coordination chemistry
1/27
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
28 Terms
three types of structural/constitutional isomerism
hydrate: ligand interchanged/replaced with water of hydration - e.g. [Cr(H2O)6]Cl3 - [Cr(H2O)5]Cl2.H2O - etc
ionisation: interchange of anionic ligands - e.g. [Co(NH3)5SO4]Br - [Co(NH3)5Br]SO4
linkage: where the same ligand can bind through different atoms (has more than one possible coordination mode - ambidentate) - e.g. nitro -NO2 vs nitrito -ONO; thiocyanate
what dominates hard-hard and soft-soft interactions?
hard-hard is ionic bonding - but dominated by ΔS - as they are well solvated (by water), when they bond, the release of water molecules is an entropic driving force
soft-soft is covalent bonding - and dominated by ΔH - orbital overlap determined by HOMO-LUMO gap
βn vs Kn
βn is the overall formation constant, = [MLn] / [M][L]n
Kn is the stepwise formation constant, = [MLn] / [MLn-1][L]
βn = K1K2…Kn (consider denominators and numerators of successive Kns)
usual trend in successive stepwise stability constants
K1 > K2 > … > Kn
statistical phenomenon: decreasing number of available sites as ligands complex
steric hindrance: if the complexing ligand is larger than the ligand displaced
Coulombic interactions: if the complexing ligand is more charged than the ligand displaced
Kn trend for Cu(II) / NH3
abnormality is K5 and K6 very small due to Jahn-Teller distortion; weak bonding at the axial 5th and 6th coordination sites
Kn trend for Cd(II) / I-
abnormality is K3 large (> K2) - due to stereochemical change:
CdI2(OH2)4 → [CdI3(OH2)]-
octahedral → tetrahedral due to steric factors (Cd2+ group 12, I- large)
loss of 3 H2O - entropically favoured
Kn trend for Fe(II) / phen
abnormality is K3 large (> K2) due to spin state change
[Fe(phen)2]2+ is still high spin but → [Fe(phen)3]2+ is low spin; big increase in LFSE stabilising (d6 !)
Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+ trend in (log)K(1)
Irving-Williams series; insensitive to choice of ligand
across the series Zeff increases (because …)
(increased Lewis acidity;) 3d AOs are lower in energy and are a better match for ligand AOs
LFSE stabilisation
increases across the period to Ni2+ and is zero at Zn2+
but Cu2+ largest?
Jahn-Teller distortion - stronger bonding of equatorial ligands increases the value of K (definitely for K1)
Irving-Williams for M2+ / en
normal for K1 and K2 (so K1 > K2 > K3) but for Cu2+, K3 is (very) low due to rigid en
rigid bidentate ligand prevents significant J-T distortion for Cu(en)32+ (fine for Cu(en)22+ as can bind in the equatorial plane)
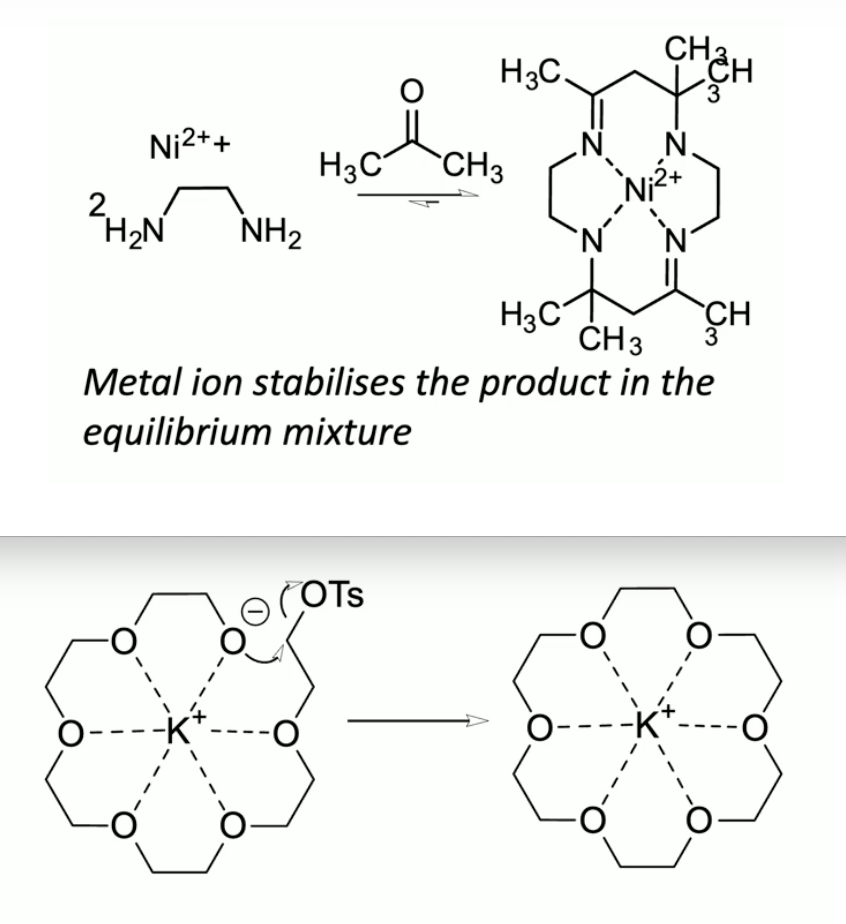
naming of cryptands? try drawing [2.1.1], [2.2.1], [2.2.2], [3.2.2]. which ion best fits [2.2.2]?
K+ for [2.2.2] cryptand
note also K+ best for 18-crown-6
entropic driving force for binding for all: release of solvation (H2O) molecules
![<p>K<sup>+</sup> for [2.2.2] cryptand</p><p><u>note also K<sup>+</sup> best for 18-crown-6</u></p><p>entropic driving force for binding for all: release of solvation (H<sub>2</sub>O) molecules</p>](https://assets.knowt.com/user-attachments/5931aad2-c2d8-4bf2-9344-4addbe4e341d.png)
factors affecting binding affinity
size match (geometric complementarity):
if too small, bonds are too long (on average - maybe some are lost entirely if metal does not sit in the centre) are therefore weaker
if too large, cage has to distort and so strain in the system is not enthalpically favourable
chelate effect: enhanced stability of chelated complexes compared with less chelated or non-chelated complexes
factors contributing to the chelate effect
mainly entropic: chelation results in a net increase in the number of independent molecules in solution
enthalpic contributions:
polar donor groups are already covalently linked together in the chelating ligand
no increase in lp-lp repulsion, compared to lp being brought closer together upon binding for a non-chelating ligand
chelating ligand usually a better donor (more basic) - due to electron donating alkyl groups
greater effective concentration of chelating ligands
once one donor group binds, it becomes more probable that another donor group of the chelating ligand binds as it is held in close proximity to the metal ion. (diagram…)
is obviously kinetic but also thermodynamic with an entropic effect - probability - can be considered in terms of microstates…
effect of chelate ring size
chelate effect generally decreases with increasing ring size
except for 5- vs 6- membered chelate rings:
bite size is larger in a 5-membered ring compared to 6-membered (draw hexagon to demonstrate)
the bond length between donor atom and metal is longer in a 5-ring vs 6-. ideal 6- rings (with their smaller bond lengths) have less strain (cf. chair cyclohexane), but for ions which are too large then this does not hold.
large ions favour 5-membered rings, small ions favour 6-membered rings. Pb2+ vs Cu2+
origins of the macrocyclic effect
enhanced stability of macrocyclic ligand complexes compared with open-chain analogues
mainly enthalpic effect:
size complementarity - stronger M-L bonds
solvation - macrocycles are usually less solvated than open-chain analogues (diagram?), so fewer ligand-solvent bonds to break before binding to metal
ligand pre-organisation: favourable conformational enthalpy is already present in the synthesised molecule, which overcomes repulsive forces between polar donor groups (e.g. lp-lp repulsion)
no increase in repulsion (strain) upon binding to the metal ion, vs open-chain which does have increase in e.g. lp-lp repulsion
entropic effect:
pre-organisation: conformational entropy - macrocycles lose fewer degrees of freedom upon complexation as they are inherently less flexible
solvation effects are harder to analyse, as although solvent molecules released from metal ion, they are also released from the centre of the cavity
choice of solvent (how polar?) can effect binding affinity - how strong are the ion-solvent, ligand-solvent bonds that need to be broken?
kinetics of macrocyclic effect
K is much greater for macrocycles than open-chain
but this is due to (very) low dissociation rate for macrocycles
mutliple coordinate bonds must be broken at once; for acyclic can do one at a time (see below) due to greater conformational flexibility
formation rate usually a bit faster for acyclic
Eigen-Wilkins: form an outer-sphere complex first, then exchange one S for L (reversible!)
any individual step could be rate determining.
for macrocycles, usually due to strain/high energy conformation required to put donor atom in the right position; free rotation in acyclic can avoid this
how would you promote macrocycle dissociation?
compete with the metal, e.g. use H+ to compete with M for N based ligands, EDTA…
anion coordination chemistry?
often bigger and more charge diffuse; need bigger receptor ligands
geometries of anions vary; spherical to linear to ??
e.g. elongated cryptand-like (protonated) macrocycle prefers linear azide
preorganisation, macrocycles helps, as with cations…
synthesis of macrocycles… how?
template effect:
(e.g.) metal-ligand interactions used to pre-organise components into the desired geometry for reaction
especially useful when desired reaction conformation is not kinetically and/or thermodynamically favoured
thermodynamic and kinetic; see other flashcard
problems:
demetallation can be difficult: may need to add competing ligand (EDTA, CN) or protonate (Ns) or redox…
hard to identify correct M
if templating not possible, use high dilution synthesis (dropwise addition of reactants over many hours)
to promote intramolecular cyclisation over oligomerisation
thermodynamic and kinetic template effects
thermodynamic: metal ion stabilises (part of) the RHS of the equilibrium, driving it
in this case Ni2+ back bonding in to C=N π*, making it less electrophilic
kinetic: coordination of precursor to macrocycle holds reactive groups in close proximity, and in the correct geometry
beware [Co(en)2Cl2]+
cis/trans isomerism - but cis isomer also has optical isomers
Schiff base condensation (diketone with diamine)
size of metal ion template can determine which product is formed [1+1] vs [2+2]
![<p>size of metal ion template can determine which product is formed [1+1] vs [2+2]</p>](https://assets.knowt.com/user-attachments/a107fb83-f5b9-4678-9c88-ae055188d0d7.png)
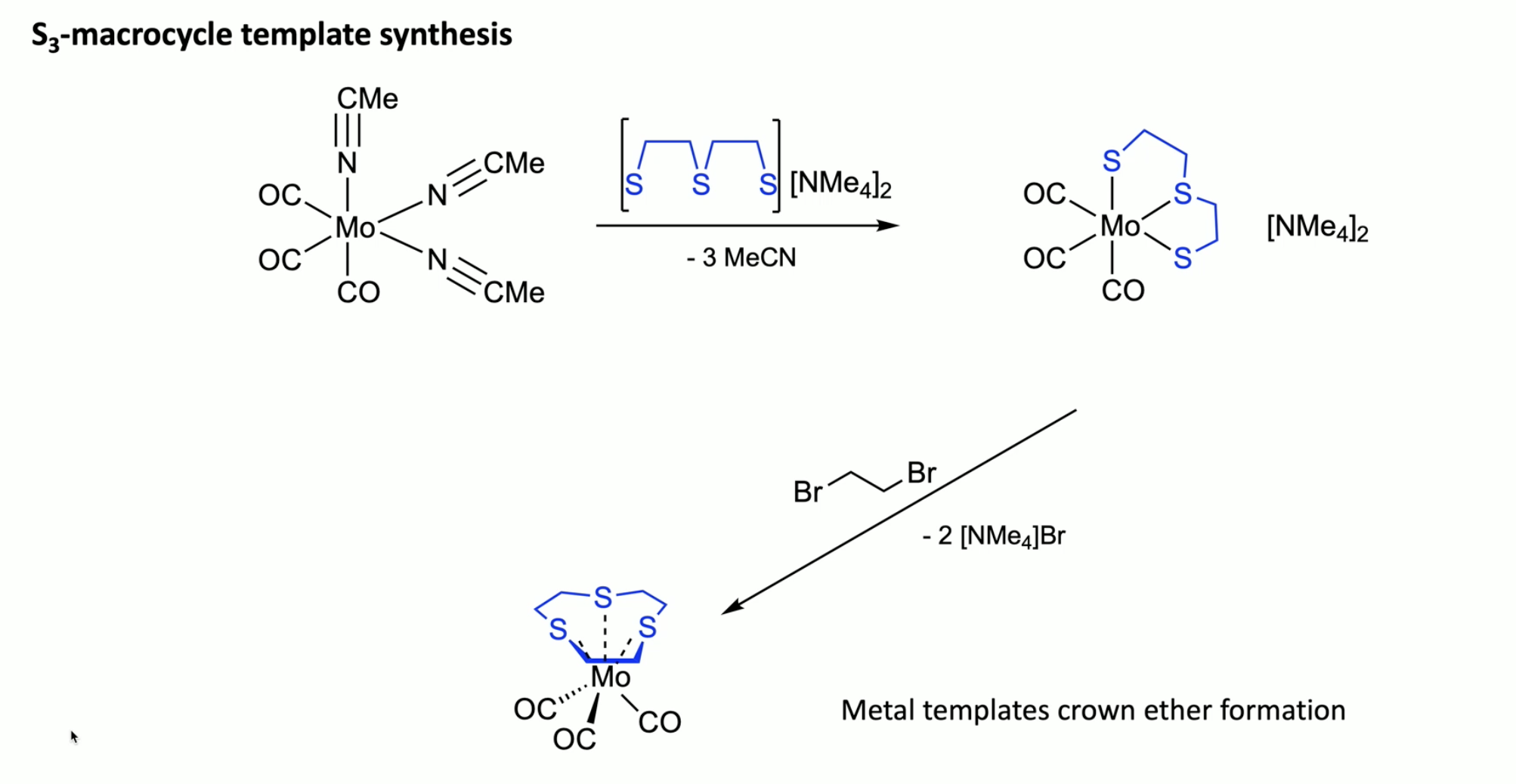
S3-macrocycle synthesis
MeCN a weakly coordinating ligand
Mo(0) favours octahedral coordination…?
point of the templating is to allow the S- (which are S-Mo) close to each other (would otherwise repel…) then can form the macrocycle with the dibromo, rather than oligo
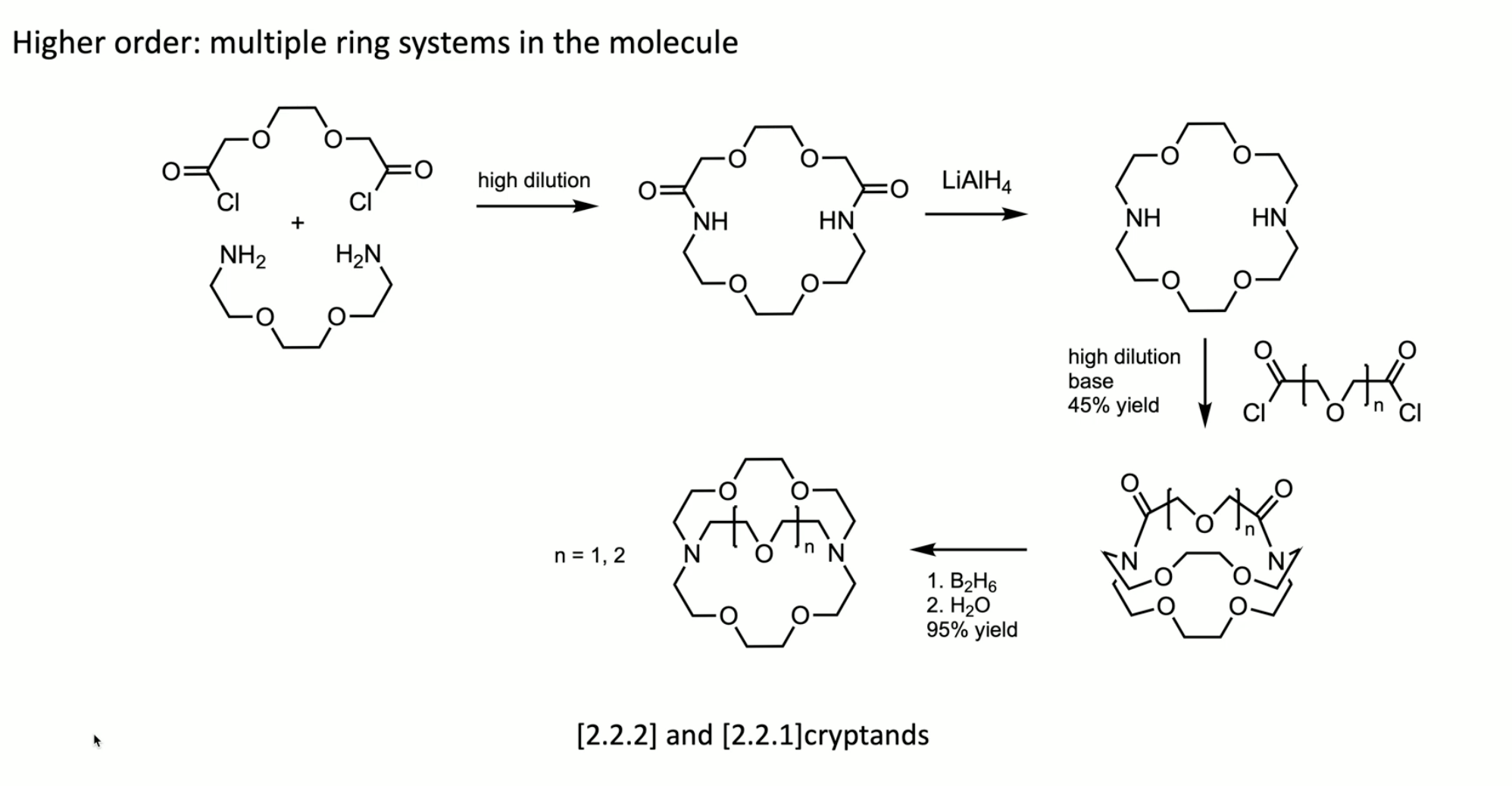
high dilution synthesis of cryptands
high dilution x 2
why? metal ions will not template well to the bis acid chlorides (electrophiles…); use high dilution
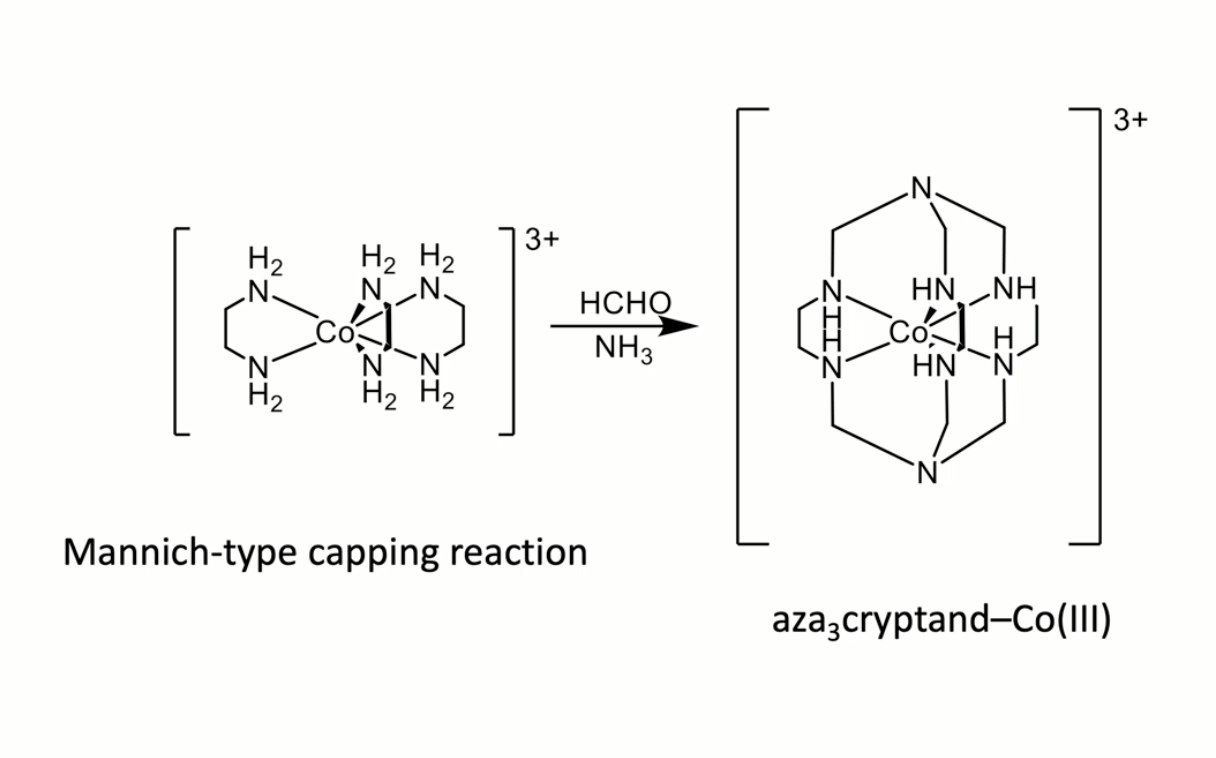
Co(en)3+ with ammonia and formaldehyde
good LFSE of Co3+ in Oh geometry; then that geometry allows capping vs oligomerisation
the point is that by holding in the right geometry, rate of intra > rate of intermolecular - kinetic template
can also happen with Fe3+ with triamine + diester
Pd2+, Fe2+ templating
Pd2+ square planar, can form ‘squares’ with en + 4,4’-bipy
Fe2+ octahedral, can form tetrahedral cage
catenane synthesis… how? (bis phenols + I-C-(C-O-C)4-C-I)
‘catenane’ = chain
need Td coordination: Cu(I) is used - Cu(MeCN)4+ - MeCN weakly binding
to demetallate -CN used (could use EDTA perhaps)
the reaction has the two ligands (bis phenols) orthogonal (Td coordination)
then with the deprotonated phenols SN2 on I-C-(C-O-C)4-C-I
[2]-rotaxane, Schiff base chemistry; both components have 3 Ns
so we need Oh coordination since 6-coordinate
use first row TMs; Co(II) especially good
always need to demetallate
[2]-rotaxane; not yet macrocycle (dangling alkenes) has 3 Ns, axle has only 1N; 3 N component has dangling alkenes
use Pd(II) - e.g. Pd(OAc)2 - for square planar geometry - coordinate to 3N component
(the fourth coordination orginally from solvent, e.g. MeCN) then the axle coordinates
then need to use Grubbs catalyst (Ru based carbene) to clip - two alkenes into one, lose ethene - ring closing metathesis
then add competing ligand to demetallate (not specified, -CN perhaps for square planar)
catenane; components have only one donor atom each (N), have dangling alkenes
need a linear template - use Au(I) (d10)
this forms the orthogonal precursor complex (much like the Td case)
then clip alkenes with Grubbs catalyst (Ru based carbene) - lose ethene, ring closing metathesis
need to demetallate - e.g. H+ for N ligands
can reduce the alkene to alkane with H2, Pd/C