GW BGZ2026 Practical - Computer lab assignment pharmacokinetics
1/54
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
55 Terms
What is pharmacokinetics?
Pharmacokinetics describes what the body does to a drug over time. It includes the processes of absorption, distribution, metabolism, and excretion (ADME). These processes determine the drug concentration in the blood and tissues and therefore influence onset, intensity, and duration of action.
What are the two core pharmacokinetic parameters?
Volume of distribution (Vd) – describes how extensively a drug distributes into body tissues compared to plasma.
Clearance (CL) – describes the body's ability to eliminate a drug per unit time.
Together, they determine drug concentration, half-life, and dosing requirements.
What is the difference between pharmacokinetics and pharmacodynamics?
Pharmacokinetics (PK): What the body does to the drug (ADME).
Pharmacodynamics (PD): What the drug does to the body (receptors, effects, MEC, toxicity thresholds).
What is a decadic (base-10) logarithm?
A decadic logarithm answers: “To what power must 10 be raised to obtain a number?”
log₁₀(x) = y means 10ʸ = x.
Example:
log₁₀(100) = 2 because 10² = 100.
Why are logarithmic scales important in pharmacology?
Many biological and drug-related variables span several orders of magnitude (e.g., plasma concentrations). A log scale compresses this wide range into manageable numbers. It also makes exponential processes (like drug elimination) appear linear.
What is the pharmacological significance of pH being logarithmic?
pH = −log₁₀[H⁺]
This means:
A 10× change in hydrogen ion concentration equals 1 pH unit.
Small pH changes represent large chemical changes affecting drug ionization and absorption.
What is the relationship between log values and fold changes?
+1 log unit = 10× increase
+2 log units = 100× increase
−1 log unit = 10× decrease
This is crucial for interpreting drug concentration changes.
What is drug absorption?
Absorption is the movement of a drug from its site of administration into the systemic circulation. It determines how much and how fast a drug reaches the blood.
What factors affect drug absorption?
Lipid solubility (higher → better absorption)
Ionization (non-ionized form absorbs better)
Surface area (small intestine > stomach)
Blood flow
Gastric emptying rate
Food and drug interactions
Formulation and stability
What is bioavailability (F)?
Bioavailability is the fraction of an administered dose that reaches systemic circulation unchanged.
F = (amount reaching circulation) / (administered dose)
IV: F = 1 (100%)
Oral: F < 1 due to incomplete absorption and first-pass metabolism
What is the difference between rate and extent of absorption?
Extent: Total amount absorbed → determines bioavailability (AUC).
Rate: Speed of absorption → determines onset and peak concentration (Cmax, Tmax).
What are key differences between IV and oral administration?
IV: immediate effect, 100% bioavailability, no absorption phase
Oral: slower onset, variable absorption, first-pass metabolism reduces F
IV produces higher and earlier Cmax than oral administration
What is drug distribution?
Distribution is the reversible movement of a drug between blood and tissues. It depends on blood flow, tissue binding, and physicochemical properties of the drug.
What is volume of distribution (Vd)?
Vd = Amount of drug in body / Plasma concentration
It is an apparent volume that describes how extensively a drug distributes.
High Vd → extensive tissue distribution
Low Vd → stays in plasma
Why can Vd exceed total body water?
Because Vd is not a real physical volume. It reflects how much drug leaves the bloodstream and binds in tissues. Lipophilic drugs can accumulate in fat and tissues, producing very large Vd values.
What is drug metabolism?
Drug metabolism is the enzymatic conversion of lipophilic drugs into more polar, water-soluble metabolites to facilitate excretion.
Why is metabolism necessary?
Because lipophilic drugs:
Are reabsorbed in renal tubules
Are poorly excreted
Would accumulate without transformation
Metabolism prevents prolonged drug action and toxicity.
What are the main sites of drug metabolism?
Liver (primary site)
Intestinal wall
Kidney
Lung
Skin (minor)
What are Phase I reactions?
Phase I reactions introduce or expose functional groups:
Oxidation
Reduction
Hydrolysis
They increase polarity and prepare drugs for Phase II metabolism.
What are Phase II reactions?
Phase II reactions conjugate drugs with endogenous molecules (e.g., glucuronic acid, sulfate), producing highly water-soluble metabolites that are usually inactive.
What is the cytochrome P450 system?
A major enzyme system in the liver responsible for Phase I oxidation reactions. It metabolizes most drugs and is a major source of drug interactions.
Key isoenzymes: CYP3A4, CYP2D6, CYP2C9.
What is drug excretion?
Excretion is the removal of drugs and metabolites from the body via urine, bile, lungs, sweat, saliva, or breast milk.
What are the renal excretion processes?
Glomerular filtration (free drug only)
Tubular secretion (active transport)
Tubular reabsorption (depends on ionization and pH)
What is enterohepatic recycling?
Drug excreted in bile is reabsorbed from intestine back into circulation, prolonging drug half-life.
What is clearance?
Clearance is the volume of plasma from which a drug is completely removed per unit time.
CL = rate of elimination / plasma concentration
It determines:
Maintenance dose
Steady-state concentration
Drug elimination rate
Higher clearance → lower drug levels and shorter half-life.
What is half-life (t½)?
Half-life is the time required for drug concentration to decrease by 50%.
t½ = 0.693 × Vd / CL
It determines:
Time to steady state (~4–5 half-lives)
Duration of drug action
Dosing interval
Time for drug elimination
What is the relationship between clearance, volume of distribution, and half-life?
t½ = (0.693 × Vd) / CL
Large Vd → longer half-life
High clearance → shorter half-life
What is steady-state concentration?
Css = Dose rate / Clearance
At steady state, drug input equals drug elimination.
What determines AUC?
AUC = (F × Dose) / CL
AUC reflects total systemic drug exposure.
What determines dosing rate?
Dose rate = Css × CL
Used to design maintenance dosing regimens.
What determines initial IV concentration?
C₀ = Dose / Vd
High Vd → lower initial plasma concentration.
How do you calculate dose in IV administration and what does it represent?
Dose is the total amount of drug administered to the body. In IV administration, it is calculated as:
Dose = Volume × Concentration
Example:
0.5 mL × 2 mg/mL = 1 mg
This represents the exact amount of drug entering systemic circulation immediately, since IV administration bypasses absorption barriers and first-pass metabolism.
Why does intravenous administration result in an immediate peak concentration (C₀)?
IV administration delivers the drug directly into systemic circulation, meaning:
No absorption phase is required
Bioavailability is 100% (F = 1)
Drug instantly distributes into plasma and tissues
Thus:
C₀ = Dose / Vd
The only factor limiting initial concentration is the apparent volume into which the drug distributes, not absorption speed.
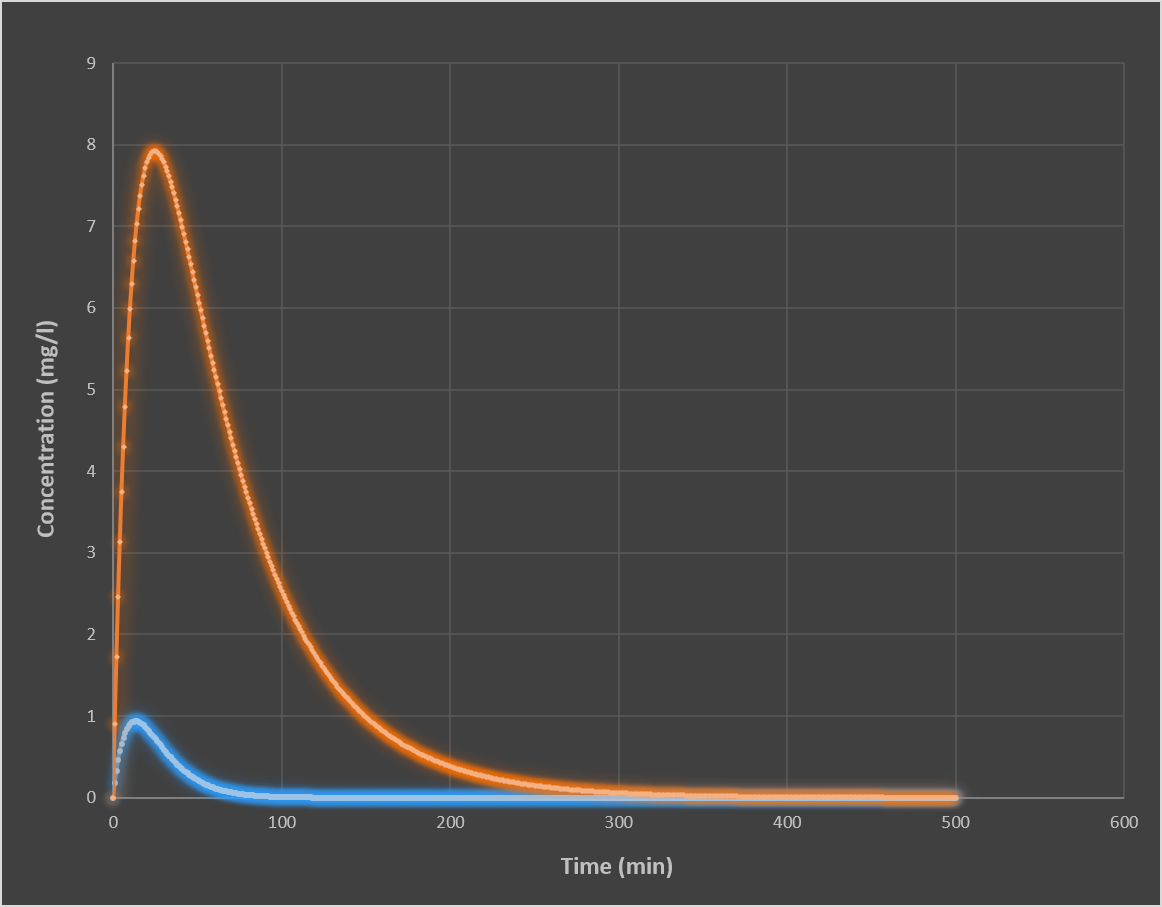
What happens to the pharmacokinetic curve when dose increases?
Increasing dose causes a proportional vertical shift in the concentration-time curve:
C₀ increases
Cmax increases
AUC increases proportionally
However:
Clearance (CL) remains unchanged
Half-life (t½) remains unchanged
Elimination rate constant (ke) remains unchanged
This is because dose changes input, not elimination capacity.
Thus, the entire curve becomes “higher,” but retains identical shape and slope.
Why does increasing dose increase AUC but not clearance or half-life?
AUC represents total systemic exposure:
AUC = (F × Dose) / CL
When dose increases:
More drug enters the system
Clearance does not change because it is a physiological constant
Therefore:
AUC increases proportionally to dose
Half-life remains unchanged because it depends on Vd and CL only
Thus, AUC reflects exposure, not elimination speed.
What happens when volume of distribution increases?
Increasing Vd means drug distributes more extensively into tissues.
Effects:
C₀ decreases (drug leaves plasma more rapidly)
Plasma concentration becomes lower
Half-life increases (drug is “hidden” in tissues)
Elimination constant (ke) decreases
AUC remains unchanged
Because:
Total drug in body is unchanged
Only distribution between compartments changes
Clinically:
Drugs with high Vd often have long duration of action due to tissue storage.
What happens when clearance increases?
Clearance determines how efficiently the body eliminates drug.
When CL increases:
Drug is removed faster
AUC decreases
Half-life decreases
ke increases
Steady-state concentration decreases
C₀ remains unchanged because it depends only on dose and Vd.
Clinically:
Increased clearance often leads to therapeutic failure unless dose is adjusted.
Why is half-life determined by both volume of distribution and clearance?
Half-life reflects both:
How widely the drug distributes (Vd)
How quickly it is eliminated (CL)
Formula:
t½ = (0.693 × Vd) / CL
Interpretation:
Large Vd → drug remains in body longer → longer t½
High CL → faster elimination → shorter t½
Thus:
half-life is not a direct property of dose, but of body handling of drug.
Why is clearance considered constant while elimination rate is not?
Clearance is a physiological constant representing organ efficiency (kidney, liver, etc.).
However:
Rate of elimination = CL × C
So:
When concentration is high → elimination rate is high
When concentration is low → elimination rate is low
Thus:
CL is constant
Elimination rate is concentration-dependent
This produces exponential decay rather than linear elimination.
Why does drug concentration decrease in a curved (not linear) pattern?
Because elimination depends on current concentration:
Rate of elimination = CL × C
As C decreases:
Elimination slows automatically
Each time interval removes a smaller absolute amount
This creates:
Rapid early decline
Slower later decline
Exponential decay curve
On a semi-log plot, this becomes linear.
Why do IV and oral administration produce different Cmax and AUC values?
IV administration:
Immediate entry into blood
F = 1 (100% bioavailability)
Highest possible Cmax
Full AUC exposure
Oral administration:
Requires absorption (ka-dependent)
First-pass metabolism reduces F
Lower Cmax due to slower entry
Lower AUC if F < 1
Key principle:
IV = dose-limited exposure
Oral = absorption + metabolism-limited exposure
What is the effect of changing absorption rate constant (ka)?
ka determines how quickly drug enters systemic circulation.
High ka:
Rapid absorption
Sharp rise in concentration
High Cmax
Early Tmax
Low ka:
Slow absorption
Blunted peak
Delayed Tmax
Lower Cmax
Prolonged exposure
ka does NOT change AUC (if dose and F unchanged), only curve shape.
Why does IV infusion reach a plateau (steady state)?
During infusion:
Drug enters body at constant rate
Drug is eliminated continuously
Initially:
Input > elimination → concentration rises
Over time:
Higher concentration → higher elimination (CL × C)
Eventually:
Input = elimination → steady state reached
Thus:
Css = Dose rate / CL
Why does volume of distribution not affect steady-state concentration?
At steady state:
Css = Dose rate / CL
Vd is not part of this equation because:
It affects distribution equilibrium
It does not affect elimination capacity
Therefore:
Vd affects time to reach Css
But not the value of Css itself
What happens when dose rate increases during infusion?
Css increases proportionally
Curve rises to a higher plateau
Early slope becomes steeper
Because:
more drug enters per unit time while clearance remains constant.
What happens when clearance increases during infusion?
Css decreases
Steady state reached at lower concentration
Elimination is faster
Because:
drug is removed more efficiently per unit time.
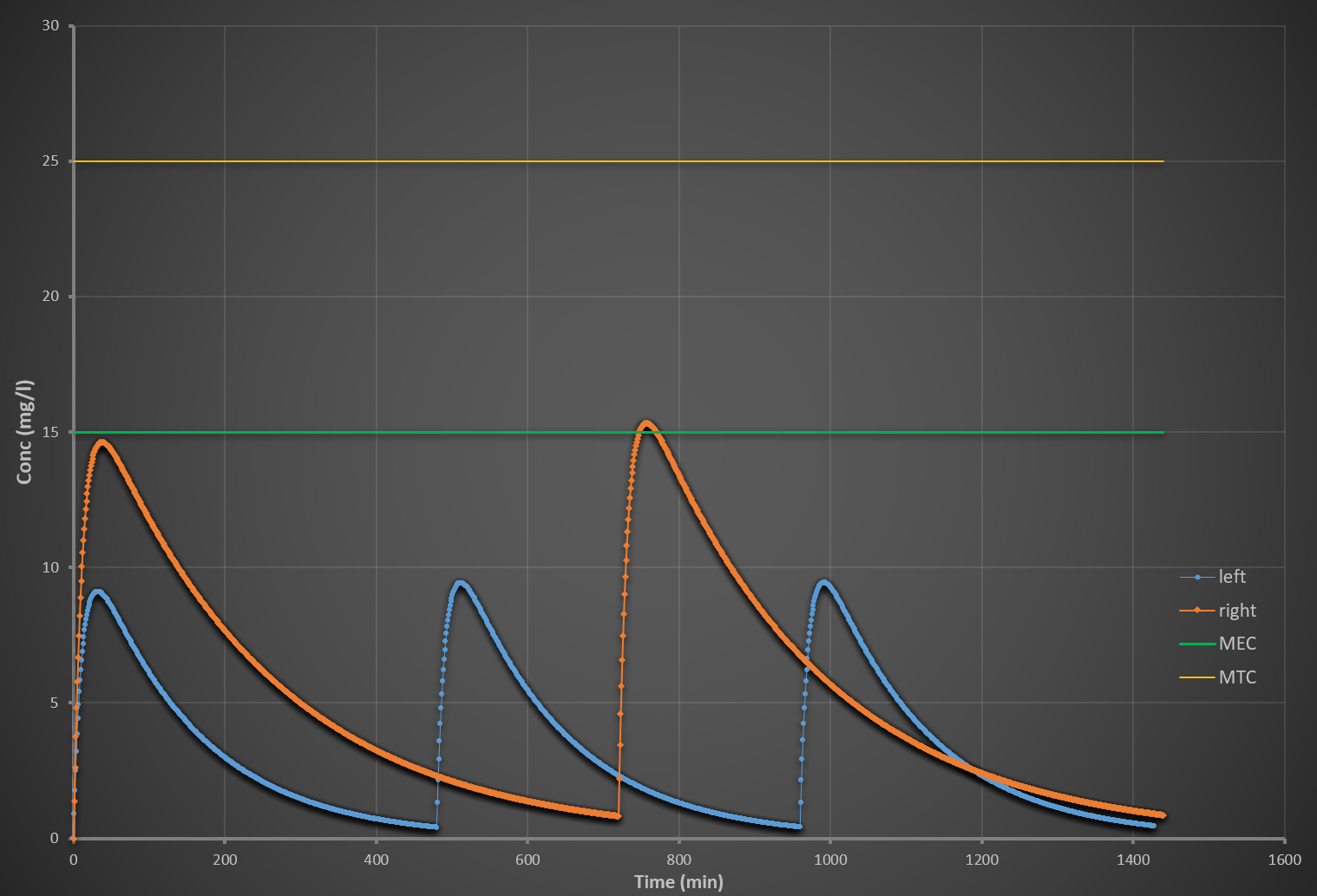
What are MEC, MTC, and the therapeutic window?
MEC (Minimum Effective Concentration): lowest concentration producing therapeutic effect
MTC (Minimum Toxic Concentration): lowest concentration causing toxicity
Therapeutic window: range between MEC and MTC
Clinical goal:
Maintain drug concentration within this window for efficacy without toxicity.
Are MEC and MTC pharmacokinetic or pharmacodynamic parameters?
They are pharmacodynamic (PD) parameters because they describe:
Drug effect on the body
Therapeutic and toxic thresholds
Not drug movement or concentration changes
PK determines concentration; PD determines effect thresholds.
What happens when dose is increased in repeated dosing?
Higher peak concentrations (Cmax ↑)
Higher trough concentrations
Greater accumulation
Increased risk of toxicity (exceeding MTC)
Steady state still occurs, but at a higher average concentration.
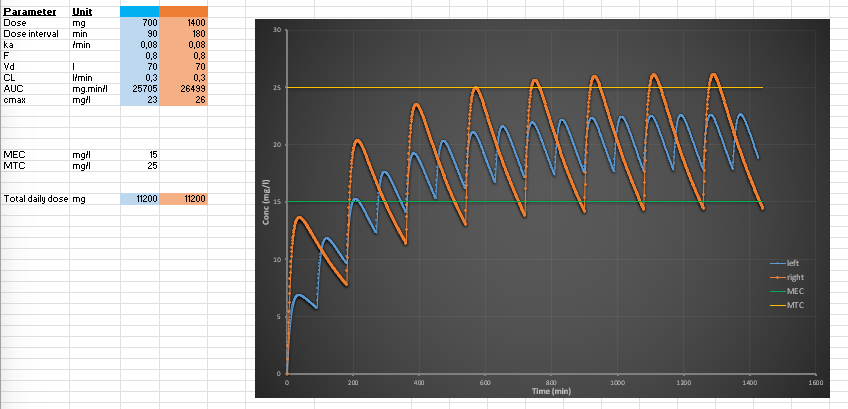
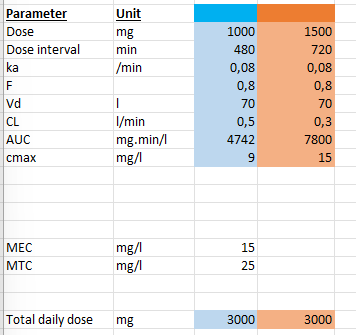
What happens when dosing interval is shortened while keeping total daily dose constant?
Example:
100 mg every 12h → 50 mg every 6h
Effects:
Lower peak concentrations
Higher trough concentrations
Reduced fluctuations
More stable plasma levels
Conclusion:
Smaller, more frequent dosing smooths concentration curves.
What do repeated oral dosing and IV infusion have in common?
Both systems:
Reach steady state after ~4–5 half-lives
Show accumulation before steady state
Achieve equilibrium when input = elimination
Are governed by clearance
Difference is only in input pattern:
Continuous (infusion)
Intermittent (oral dosing)
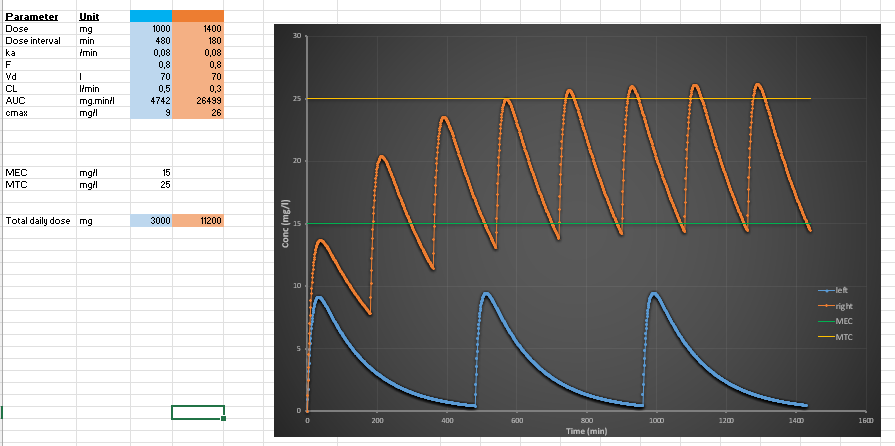
How do you design a dosing regimen to stay within the therapeutic window?
Goal:
Maintain concentration between MEC and MTC.
Strategy:
Increase dose if below MEC
Decrease dose if above MTC
Shorten dosing interval to reduce fluctuations
Use loading dose for rapid effect
Use maintenance dose for steady-state control
Best approach:
Smaller, more frequent doses → smoother curve → safer therapy.
What happens to drug levels when clearance decreases due to kidney failure?
Clearance decreases
Half-life increases
Drug accumulates
Css increases if dose is unchanged
Higher risk of toxicity
Clinical response:
Dose reduction or increased dosing interval is required.
How do you calculate rate of elimination?
Rate of elimination (mg of drug/min) = clearance x Cpl
Cpl is plasma concentration
CL is a constant
High Cpl → high rate of elimination and the other way around.
What does half-life determine?
1. Time to reach steady state (during repeated dosing or infusion)
When a drug is given repeatedly or by continuous infusion, it gradually accumulates until input = elimination.
After 1 half-life → ~50% of steady state reached
After 2 half-lives → ~75%
After 3 half-lives → ~87.5%
After 4 half-lives → ~93.75%
After 5 half-lives → ~96–97% (practically steady state)
So, half-life determines how long it takes for the drug to “build up” to a stable level in the body.
2. Time to eliminate a drug after stopping it
When dosing stops, the same rule applies in reverse:
After 1 half-life → 50% remains
After 2 half-lives → 25% remains
After 3 half-lives → 12.5% remains
After 5 half-lives → ~3% remains (clinically negligible)
So, half-life determines how long a drug “stays in the body” after stopping treatment.
3. Dosing interval (clinical scheduling)
Half-life helps decide how often a drug should be given:
Short half-life → frequent dosing (e.g., multiple times per day)
Long half-life → once daily or even less frequent dosing
4. Degree of accumulation
If dosing happens before the previous dose is fully eliminated:
Short half-life drugs → little accumulation
Long half-life drugs → significant accumulation