Musculoskeletal disorders
1/21
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
22 Terms
Types of muscular dystrophy
1. DUCHENNE MUSCULAR DYSTROPHY (DMD)
2. BECKER MUSCULAR DYSTROPHY (BMD)
3. FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY (LANDOUZY-DEJERINE DISEASE)
4. EMERY-DREIFUSS MUSCULAR DYSTROPHY
5. MYOTONIC MUSCULAR DYSTROPHY
6. MYOTONIA CONGENITA (THOMSEN DISEASE)
7. CONGENITAL MUSCULAR DYSTROPHIES
What is muscular dystrophy?
inherited disease with progressive muscle damage
Duchene muscular dystrophy
What is DMD? → X-linked recessive muscular dystrophy due to dystrophin gene mutation (Xp21).
Most common hereditary neuromuscular disease? → Duchenne muscular dystrophy.
Gene affected in DMD? → Dystrophin gene (Xp21).
Inheritance of DMD? → X-linked recessive.
Major clinical features of DMD? → Progressive weakness, developmental delay, intellectual impairment.
Most characteristic muscle enlargement in DMD? → Calf pseudohypertrophy.( with wasting oof thifh muscles)
Most common site of hypertrophy after calves? → Tongue.
Age of appearance of Gowers sign in DMD? → Evident by 3 years, fully expressed by 5–6 years.
Characteristic gait in DMD? → Trendelenburg gait (hip waddle).
CK level in DMD? → Markedly elevated (15,000–35,000 IU/L). Normal - <160 IU/L
Confirmatory test for DMD? → PCR for dystrophin mutation.
If PCR is negative but suspicion is high? → Muscle biopsy with dystrophin immunocytochemistry.
Complications of DMD? → Contractures, cardiomyopathy, malignant hyperthermia.
Treatment of DMD? → Physiotherapy, nutrition, glucocorticoids, treatment of cardiac and pulmonary complications.
Cause of death in DMD? → Respiratory failure, heart failure, pneumonia, aspiration.
Life expectancy in DMD? → Usually 18–20 years.
Gowers sign
What is Gowers sign? → Use of hands to climb up thighs while standing due to proximal muscle weakness.
Seen in which disorders? → Duchenne, Becker, myotonic dystrophy, centronuclear myopathy.
Bekers muscular duystrophy
Inheritance of BMD? → X-linked dystrophinopathy.
Difference between DMD and BMD? → BMD is milder with later onset.
Ambulation in BMD? → Maintained till late adolescence.
Features of BMD? → Calf pseudohypertrophy, cardiomyopathy, elevated CK.
Death in BMD usually occurs at? → Mid to late 20s.
FACIOSCAPULOHUMERAL DYSTROPHY
Inheritance? → Autosomal dominant.
Characteristic muscles involved? → Face and shoulder muscles.
Classic sign? → Scapular winging.
Associated features? → Hearing loss, retinal vasculopathy.
Grovers sign seen, flattened deltoid, wasted biceps-triceps
Calf hypertrophy present? → No.
Complications? → Kyphoscoliosis, lumbar lordosis.
Unique feature among muscular dystrophies? → Asymmetrical weakness.
Mouth rounded and appears puckered because the upper and lower lips protrude
Cardiac involvement in muscular dystrophy

Emery-dreifuss muscular dystrophy
Other name? → Scapuloperoneal muscular dystrophy.
Inheritance? → X-linked recessive.
Characteristic feature? → Early elbow and ankle contractures
Myotonic muscular dystrophy
Second most common muscular dystrophy? → Myotonic dystrophy.
Inheritance? → Autosomal dominant.
Mutation? → CTG trinucleotide expansion on chromosome 19q13.
What is myotonia? → Delayed muscle relaxation after contraction.
Multisystem involvement? → GIT, uterus, endocrine system, cataracts, malignancy.
Characteristic facial appearance? → Inverted V-shaped upper lip.
Myotonia congenita
Other name? → Thomsen disease.
Basic defect? → Chloride channelopathy.
Characteristic appearance? → Generalized muscular hypertrophy (“bodybuilder” appearance)
Congenital muscular dystrophy
Inheritance? → Autosomal recessive.
Sevefre damagde at birth mostly to brain
Associated brain abnormalities? → Lissencephaly, pachygyria, polymicrogyria.
Complication? → Severe epilepsy.
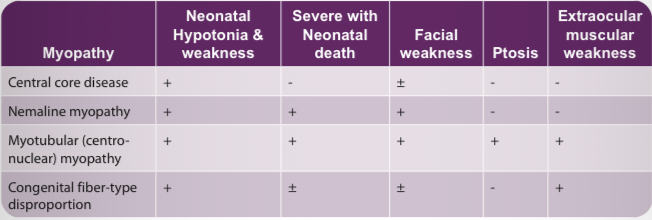
Congenital myopathy
Definition? → Non-progressive congenital neuromuscular disorders diagnosed by muscle biopsy.
Central core disease findings? → Neonatal hypotonia and weakness.
Nemaline myopathy finding? → Severe neonatal disease with facial weakness.
Myotubular myopathy hallmark? → Ptosis and extraocular muscle weakness
Spinal muscular atrophy (SMA)
What is SMA? → Degeneration of anterior horn motor neurons.
Inheritance? → Autosomal recessive.
Gene affected? → SMN gene on chromosome 5q.
Function of SMN gene? → Prevents apoptosis of motor neuroblasts.
SMA Type 0? → Severe fetal form in perinatal period
SMA Type 1? → Werdnig-Hoffmann disease- severe form infantile period
SMA Type 2? → Late infantile form.(slowly progressive)
SMA Type 3? → Kugelberg-Welander disease.(chronic juvenile)
Clinical features of SMA? → Hypotonia, weakness, absent reflexes.
Tongue finding in SMA? → Fasciculations.(denervation of muscles)
Extraocular muscles in SMA? → Spared.
CK level in SMA? → Usually normal.
Definitive diagnosis of SMA? → SMN gene testing.
Rickets
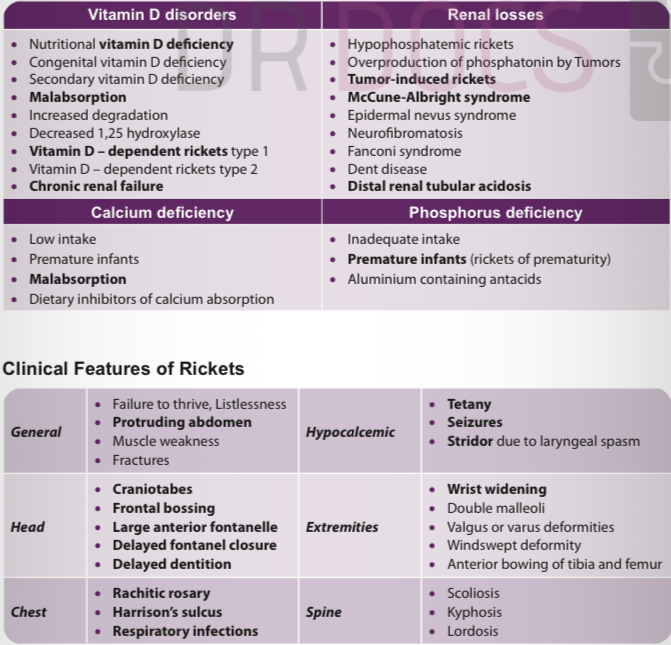
Definition of rickets? → Defective mineralization of growth plate before epiphyseal fusion.
Most common nutritional cause? → Vitamin D deficiency.
Causes of rickets? → Vitamin D deficiency, calcium deficiency, phosphate deficiency, renal disease.
General features of rickets? → Failure to thrive, protruding abdomen, muscle weakness.
Hypocalcemic manifestations? → Tetany, seizures, laryngeal stridor.
Head signs of rickets? → Craniotabes, frontal bossing, delayed dentition and fontanelle closure
Chest signs? → Rachitic rosary, Harrison sulcus.
Extremity signs? → Wrist widening, double malleoli, bow legs.
Radiological findings of rickets? → Fraying, cupping, widening, splaying of metaphysis.
Best X-ray to diagnose rickets? → AP wrist radiograph.
Biochemical findings in nutritional rickets? → Low phosphate, variable calcium, high ALP.
Stoss therapy dose? → 300,000–600,000 IU vitamin D single dose.
Alternative therapy - 2000-5000 IU vit D/day for 4-6 weeks
Maintenance vitamin D after treatment? → 400 IU/day.
Rx for nutritional rickets in malnutrition - Choecalciferol
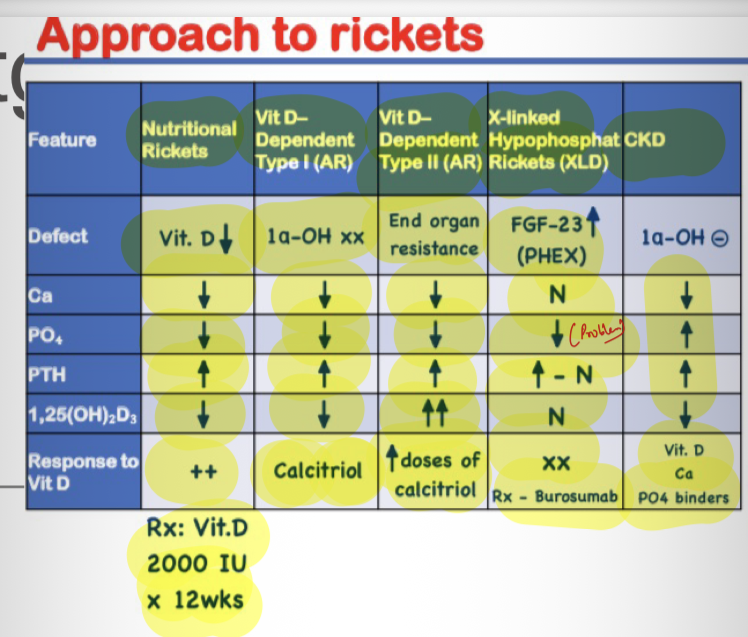
X-linked hypophosphatemic rickets
Most common inherited hypophosphatemic rickets? → X-linked hypophosphatemic rickets.
Gene affected? → PHEX.
Inheritance? → X-linked dominant.
Pathophysiology? → Gene mutation →Increased FGF-23 → inhibit phosphate reabsorbtio by PCT →increased phosphate wasting.
Lab findings? → Hypophosphatemia, high ALP, normal calcium, high phosphate renal excretion
Treatment? → Oral phosphate + calcitriol.
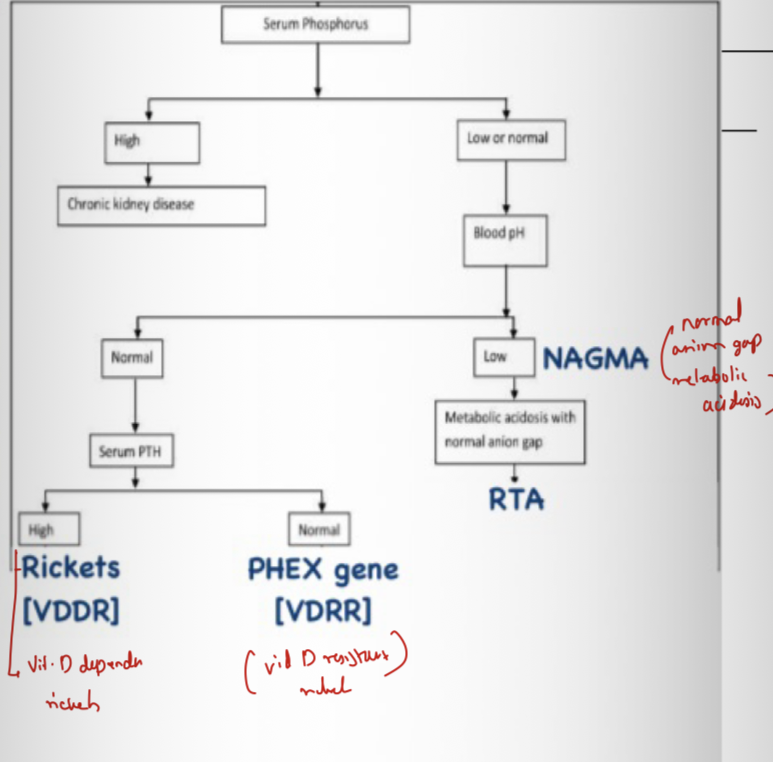
Fanconi syndrome
Basic defect? → secondary to Generalized proximal tubular dysfunction.
Substances lost in urine? → Phosphate, glucose, bicarbonate, amino acids, urate.
Skeletal manifestation? → Hypophosphatemic rickets +RTA (renal tubular acidosis
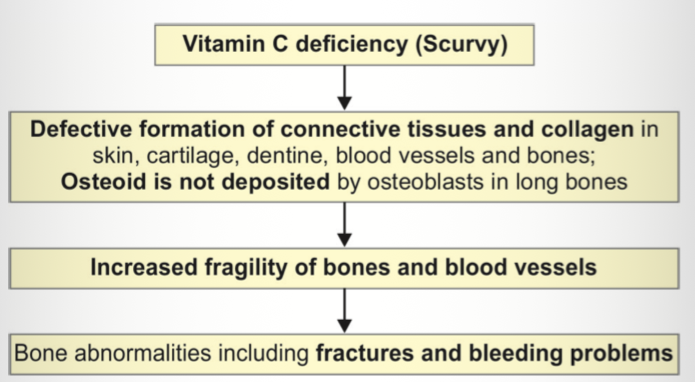
Scurvy ( barlow’s disease)
Cause of scurvy? → Vitamin C deficiency.
At-risk children? → Heat-treated cow milk-fed infants.
Basic defect? → Defective connective tissue and collagen formation.
Clinical features? → Pallor, irritability, infections.
Painful limb manifestation? → Subperiosteal hemorrhage causing pseudoparalysis.
Chest finding? → Scorbutic rosary.
Radiological findings -
• Ground-glass appearance of long bones - due to trabecular atrophy with pencil thin cortex
• White line of Frãnkel: Seen at metaphysis, represents well-calcified cartilage
• Wimberger sign: Sclerotic ring around epiphyseal centers of ossification
• Trumerfeld zone: A zone of rarefaction under white line at metaphysis; more specific;
• Pelkan spur: Lateral prolongation of the white line and maybe present at cortical ends
• Subperiosteal hemorrhages; dumbbell shape of affected bone
White line of Frankel? → Dense metaphyseal line.
Wimberger sign? → Sclerotic ring around epiphysis.
Trummerfeld zone? → Zone of rarefaction beneath white line.
Pelkan spur? → Lateral metaphyseal spur.
Treatment of scurvy? → Vitamin C 100–200 mg/day.
Osteoporosis
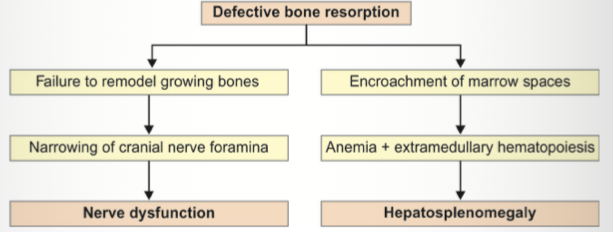
Other name? → Marble bone disease.
Gene mutation? → CLCN7.
Basic defect? → Defective osteoclast-mediated bone resorption.
Clinical features? → Macrocephaly, anemia, hepatosplenomegaly.
Neurological complications? → Deafness, blindness.
Radiological appearance? → Bone-within-bone appearance, bone sclerosis
Vertebral appearance? → Sandwich vertebra.
Lab investigation? - Low Ca,P ; increased PTH; normal vit D
Treatment? → Hematopoietic stem cell transplantation.
Osteogenesis imperfecta
Other name? → Brittle bone disease.
Defect? → Type I collagen abnormality.
Classic triad? → Fragile bones, blue sclera, deafness.
Other features? → Dentinogenesis imperfecta, hyperextensible joints.
Inheritance? → Usually autosomal dominant.
Most common genetic cause of osteoporosis? → Osteogenesis imperfecta.
Treatment? → Bisphosphonates
Achondroplasia
Inheritance? → Autosomal dominant.
Gene affected? → FGFR3.
Most cases arise from? → New mutation.
Characteristic limb shortening? → Rhizomelic.
Hand abnormality? → Trident hand.
Head features? → Large head, frontal bossing.
Intelligence in achondroplasia? → Usually normal.
Bowing of legs, dentall crowding,delayed motor milestones
Pelvic X-ray finding? → Champagne glass pelvis.(short iliac bones, flat acetabular roofs)
Spinal radiological finding? → Decreased interpedicular distance.
Common complications? → Obesity, hearing loss, otitis media.
Limb shortning types
Micromelia → Entire limb shortened.
Rhizomelia → Proximal segment shortened.
Mesomelia → Middle segment shortened.
Acromelia → Distal segment shortened.
Phocomelia → Proximal and middle segments absent/shortened.
Juvenile idiopathic arthritis
Definition of JIA? → Arthritis before age 16 lasting ≥6 weeks.
Most common rheumatic disease in children? → JIA.
Pathogenesis? → Autoimmune T-cell mediated inflammation.
Types of JIA? → Oligoarticular, polyarticular, systemic, psoriatic, enthesitis-related.
Systemic-onset JIA fever pattern? → Quotidian fever for ≥2 weeks.
Systemic-onset JIA associated features? → Rash, lymphadenopathy, hepatosplenomegaly, serositis.
RF in systemic JIA? → Usually absent.
Most serious complication of JIA? → Macrophage activation syndrome (HLH).