BLD 424 Exam 2: hereditary and acquired hemolytic anemias
1/36
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
37 Terms
pathophysiology of hereditary hemolytic anemia
anemia of increased destruction
RBC morphology
1. >80% of cells have abnormal shape
2. membrane defects: caused by decreased deformability of the RBC
Hereditary ellipocytosis vs spherocytosis
ellipocytosis (ovalocytosis): can tell on the slide, >80% of cells are ovalocytes
spherocytosis: cannot tell on the slide
red cell energy production review
1. depends on glycolysis (anaerobic)
2. side pathways important for other cellular functions:
-hexose monophosphate shunt: keeps glutathione reduced
-methemoglobin (fe 3+)reductase pathway maintains reduced heme (fe 2+: O2 cannot bind) iron
-rapoport-leubering shunt forms 2,3 BPG
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
1. a number of variants of the enzyme mutations
2. most common form is sex linked recessive (most often african and asian desents
3. steady state of symptomatic anemia
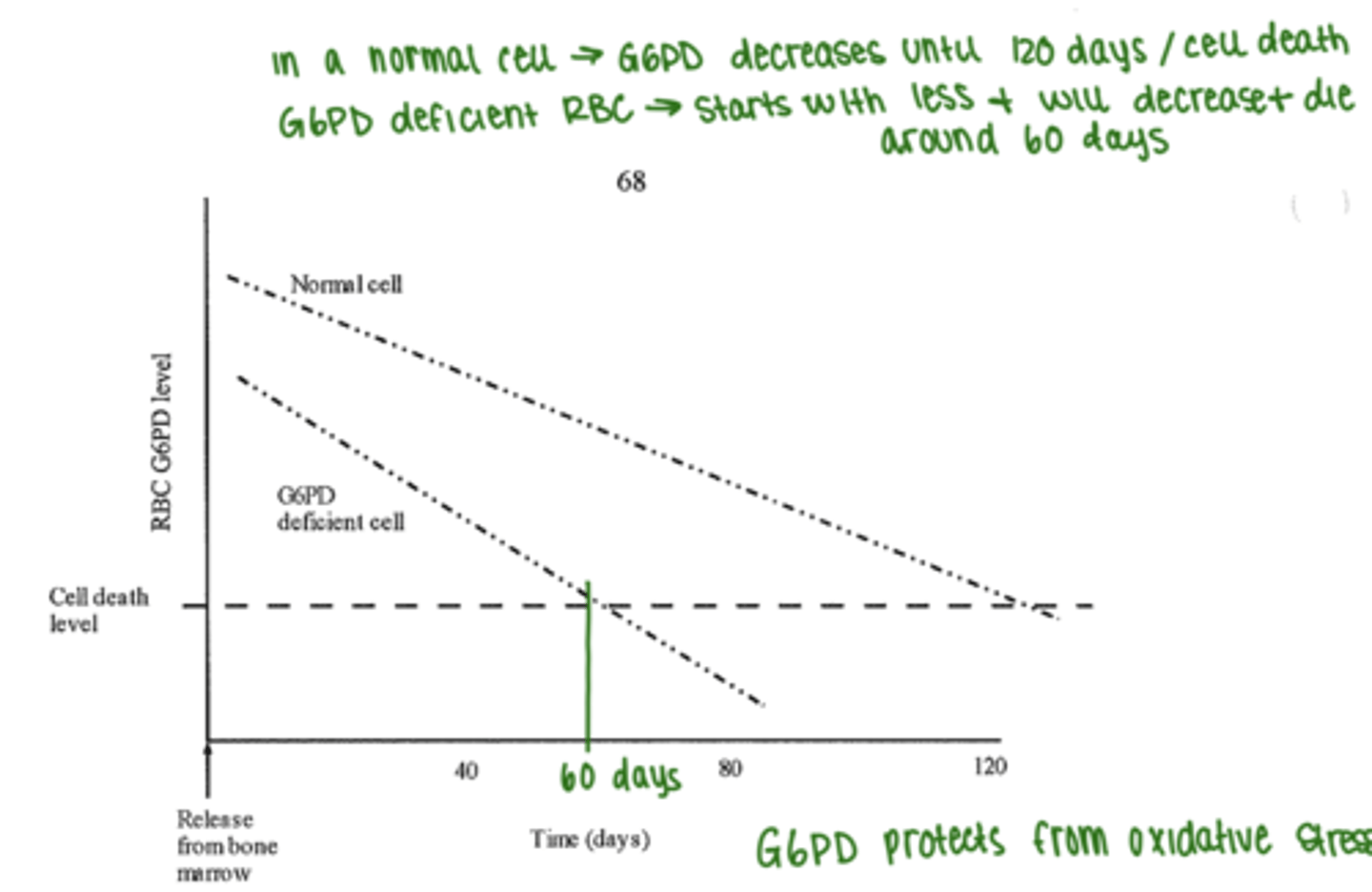
4. fully compensated state with low levels of hemolysis: RBCs have shortened lifespan (see diagram)
5. can have acute hemolytic episode: infection, drugs, fava beans
G6PD deficiency: pathogenesis of hemolysis
1. without G6PD, glutathione cannot be reduced:
-oxidant injury to hemoglobin Fe2+ --> Fe3+ and globin chains denature
-aggregation of denatured HgB - Heinz bodies adhere to RBC membrane
- hemolysis occurs by 2 mechanisms:
a. macrophages try to remove heinz bodies and RBCs lyse
b. more commonly RBCs lyse moving through the spleen --> fragmentation
-oldest RBCs lyse first
clinical presentation of G6PD deficiency during crisis
1. hemoglobinuria: Hgb in urine --> stress on kidneys
2. back pain --> stress on kidneys
3. malaise
4. jaundice
Laboratory diagnosis of G6PD deficiency WITHOUT crisis
1. normal CBC, maybe increased retics
2. quantitate G6PD in RBCs
-no reason to test unless prior hemolytic episode
-screened for defect in newborn screening
Laboratory diagnosis of G6PD deficiency WITH crisis
they have had a hemolytic event
1. blood picture: normo, normo, poik with shisto and spheros, increased RDW (aniso)
2. hemoglobinemia: free hgb in plasma
3. hemoglobinuria: brown urine
4. decreased haptoglobin
5. Quantification of G6PD in RBC may be WRR because most deficient RBCs lysed first
6. positive for heinz bodies (precipitated globin chains)
laboratory diagnosis of G6PD deficiency following a crisis
1. increased retics (MCV can get macrocytic due to reticulocytes
2. increased indirect bili
3. G6PD is WRR in retics
pyruvate kinase deficiency physiology
1. autosomal recessive: homozygosity required for clinical problems to develop
2. reduced energy production leads to rigid cells that are removed in the spleen
-cells are vulnerable once mitochondria is gone and they must rely on anerobic glycolysis (retic stage)
-increased 2,3 DPG promotes oxygen delivery to tissues so moderates the impact of the anemia
3. severe hemolysis: jaundice, splenomegaly, gall stones
pyruvate kinase deficiency laboratory diagnosis
1. blood picture: normo normo, increased RDW, reticulocytosis that may cause macrocytosis
2. elevated LD
3. elevated indirect and total bilirubin
4. decreased haptoglobin
5. negative DAT
6. PK reduced
7. heinz bodies (precipitated globin chains)
pyruvate kinase deficiency treatment
splenectomy and transfusions
quantitative hemoglobinopathy
thalassemia (cooleys): globin chains have normal structure but not correct number of them
qualitative hemoglobinopathy
normocytic anemias of increased destruction: globin chains abnormal
-sickle cell anemia
sickle cell anemia physiology
1. defect in substitution of glutamic acid for valine at the 6 position on the beta chain - HbS
-substitution of one base pair: thymidine for adenine (GAG GTG)
2. deoxygenated hgb less stable
3. cells become rigid and form sickle
4. sickles block capillaries causing tissue damage necrosis and severe pain
homozygous sickle cell disease
1. BsBs
2. severe hemolytic anemia
3. sickle crisis caused by stress, dehydration, infection, exercise
4. sickled cells block vessels throughout the body causing infarcts and thrombosis and organ damage
5. spleen can die/autosplenectomy
6. cells become irreversibly sickled and ultimately lyse or get engulfed by macrophages (hemolytic jaundice)
homozygous sickle cell disease in babies
1. disease not apparent at birth due to HbF (2xalpha) (2x gamma)
2. at 6 months bone marrow responds that there are bone deformaties from now severe
Heterozygous sickle cell disease
1.BBs
2. typically asymptomatic
3. important for family planning
4. 10% if african american population
double heterozygous sickle cell disease
1. BsB+
2. may have one sickle gene and the other beta gene is thalassemia
blood picture of sickle cell anemia
normo, normo
increased RDW
retics/NRBCs
poik: sickles, targets, shistos
basostip/howell/pappenheimers
thrombocytosis/leukocytosis
sickle cell disease evidence of hemolysis
1. increased indirect and total bili
2. increased lactate dehydrogenase
3. sometimes jaundice/hgburia
4. increased urine urobilinogen
testing for sickle cell disease
1. screening for HbS: released Hb is deoxygenated by sodium dithionite
2. confirmation: hemoglobin electrophoresis, HPLC
hemoglobin electrophoresis for sickle cell disease
1. alkaline separation first, followed by acid to separate co-migrating fractions in S region
2. acid separation to isolate HbS
HPLC for sickle cell disease
1. cation exchange: HgB binds to column with different affinities
2. co migrating hemoglobins: HbS, HbC
sickle cell disease treatment
1. supportive during crisis with fluids and oxygen
2. hydroxyurea: increases HbF levels
3. voxelotor and crizanlizumab: inhibit sickle forming
4. bone marrow transplant
5. CRISPR
hemoglobin C disease physiology
mutation in B globin chain
1. seen in the same populations as HbS
2. globin variant with lysine replacing glutamic acid at the 6 position on the beta chain
hemoglobin C clinical disease
1. occurs only in homozygotes and is mild to moderate hemolytic anemia with all attendant clinical signs and laboratory findings
2. splenomegaly
hemoglobin C blood picture
normo,normo
1. target cells
2. HbC crystals
3. reticulocytosis
4. HbC on electrophoresis
hemoglobin SC
1.BsBc
2. no normal beta chains: S and C instead
3. compne sated, mild hemolytic anemia intermediate in severity between SA(trait) and SS(disease)
4. HbS and HbC both apparent on electrophoresis
5. sickle cells and C crystals on peripheral blood
6. pocket book cells: folded target cells
immune hemolysis
acquired
1. mechanical: usually IgM antibodies that bind C and cause shistocytes
2. macrophage mediated: IgG antibodies cause spherocytosis with positive DAT
Paroxysmal nocturnal hemoglobinuria physiology
membrane defects (intrinsic)
1. defective stem cell causes membrane protein deficiency that allows cells to bind C more than usual
2. missing GPI anchored proteins
3. decreased expression of surface markers that anchor to GPI: CD55, CD59 (prevent RBC lysis by complement)
4. RBCs and WBCs have increased sensitivity to lysis by complement
mechanical (fragmentation) hemolysis caused by
1. malaria, bacteria, and other parasites
2. artificial heart valves
3. disseminated intravascular coagulation
4. venoms
5. burns: microcytic
clinical findings of paroxysmal nocturnal hemoglobinuria
1. when sleeping, blood pH drops, intravascular hemolysis (have low level of hemolysis all the time)
2. thrombosis (clots): complement binds and activates platelets
3. prone to infections: complement binds WBCs and they lyse
PNH blood picture
1. pancytopenia
2. normocytic w/ retic and increased RDW
3. severe anemia = NRBCs
4. may see shistocytes
PNH other lab diagnosis features
1. increased total and indirect bili
2. hemoglobinuria
3. decreased haptoglobin
4. increased plasma hemoglobin
5. increased LD
6. increased urobilinogen
7. negative DAT
8. urine tubular cells in sediment stain pos. for iron
FLAER for PNH
1. binds GPI directly
2. CD24 is on all normal granulocytes
3. monocytes are negative for CD14