replica plate? yes → screen for plaque morphology
- rII→nonsense codons, genetic code, gene structure
- 1950s and 1960s, Seymour Benzer
- rII mutants affect lysis inhibition and host range
r+ → plaque size = small → host range = all E.coli
rll → plaque size = large → no growth on K12 lamda (contains a lamda phage lysogen protects against T4 infection)
- rll mutant phage isolated by screening for large plaques OR negative selection; look for no growth on K12 lamda
- in order to study gene structure, Benzer first determined how many genes were responsible for the rII phenotype by complementation analysis
Identified 2 genetic loci; rllA and rllB
Constructing a genetic linkage map of a phage
- map genes/complementation groups relative to one another
- requires that most of phage genes be identified→no more complementation groups can be identified
1) select mutants in different complementation groups/genes
2) perform RECOMBINATION EXPERIMENTS/CROSSES to determine distances between genes & order
- Two-factor crosses: measure RF in phage
coinfection, growth on permissive conditions (determines total progeny), growth in restrictive conditions
Eg. am X ts. 1) sup-, low temp to measure total progeny (permissive). 2) sup+, high temp to measure recombinants (restrictive). This method only measures half of the total recombinants because double mutants are not recovered.
Use high phage titer to allow coinfection in permissive conditions. Plate in permissive → total possible progeny. Plate in restrictive → total recombinants x2
Three factor crosses
- For mutations that are very close together (very close genes/mutations in same gene), scientific error precludes using 2-factor crosses to determine order
use 3-factor crosses to determine mutation/gene order
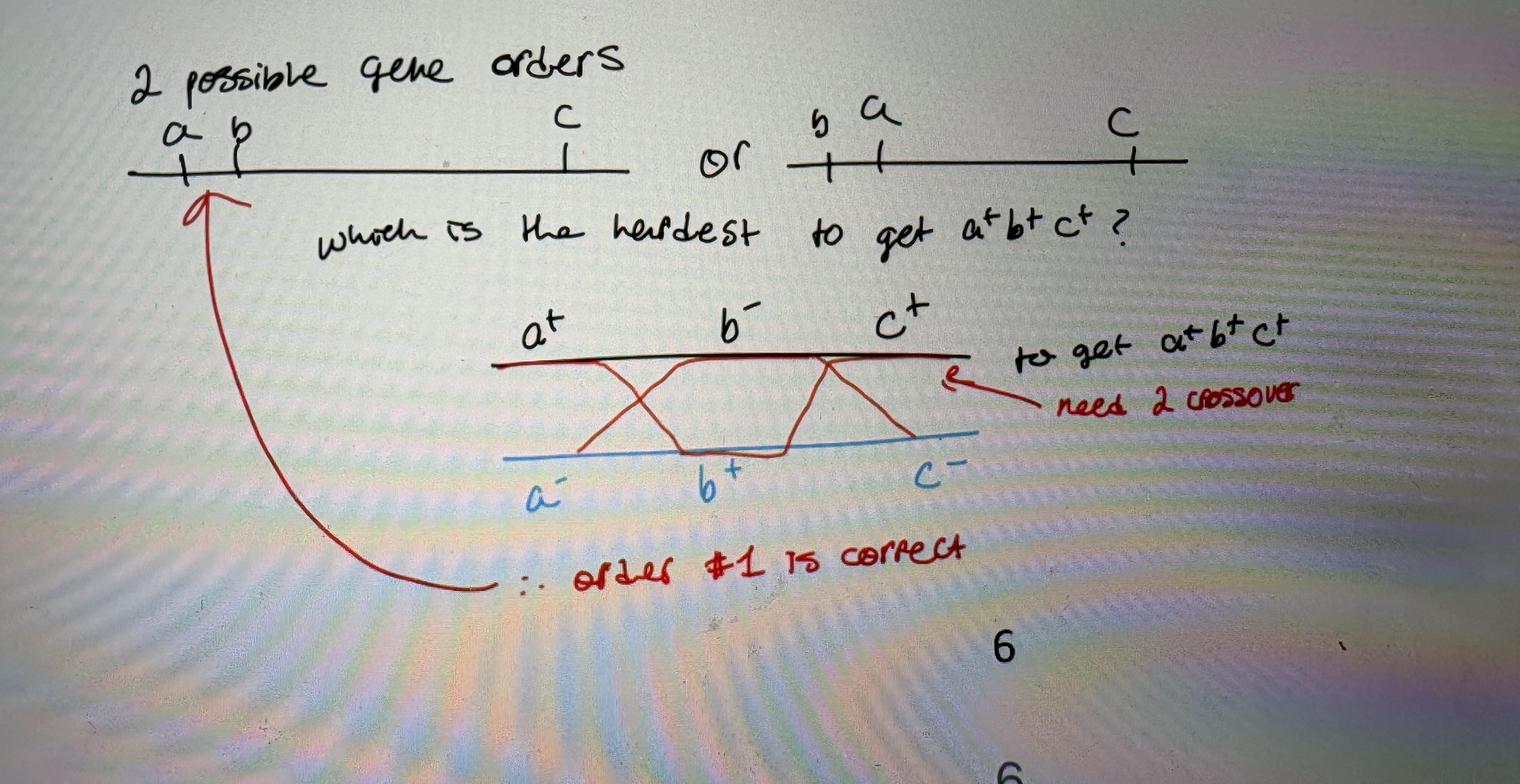
1. 2-factor crosses are used to assign two possible gene orders
- a & b are so close together (0.3%), you can’t determine which map is correct cause in realm of standard error.
2. a mutant strain with 2 mutations is crossed with a mutant with only 1 mutation and the recombinants are examined
- a+b-c+ (single mutant) X a-b+c- (double mutant)
- choose 1 phenotype to select for
- # crossovers required to generate recombinants depends on gene order:
greater # of crossover required → less frequent recombinant class
a+b+c+ = Low #, rarest class thus requires greatest # of crossover to occur
rarest recombinant class represents multiple crossover (see pic)
Constructing a fine structure map of the rll gene/Deletion mapping
- It was known that genes were organized in a linear array along DNA molecules
- structure of genes unknown → linear?
- Benzer → mapped about 2400 mutations in the rII locus with goal of understanding internal organization of a gene
- rII gene permitted observation of rare intragenic recombination because:
ease of isolation of rll mutants
high recomb freq of T4
Ability to select for rll mutants → wt = plaques on K, rll = no plaques on K
crosses are simple = coinfection
ability to easily isolate rll gene deletions
- Map mutations within genes (fine structure of gene)
select mutants from same complementation group. Initially identified 2 complementation groups; rllA & rllB
- Benzer’s important conclusions from his work:
a gene has internal structure (ia a divisible unit)
Fundamental units of mutations are diff from one another
gene is linear
- Can map extent of a deletion because areas deleted can’t be recombined (are gone so can’t engage in recomb event).
- Deletion strains can allow rapid mapping of point mutations
- If point mutation lies outside the deletion, wt will be recovered. If point mutation is w/in deleted segment no wt recovered.
- 2400 mutant strains of rll → too many to cross. Used deltetion mapping
- Deletion mutants identified by:
inability to revert (no plaques on E. coli K12 )
no wild-type progeny when crossed with many point mutants
- the extent of deletions determined by crossing the deletions against one another
- Can map extent of deletion by crossing non-revertible strains to one another
If deletions overlap = no wt progeny
- After inital ordering, endpoints defined by crossing against previously mapped rll mutants
created a suite of known deletion mutant breakpoints
Point mutants mapped using deletion mutants/Spot tests
1. use deletion mutants to quickly localize new point mutations
- crossing to known largest deletion
- wt can be formed if there is a crossover between deletion endpoint & mutation location
2. cross mutations against a smaller set of deletions within the first deletion they localized to
- Crossed rll revertible mutations to biggest known deletion first.
- allows a quiet refinement of mutation location
- a set of mutants that all mapped to large A5 region
- cross revertible rll mutants to a subset of smaller strains
- Do it again. All strains map to a small segment of rll locus
3. cross mutations against individual point mutants in two- and three-factor crosses
- now, you have a set of mutants that you know are tightly linked
- crosses reveal the relative order of alleles
- Required over 25 000 crosses. If you crossover, you can grow.
- Benzer developed a SPOT TEST →10 to 20 crosses on a single plate
- No phage will grow without a cross → no plaques
- Spot w/ rll mutant to be mapped:
Grow into a full plaque: mutant & deletion complement
Small plaques: Plaques form only if crossover thus mutant is in same complementation group as deletion (no complemenetation). Mutant doesn’t lie w/in deletion so recombinant wt phage produced
No plaques: Mutant site is w/in deletion- no complementation, no recombination, no growth
- What can grow on spot test?
rllA mutants w/ rllB mutants because complement
fewer plaques form if a crossover is required
- Spot test allowed:
rapid analysis of MANY crosses on fewer plates (10-20 crosses per plate)
Determining complementation, recombination, intradeletion mutations
Conclusions from fine structure mapping
- topology of rII gene is simple and linear; can line up mutations suggesting genes are linear in DNA
- all parts are not equally “mutable” → Hotspots (both spontaneous or induced)
- non-specific mutagens show specificity for certain sites
- varies amongst mutagens
rllB: Each box is a known mutant strain. 1 strain was mutant at this locus. Can identify each mutant & map them w/ respect to one another
rllA: Hotspot locus that was frequently mutated.
- How would you know when you had isolated 2 mutants affecting the same site?
2 revertible mutants (not deletions) that NEVER crossover
- two-factor and three-factor crosses did not agree
- eg. two-factor crosses place r, h, and ac in the order: h ac r; three-factor crosses suggested the order was r h ac
- how can this be?
Sometimes they mapped really close together (2 factor). Sometimes they were unlinked (3 factor).
Conclusion from the data was that maybe T4 phage genome was circular
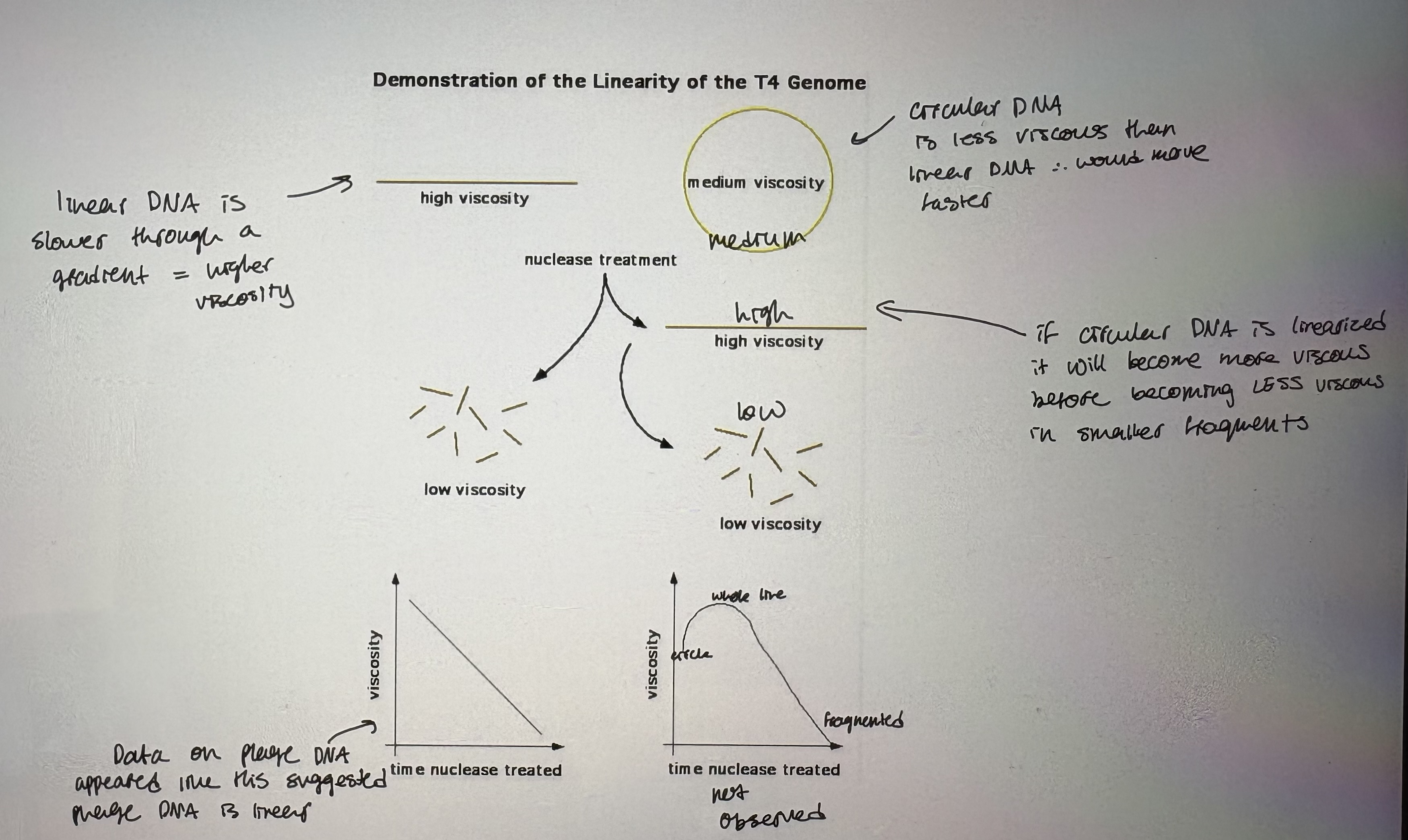
- However, viscosity experiment revealed that T4 phage heads carry linear DNA (see pic)
Linear DNA is slower through a gradient = higher viscosity
Circular DNA is less viscous than linear DNA thus would move faster
If circular DNA is linearized it will become more viscous before becoming less viscous in smaller fragments
Data on phage DNA appeared suggested phage DNA is linear
Graph: Viscosity steadily decreased overtime, indicating genome is linear
- Possible explanations for the circularity of the T4 map;
Circulization occurs after infection
Individual DNA fragments are terminally redundant; phage genome has extra DNA at ends w/ redundant sequences
- Evidence for terminal redundancy:
Genetic → phage heterozygotes
Don’t normally see heterozygosity in haploid. But coinfection w/ 2 strains carrying diff genetic markers like a heterozygote
How do phage heterozygotes arise? Cross 2 strains that have terminal redundancy & look for recombinants
Crossover allows for novel comboinations of alleles. Shows 2 copies of 1 allele on one genome
If phage genome is shortened by deletion → isolate an inc # of terminal heterozygotes
The bigger the deletion, the more genes that show heterozygosity due to terminal redundancy. Thus bigger deletions have bigger terminal redundancy suggesting a fixed genome size regardless of amount of content
Biochemical → if you denature (dsDNA to ssDNA) many genomes then allow reannealing (ssDNA to dsDNA) = resulted in concatomers (repetitive genomes joined together)
Reannealing requires complementarity. Exonuclease digestion inc cocantomerization
After exonuclease digestion and low conc of DNA, reannealing, viscosity experiments showed it reannealed as a circle
- T4 DNA is linear and terminally redundant
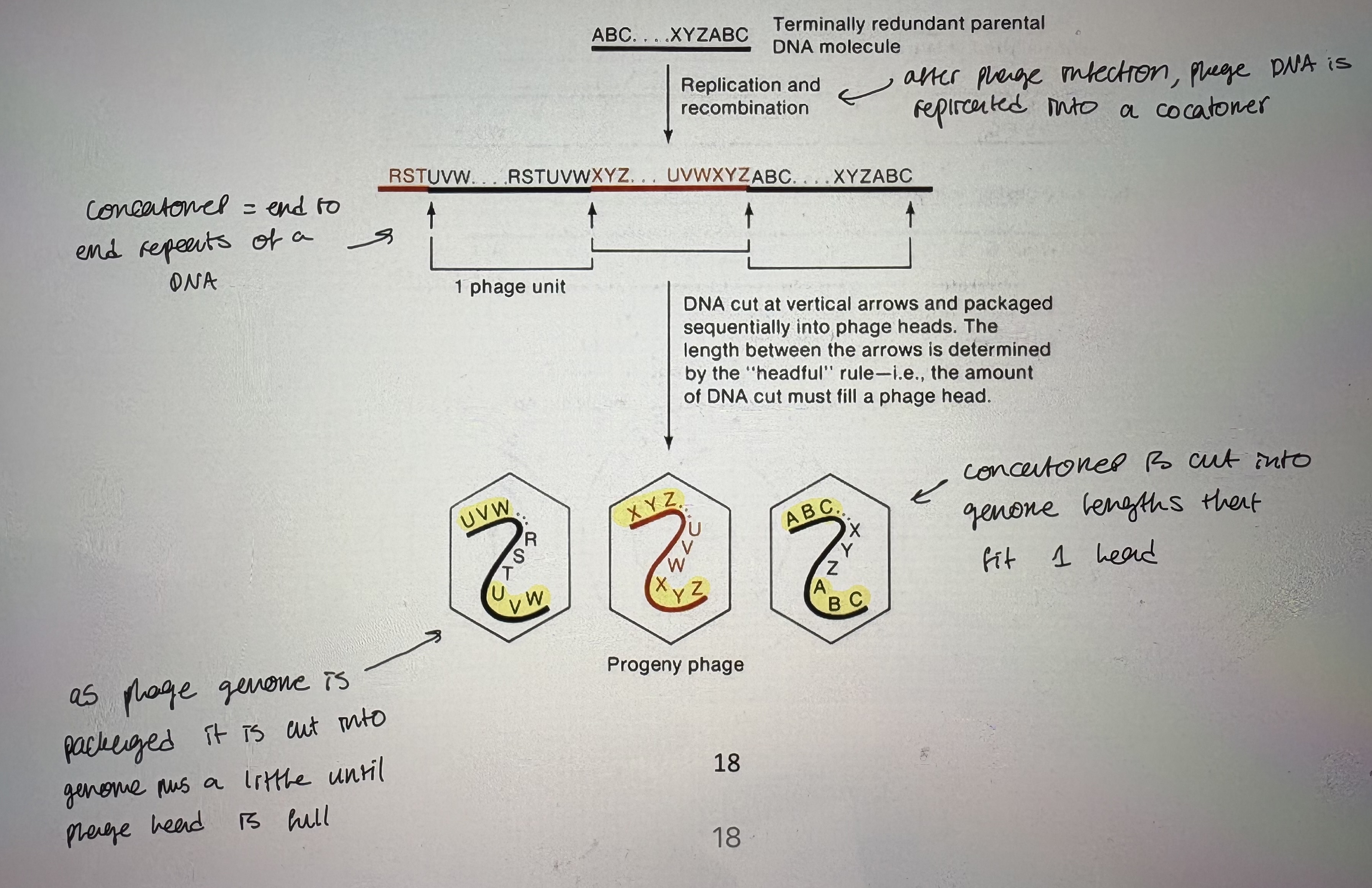
Headful Packaging of Replicated T4 DNA
- How are terminally redundant genomes generated?
- headful packaging of phage DNA into phage head explains circular map/linear genome confusion
- After phage infection, phage DNA is replicated into a concatomer (end to end repeats of a DNA)
- As phage genome is packaged it is cut into genome plus a little until phage head is full
- Concatomer is cut into genom lengths that fit 1 head
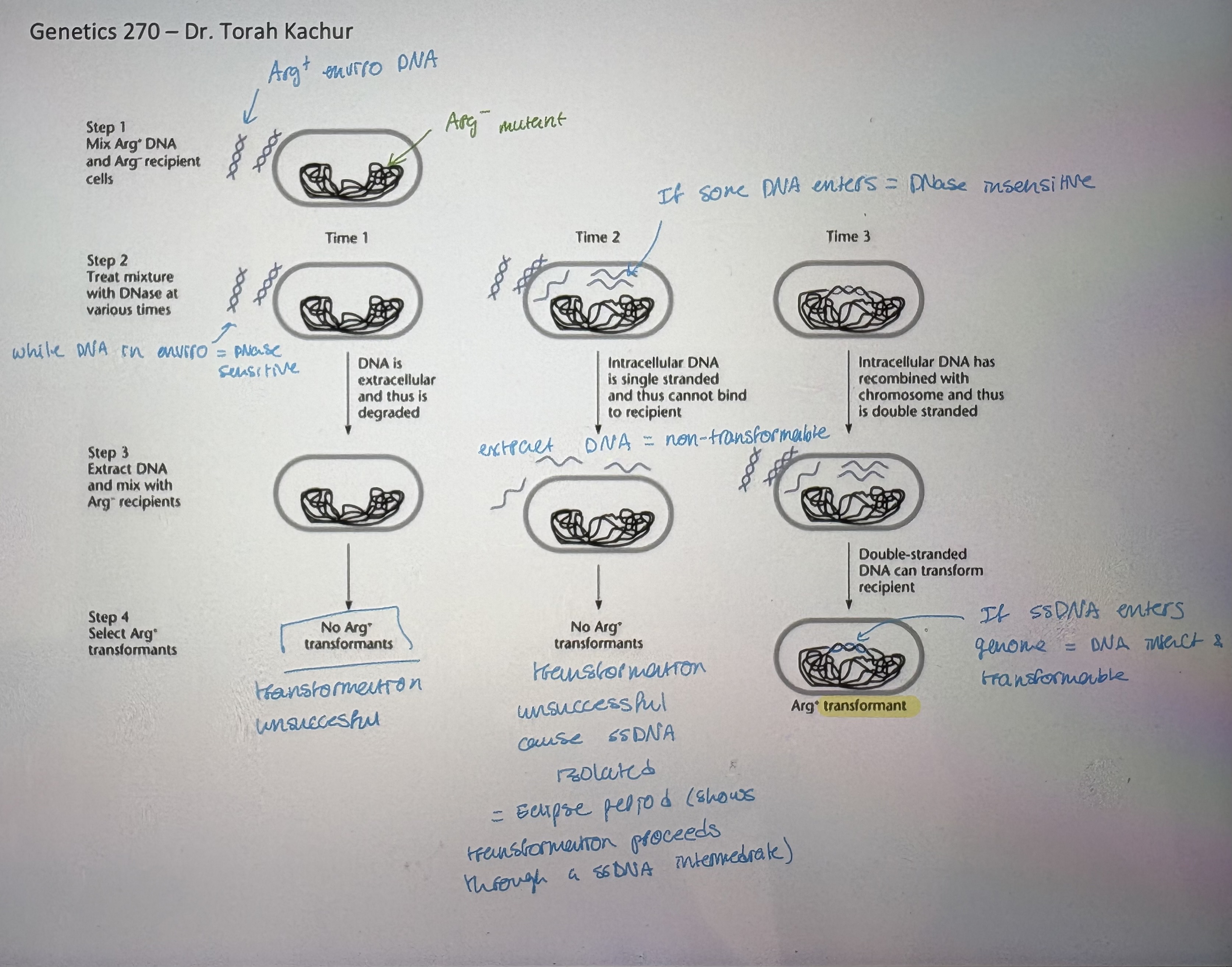
Bacterial transformation overview/Biology of transformation
- Uptake of free DNA from environment
- recipient bacterial cell aquires DNA from environment (comes from dead bacteria)
- bacteria takes up linear DNA & fate of DNA is:
Degradation = if no sequence homology
Integration into bacterial chromosome = if there is sequence similarity/homology
- Transformant carries new DNA from related donor
- first demonstration = Griffith’s transforming principle
- R Streptococci (avirulent) acquire TRANSFORMING PRINCIPLE from S Streptococci (virulent) → allows them to infect & kill mice
Used Streprococcus spp. = rare naturally cometent species
Showed an abiotic factor can “transform” a nondisease causing strain into disease causing = later shown that DNA was the transforming principle
1. Detection: require a phenotype that can be selected for/against
2. Competence: the ability to take up DNA from environment
- special state of bacteria in which they can take up exogenous DNA
- influenced by growth state, medium
- species variable
- Involves:
cell wall changes
encode & express Com proteins to induce natural competence
CSF (competence specific factor): is produced at high cell densities → helps convert other cells to competence (indirect activation of Com proteins which activate natural competence)
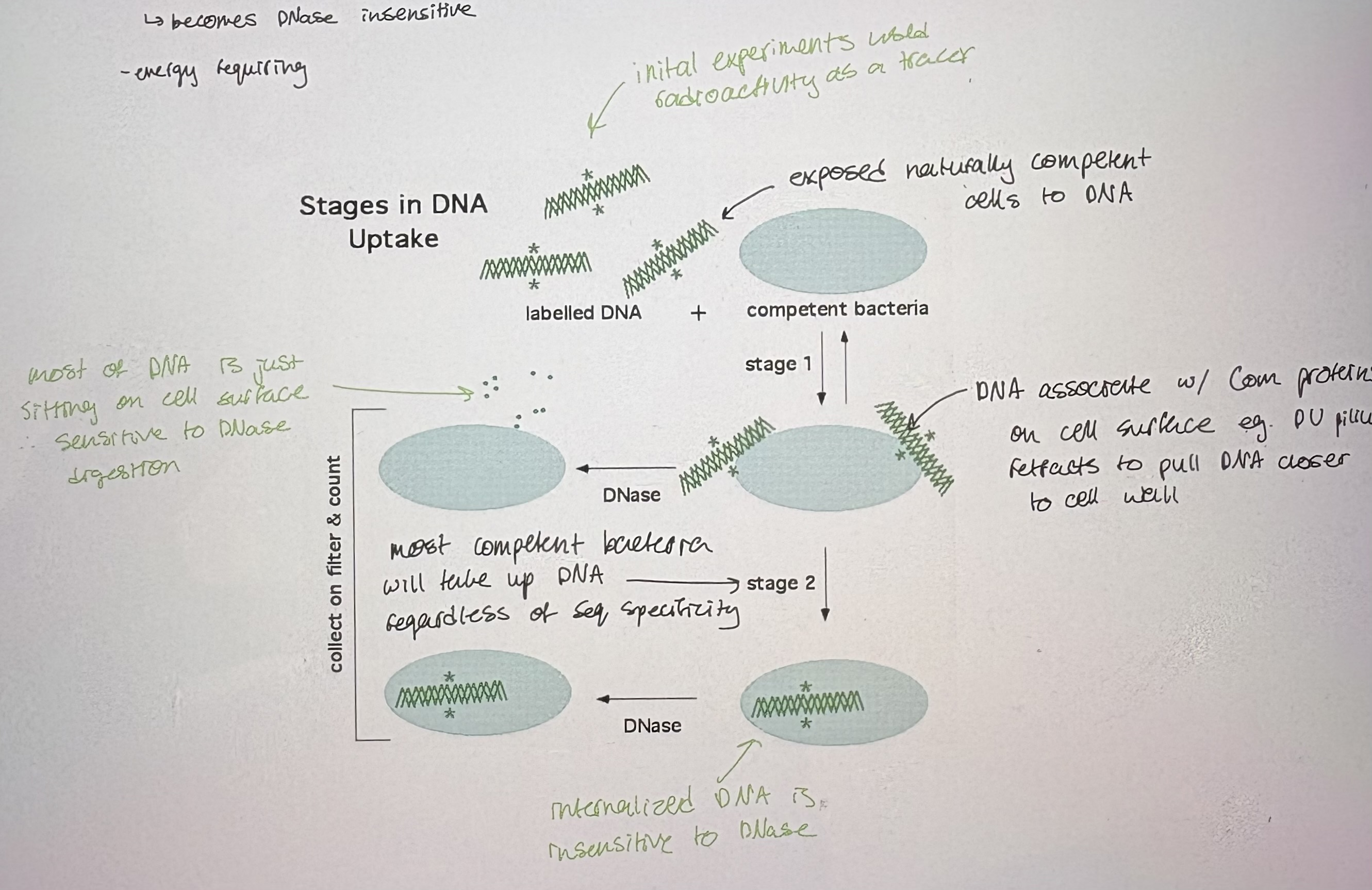
3. DNA uptake: 2 steps → revealed by DNase sensitivity experiments (see pic)
- i) Reversible binding to cells:
requires dsDNA to bind to competence receptors
DNA is still be extracellular → can be washed off, sensitive to DNase
- ii) irreversible interaction
DNA passes across cell memb; becomes DNase insensitive
energy requiring
Mechanism of transformation
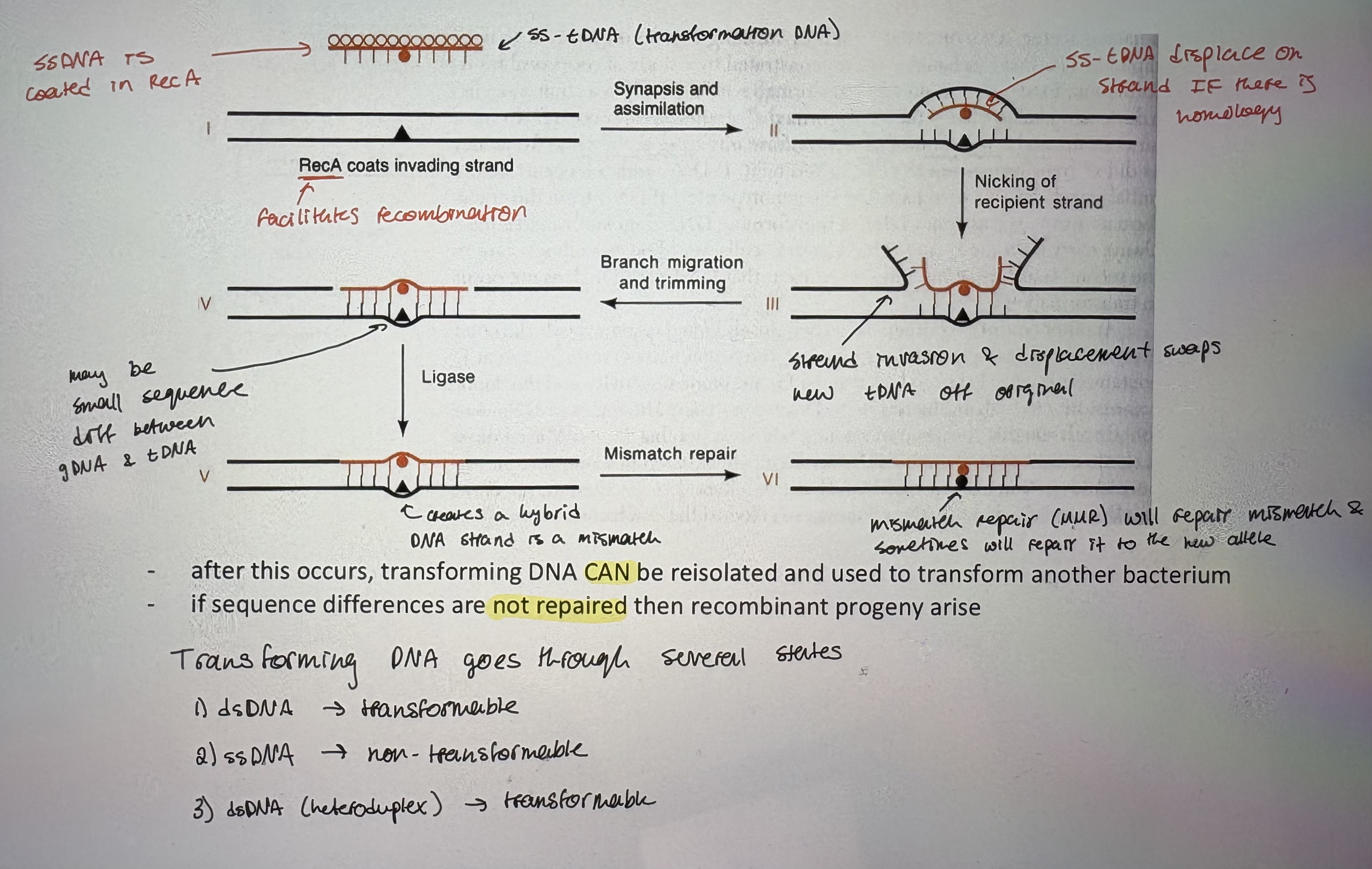
- DNA becomes incorporated stably into genome of recipient
- Shortly after DNA is taken up → DNase one strand
- ss-tDNA is coated in RecA (facilitates recombination)
- ss-tDNA displace one strand IF there is homology
- Nicking of recipient strand. Strand invasion & displacement swaps new tDNA off original
- May be small sequence diff between gDNA & tDNA
- Creates a hybrid DNA strand is a mismatch
- Mismatch repair (MMR) will repair mismatch & sometimes will repair it to the new allele
- after this occurs, transforming DNA CAN be reisolated and used to transform another bacterium
- if sequence differences are not repaired then recombinant progeny arise
- Transforming DNA goes through several states:
dsDNA → transformable
ssDNA → non-transformable
dsDNA (heteroduplex) → transformable
- Arg- example (see pic)
Natural & Artifical transformation/Uses of transformation
1. Natural Transformation
- only some bacteria; eg. Haemophilus influenzae, Neisseria gonorrhoeae, Streptococcus pneumoniae
- DNA uptake may be species specific
- DNA must have regions of homology
- may be used for: (mechanism of inc genetic diversity)
Nutrition, repair, recombination
2. Artificial Transformation
- generally used to move plasmids into bacteria
- most bacteria not naturally competent, can force competence:
i) Calcium Ion Induction
- cells treated with calcium ions will take up both ssDNA and dsDNA
- DNA is added and forms calcium-DNA complexes that bind to cell
- heating causes transport of DNA into cell
- if DNA can replicate, it becomes permanently incorporated into cell
- requires SELECTION in order to detect
ii) Electroporation
- cells are exposed to an electric field
- small pores transiently form in membrane, making cell permeable to exogenous molecules, like DNA
- much more efficient than calcium transformation
- Mapping: based on principle that 2 markers will only transform together if they are physically linked. The closer 2 loci are, the more likley they will transform together.
If the 2 loci are far apart → single transformants recovered, no double transformants
If 2 loci are close together → should transform together
- When setting up a cotransformation experiment; select for one donor genotypes tp test for successful transformation then select for any cotransformants of other locci potentially linked to one donor genotype
Donor = wt. Ideally take wt donor DNA (plus at loci being tested) beacuse doing positive selection is easier
The recipient should be mutant for loci being tested
- Co-transformation frequency (COTF): the % of total transformants selected for 1 marker also recombinant for another marker
# x+y+ cotransformants/ # x+ transformants
numerator shows succesful cotransformation, denominator shows succesful transformation
Thus, higher frequency = loci are CLOSER together
- Total x+ tranformants includes x+y+ AND x+y- transformants
- Identification of chemical agents that act on DNA: pure DNA is exposed to chemical and the progeny of a transformation experiment are examined for mutants (see pic)

Bacterial conjugation overview
- first observed in 1947 by Lederberg and Tatum – “gene recombination in E. coli”
Took 2 diff auxotrophic strains → added strains into same media → got non-parental gene combinations
- mixed together different auxotrophic strains and got strains unlike either of parents
- interpreted data as “assortment of genes in new combinations”
- possible explanations:
Cell fusion
transforming factors in media promoted competence (generally non-competent species)
direct transfer of genetic material → conjugation
- Bacterial plasmids: replicate independent of host genome, free floating in cytoplasm
small, circular, supercoiled DNA molecules
depend on host cell for replication functions
number of copies per cell varies among plasmids
bacteria can harbour more than 1 plasmid at a time
diff types of plasmids. Eg F plasmid → conjugative plasmid
- Most plasmids confer a benefit to bacteria under certain conditions
- Transfer of plasmid DNA is a multi-step process involving several functions
some plasmids are self-transmissible (they encode all the functions needed to move among strains)
Ex. F-plasmid (fertility factor): donor strains = F+ (“male”). Recipient strains = F-, lack of a transmissible plasmid (“female”)
Very low copy # of F plasmid
- Genes on F plasmid encode proteins for conjugation & DNA transfer process.
“selfish” insertion element (IS) = allow integration into genome
During conjugation, F plasmid can be fully transferred from F+ to F- cell
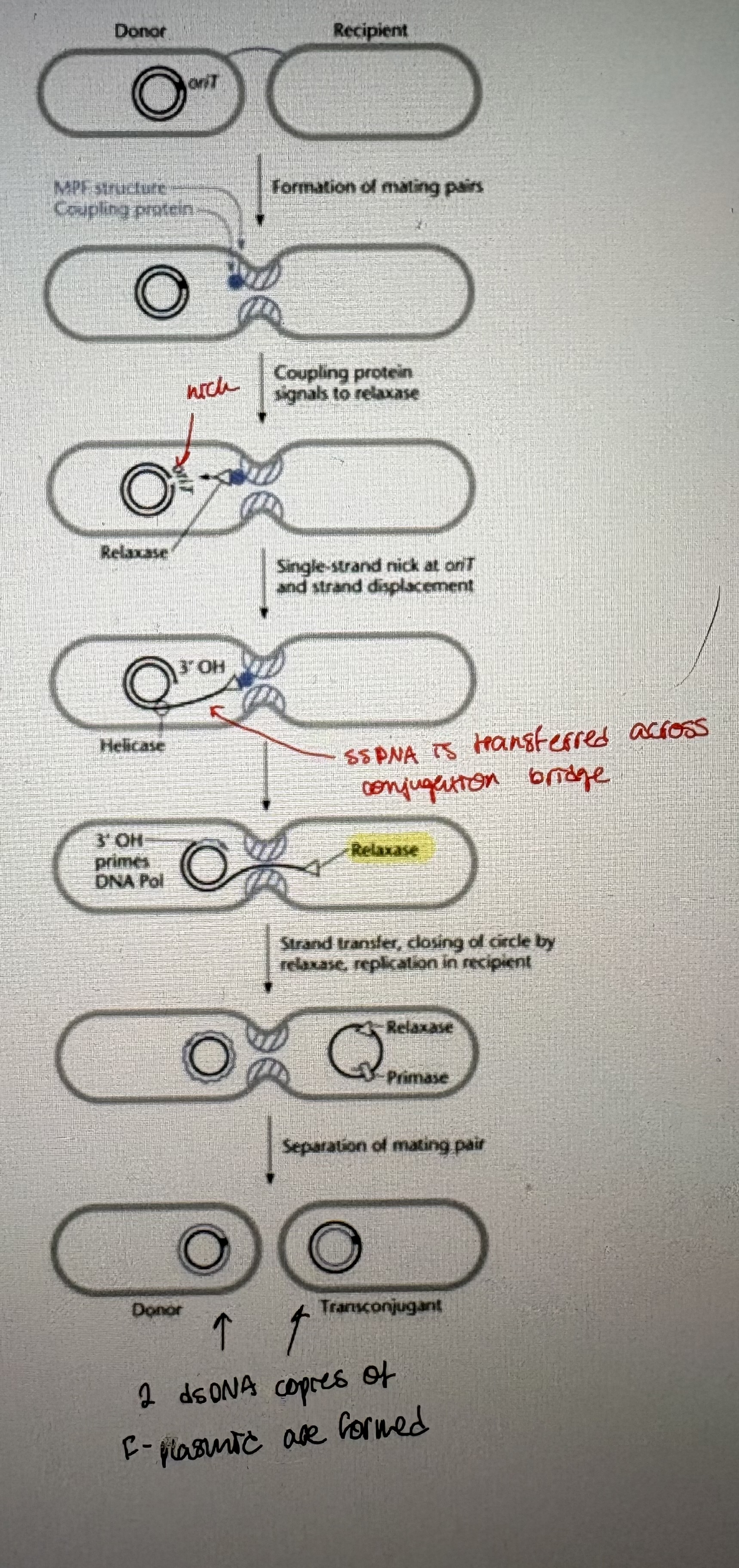
Plasmid transfer steps/Chromosome transfer by plasmids
1. Formation of mating pairs
- requires cell-to-cell contact between donor and recipient
- mediated by a sex pilus on donor cell (F pilus for the F plasmid)
- pilus is made up of polymer of pilin (protein)
- tip of pilus binds to recipient cell to initiate mate pairing.
- Note: DNA has not been shown to move through pilus
2. Preparation for DNA transfer
- cells contact each other after pilus retraction
- form conjugation bridge = allow direct transfer of DNA between bacterial cell
- Nicking of F plasmid at OriT (origin of transfer)
3. DNA transfer
- direct between cells
- ssDNA is transferred after nicking by relaxase
4. Formation of replicative plasmid in recipient
- ssDNA in both the recipient & donor is replicated to dsDNA → rolling circle replication (RCR)
- Conjugation functions are encoded by tra genes (all encoded on F plasmid)
traA = pilin subunit. traY = relaxase.
- other plasmids may encode some, none, or all of functions required to be self-transmissible
Self transmissible: mating pair formation DNA transfer (F plasmid)
Conjugative: mating pair formation
Mobilizeable: DNA transfer
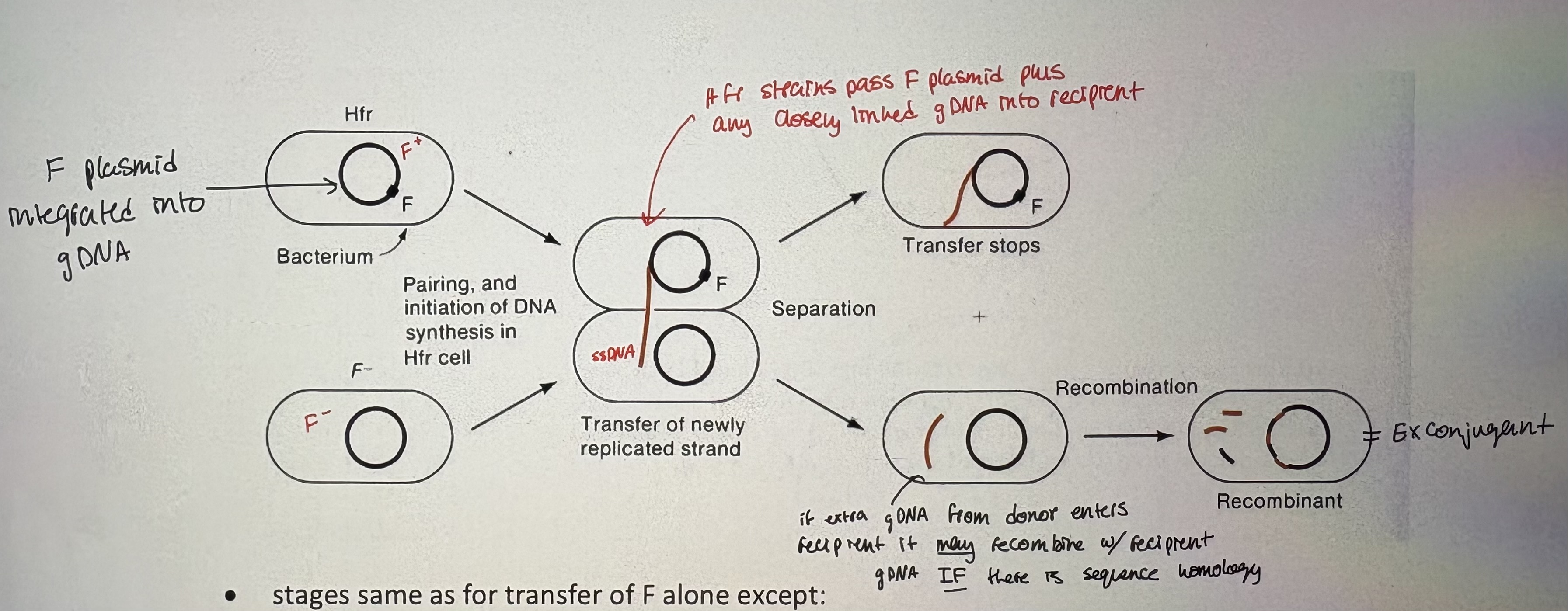
- F can integrate into the bacterial chromosome and mediate transfer of the chromosome to a recipient
• these strains are called Hfr strains (for high frequency of recombination) → many recombinants formed when Hfr strain mixed with recipient strain
- Insertion of F into the E.coli chromosome: occurs by recombination between homologous insertion elements on the F plasmid and the chromosome
F plasmid can integrate inro gDNA (genomic DNA)
~>20 typical insertion sites (have homology to IS sequence on F plasmid)
BUT = integrated F plasmid reatins self-transmissible properties creating Hfr strain
- Creating Hfr strain:
relaxase nicks the oriT & pulls ssDNA across conjugation bridge. Orientation of OriT dictates direction of transfer
IS element provide sequence to allow recombination
Hfr strains can integrate in diff location & orientations
- upon integration, F behaves as part of chromosome but retains self-mobilization ability (see pic)
- stages same as for transfer of F alone except:
take ~100 min of stable conjugation to transfer F plasmid plus entire E.coli genome into a recipient
mating pair usually breaks apart before 100 min (can also experimentally break up mating by agitation = “interrupted mating”)
some donor DNA can recombine recipient to create novel allelic combos
- Detection of recombinants: can select for successful conjugation by testing presence of donor DNA alleles

Hfr mapping
- an Hfr strain contains a single transfer origin
- gene on gDNA will be transferred in order they appear after OriT = measured in min (pinch & pull arrowhead)
- methods of selection (for conjugant = donor DNA recieved into recipient) & counterselection (against donor)
Eg. Pro+ StrR recombinants are plated out after various times of mating between Hfr pro+ strS (donor) and F- pro- strR (recipient)
donor is steptomycin sensitive thus growing conjugants in presence of strep selects against donor → counterselection
Pro+ selects for successful conjugation, StrR selects for recipient cells
When does pro+ appear in recipient? ~20 min thus pro gene is 20 min away from Hfr site
- When does the pro gene enter the recipient?
to figure this out, the time is graphed on the X axis and the number of recombinants on the Y axis
extrapolating back to zero yields the time of entry of the pro+ gene (actual value will be on X-axis)
the pro gene entered at approximately 15 minutes
- Hfr Mapping with collections of Hfrs: used to localize a gene/mutation to a small area of the chromosome
use collections of Hfr strains
each Hfr strain: 1) contains selectable marker that is transferred early = allows selection for conjugation (allows recipient to grow ie positive selection) 2) contains selectable marker that is far away from OriT = allows selection against donor (kills donor)
Method: 1) 4 diff known Hfr strains allows refined mapping of chromosome loci. 2) do interupted mating experiments for each Hfr strain 3) more frequent apperance of selected trait → the closer its to that Hfr site (see pic)
- Note: only Hfrs located near the chromosomal position of the mutant gene and in the correct orientation will transfer the corresponding wildtype gene at a high frequency