DRUGS TARGETING BACTERIA
1/210
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
211 Terms
Chemistry behind immune response
Paul Ehrlich (Father of Chemotherapy)
Discovery of Penicillin
Alexander Fleming
Discovered penicillin from
Discovered penicillin from
Penicillin as Medical Treatment
Penicillin as Medical Treatment
Streptomycin discovery
First to discover antibacterial agent from, or antimicrobial agent from soil microorganisms, Streptomycin
Selman Waksman
Define Antibiotics
“... a substance produced by microorganisms, which have the capacity of inhibiting the growth and even of destroying other microorganisms.” - S. Waksman
Product of metabolism
Synthetic product produced as structural analogues of naturally occurring antibiotics
Antagonizes the growth or survival of one or more species of microorganisms
Effective in low concentrations
“... a substance produced by microorganisms, which have the capacity of inhibiting the growth and even of destroying other microorganisms.” - S. Waksman
Antibiotics
Product of metabolism
Antibiotics
Synthetic product produced as structural analogues of naturally occurring antibiotics
Antibiotics
Antagonizes the growth or survival of one or more species of microorganisms
Antibiotics
Effective in low concentrations
What sets antibiotics apart from antiseptics or disinfectants is that antibiotics are structurally specific where they target a particular molecular drug target.
So in antibiotics, even in synthetic types, it works at a very small, specific dose to kill bacteria or other microorganisms.
Antibiotics
ATTRIBUTES OF AN IDEAL ANTIBIOTIC
Selective toxicity
Chemically stable
Acceptable rate of biotransformation
COMMERCIAL PRODUCTION OF ANTIBIOTICS
Natural: fermentation
Semisynthetic
Synthetic
If we only extracted them from microbes, we'd likely face supply shortages. Still, some antibiotics are still produced that way, by
Natural: fermentation
The precursor molecule is produced by fermentation, but to convert it into a fully functional antibiotic, you need one to three steps of chemical synthesis. So the precursor is natural, but the final product involves synthesis and manipulation.
Semisynthetic
We also have fully synthetic antibiotics, but that only happens if the synthesis is easy
Synthetic
Prokaryotic Not defined Nucleus
Bacterial Cell
Not as compartmentalized as that of eukaryotic cells (Found in cytoplasm)
Simple Structures
Bacterial Cell
DNA replication and transcription happen in the
nucleus
Only translation happens at the
ribosome
mRNA undergoes what before leaving the nucleus
post-transcriptional modification (capping, poly-A tail, splicing)
Then translation occurs at the
rough ER.
post-transcriptional modification for folding and modification (adding fats or carbohydrates) (post-translational modifications) before being released by the cell OCCURS IN
Golgi apparatus
replication, transcription, and translation can happen simultaneously in what type of cell
bacterial cell
have no defense mechanisms when it comes to mutations in their DNA.
They dont have DNA proofreading, repair mechanisms kaya sila very resilient
bacterial cell
With cell membrane and cell wall
Bacterial cell
synthesizes essential vitamins
Bacterial cell
50s and 30s (70s)
This could be used as a specific drug target that allows us to not affect 60s and 40s
Bacteria
60s and 40s (80s)
humans
proposes that eukaryotic cells evolved when large prokaryotic cells engulfed smaller, energy-producing prokaryotic cells (like aerobic bacteria) rather than digesting them. These engulfed cells became endosymbionts, eventually evolving into organelles like mitochondria and chloroplasts, providing the host cell with ATP in a symbiotic relationship.
The Endosymbiotic Theory
Ribosomes, Dna similar to bacteria. Endosymbiotic Theory
Kaya yun pinanghahawakan na pwedeng ganun yung nature ng evolution ng eukaryotics from prokaryotic. And so if that is the case, those antibiotics affecting human and bacteria ribosomes, baka kung minalas, pwede niya maapektuhan mitochondrial makeup
presence of a lipopolysaccharide layer and thin peptidoglycan
are also much more resistant to other types of antibiotics.
Gram negative
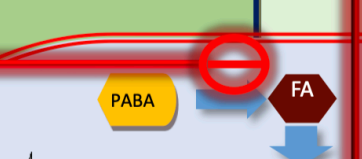
act as competitive inhibitors of the enzyme dihydropteroate synthase, which bacteria use to synthesize folic acid from para-aminobenzoic acid (PABA). Because these drugs are structural analogs of PABA, they "trick" the enzyme into binding with them instead, effectively halting the production of folic acid. Since folic acid is a vital cofactor for converting dUMP to dTMP, its absence leaves the bacteria without enough thymine to replicate their DNA, leading to inhibited growth and DNA strand breaks. Because humans absorb folic acid from food rather than synthesizing it themselves, this pathway is a selective target that leaves human cells unharmed.
SULFONAMIDES
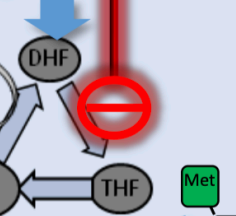
is a competitive inhibitor of the enzyme dihydrofolate reductase (DHFR), which is responsible for converting dihydrofolate (DHF) into tetrahydrofolate (THF). By blocking this specific step, the drug prevents the formation of the active form of folic acid needed for the synthesis of thymidine. B
ecause this occurs right after the step blocked by sulfonamides, the two drugs are often combined (e.g., Co-trimoxazole) to create a sequential blockade that is much more powerful than either drug alone.
a trait it shares with the chemotherapy drug Methotrexate.
Trimethoprim
Problem with Trimethoprim
While humans also possess DHFR, Trimethoprim has a significantly higher affinity for the bacterial version of the enzyme, though it can still occasionally impact human cells
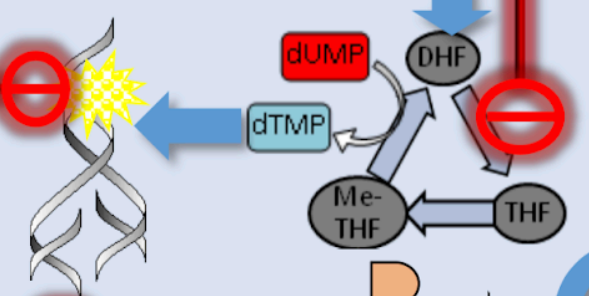
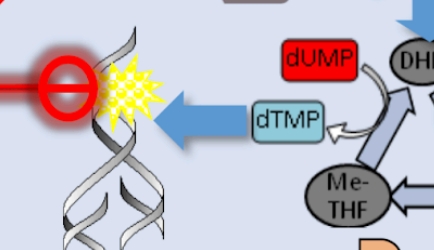
Explain this
The process shown is Thymidylate Synthesis. First, Me-THF (Methyl-tetrahydrofolate) acts as a methyl donor; it transfers its methyl group to dUMP (deoxyuridine monophosphate) via the enzyme thymidylate synthase. This reaction converts dUMP into dTMP (deoxythymidine monophosphate) and leaves behind DHF (dihydrofolate), which must then be recycled back to THF to keep the cycle moving. The dTMP is subsequently phosphorylated twice more to become dTTP (not ATP), which is the specific "T" nucleotide used as a building block for DNA replication.
is only used to pertain to the topoisomerase II in bacterial cells
DNA gyrase
Topoisomerase or DNA gyrase job
remove the supercoiling that comes as a result of the uncoiling done by DNA helicase
T/F In the nomenclature of these enzymes, bacteria use Roman numerals while in eukaryotes, it's Roman letters.
F: bacteria use Roman letters while in eukaryotes, it's Roman numerals.
inhibit DNA gyrase, allowing for the DNA to break from increased tension.
inhibit bacterial DNA synthesis by targeting two key enzymes: DNA gyrase (Topoisomerase II) and Topoisomerase IV. During replication, DNA helicase unwinds the double helix, which creates intense "supercoiling" or physical tension further down the strand. DNA gyrase normally travels ahead of the replication fork to cut, rotate, and reseal the DNA to relieve this stress. These drugs bind to these enzyme-DNA complexes, locking them in place and preventing the DNA from being resealed. This results in permanent double-stranded breaks and massive tension that causes the DNA to snap, leading to rapid bacterial cell death.
Quinolones
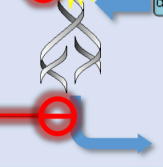
Inhibits RNA polymerase, which is responsible for the transcription process of DNA to RNA.
inhibits bacterial protein synthesis at the very first step by targeting DNA-dependent RNA polymerase. It binds specifically to the beta subunit of this enzyme, preventing it from transcribing DNA into messenger RNA (mRNA). Without mRNA, the "instructions" for building proteins never reach the cell's machinery, effectively starving the bacteria of the enzymes and structural components they need to survive. Because Rifampicin targets the bacterial version of RNA polymerase and not the human version, it is highly effective at stopping bacterial growth while leaving our transcription process alone.
Rifampicin
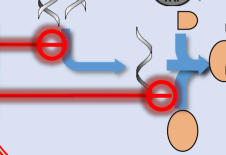
Yung image sa baba
Targets the association of the large and small ribosomal subunits, inhibiting them from joining together and inhibiting the translation process.
Linezolid is an example of this drug class.
are unique protein synthesis inhibitors that act at the very beginning of the translation process. They bind to the P site of the 50S ribosomal subunit, which prevents the formation of the 70S initiation complex. By blocking the association of the large (50S) and small (30S) subunits, the drug ensures that the "machinery" never gets put together to read the mRNA. Because this mechanism is different from other antibiotics like macrolides or tetracyclines, there is often less cross-resistance with this class.
OXAZOLIDINONES

These bind to the large ribosomal subunit after it has merged with the small subunit.
inhibit bacterial protein synthesis by binding to the 50S ribosomal subunit. Unlike Oxazolidinones, which prevent the "burger bun" from closing, these drugs act after the ribosome has already assembled. They specifically block translocation, the step where the ribosome slides down the mRNA strand to add the next amino acid to the growing peptide chain. By physically "plugging" the exit tunnel or interfering with the movement of tRNA, they cause the assembly line to grind to a halt, preventing the bacteria from producing functional proteins.
MACROLIDES AND LINCOSAMIDES
Macrolides (like Erythromycin) and Lincosamides (like Clindamycin)

Similar to macrolides and lincosamides, but they bind to the small ribosomal subunit instead.
both inhibit protein synthesis by binding to the 30S ribosomal subunit (the "small" half of the ribosome).
AMINOGLYCOSIDES AND TETRACYCLINES
Aminoglycosides (e.g., Gentamicin) and Tetracyclines (e.g., Doxycycline)
act as bacteriostatic agents by blocking the attachment of aminoacyl-tRNA to the A-site, effectively stopping the addition of new amino acids.
TETRACYCLINES
are often bactericidal because they cause the ribosome to misread the mRNA code; this leads to the production of misprioritized or "junk" proteins that insert into the bacterial cell membrane, causing it to leak and the cell to die. While they can work on many bacteria, their biggest hurdle is penetrating the complex, lipophilic outer membrane of Gram-negative organisms.
Aminoglycosides
are so lipophilic; this is why, even though these agents can work on both types of bacteria, there is a larger amount of rejection when used on these types of bacteria. All it needs is to find a way to somehow penetrate the cell membrane
Gram negative bacteria
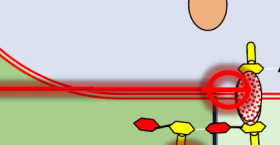
Target protein involved in translocating NAM and NAG
Remember that NAM and NAG are synthesized in the cytoplasm and then brought out and form the bacterial cell wall.
This drug does not allow the transportation of these NAM and NAG chains from the cytoplasm and through the cell membrane, thus inhibiting the synthesis of the cell wall.
NAG is N-acetylglucosamine, while NAM is N-acetylmuramic acid
inhibits bacterial cell wall synthesis by interfering with the recycling of bactoprenol, the lipid carrier molecule. Bacterial cell wall building blocks (NAM and NAG) are synthesized inside the cytoplasm but must be transported across the inner membrane to the exterior to form the peptidoglycan layer. THis drug binds to the pyrophosphate on bactoprenol, preventing it from dephosphorylating and returning to the inner side of the membrane to pick up another precursor. By blocking this "shuttle" system, the cell runs out of materials to build its protective outer wall, leading to cell lysis.
BACITRACIN
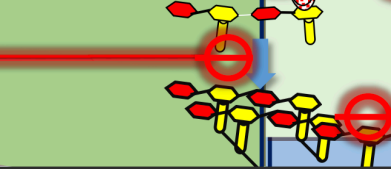
The beta-lactams inhibit the cross-linking of these NAM and NAG chains.
While these are generally not as effective in Gram (-) bacteria, the later generations of the other drug we see that they also have some effectiveness against gram negative bacteria.
(collectively known as Beta-lactams) inhibit the final stage of bacterial cell wall synthesis. They act by binding to and inhibiting (PBPs), specifically the transpeptidase enzyme. This enzyme is responsible for "cross-linking" the peptidoglycan chains (the NAM and NAG strands). Without these cross-links, the cell wall loses its structural integrity and cannot withstand the high osmotic pressure inside the bacteria. This causes the cell to swell and eventually burst, a process known as osmotic lysis.
PENICILLINS AND CEPHALOSPORINS
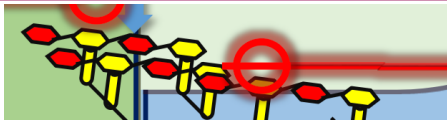
These agents target the outer wall of the bacteria, specifically in the D-Ala-D-Ala terminus. This causes steric hindrance and weakens the cell wall.
There are also glycopeptides that are non-ribosomal peptides, meaning that they are not formed in the ribosomal pathway. They are short and cyclic, and because of that, they have a lipophilic head and hydrophilic tail, which allows them to penetrate the cell membrane.
inhibit bacterial cell wall synthesis through a high-affinity binding to the D-Ala-D-Ala terminus of the peptidoglycan precursor. Unlike Beta-lactams that target the enzyme, Glycopeptides target the substrate itself. By capping the D-Ala-D-Ala end, they create "steric hindrance"—physically blocking the transpeptidase enzyme from accessing the site to create cross-links. This results in a weak, unstable cell wall that leads to bacterial death. Because these molecules are very large and bulky, they generally cannot pass through the outer membrane of Gram-negative bacteria, making them primarily effective against Gram-positive organisms like MRSA.
GLYCOPEPTIDES
Vancomycin etc.
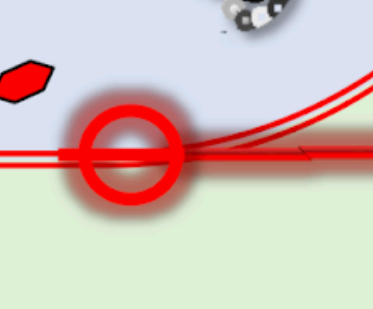
are non-ribosomal antibiotics composed of a lipid (fatty acid) linked to a short peptide chain that binds to bacterial cell membranes and increases permeability, leading to cell death.
function by directly disrupting the bacterial cell membrane rather than inhibiting a specific metabolic enzyme or the cell wall. They consist of a lipid tail that inserts itself into the cytoplasmic membrane in a calcium-dependent manner. This insertion causes rapid depolarization of the membrane, leading to a massive efflux of intracellular ions (especially potassium). This loss of membrane potential halts all synthetic processes (DNA, RNA, and protein synthesis) and triggers cell death. Because they work on the membrane itself, they are effectively bactericidal even against resting or slow-growing bacteria that might resist other antibiotics.
Lipopeptides
Daptomycin

are cyclic polypeptide antibiotics that bind to lipopolysaccharides (LPS) in the outer membrane of Gram (-) bacteria, disrupting membrane integrity and causing leakage and cell death.
(CELL WALL)
are "detergent-like" antibiotics that specifically target Gram-negative bacteria. They work by binding to lipopolysaccharides (LPS) and phospholipids in the outer bacterial membrane. The drug's cationic (positively charged) peptide portion displaces calcium and magnesium ions that normally stabilize the LPS molecules. This insertion physically disrupts the membrane's integrity, creating holes that increase permeability. The resulting leakage of intracellular contents leads to rapid cell death. Because they target the outer membrane, they are often a "last resort" for multi-drug resistant Gram-negative infections.
POLYMYXINS
Reasons for MICROBIAL RESISTANCE
Impermeability
Modification
Pumping out
Inactivation
MICROBIAL RESISTANCE
Modified cell wall protein
Impermeability
MICROBIAL RESISTANCE
Modified drug target
Modification
MICROBIAL RESISTANCE
Increasing active efflux of the drugs
Pumping out
MICROBIAL RESISTANCE
Add a phosphate group on the antibiotic, which will reduce its ability to bind to the bacterial ribosomes
Inactivation
In Gram (-) bacteria,beta-lactamases can be overexpressed and hydrolyze the beta-lactam ring of what drug
penicillin in the periplasmic space.
To counter this, some drugs inhibit beta-lactamases instead of acting as antibacterials. An example is
clavulanic acid
Explain SYNTHESIS OF CELL WALL
Peptidoglycan monomeric unit
Transglycosylation
Crosslinking by transpeptidase (catalyzed by the enzyme transpeptidase)
cell wall synthesis
The precursor disaccharide units are formed in the cytoplasm. Peptides are attached, and several steps follow before the structure is transported outside. The nascent peptidoglycan chain refers to the newly formed chain.
Peptidoglycan monomeric unit
cell wall synthesis
After translocation to the periplasmic space, the disaccharide units are linked to the existing peptidoglycan chain through WHAT STEP. This extends the structure of the cell wall.
Transglycosylation
Cell wall synthesis
For the cell wall to become strong, THE WHAT is required. This process is catalyzed by the enzyme transpeptidase, also known as penicillin-binding protein. It links the D-alanine–D-alanine residues. The presence of D-amino acids is unique to bacteria and contributes to selectivity.
Crosslinking by transpeptidase
DRUGS ACTING ON CELL WALL SYNTHESIS
Penicillins
Cephalosphorins
Β-lactams
Vancomycin
Carbapenems
Cycloserine
Bacitracin
Nonribosomal peptides - produced ng other microbes
Cycloserine, Bacitracin, Vancomycin
It’s a transpeptidase inhibitor; it particularly inhibits the peptide linkage between the peptidoglycan monomers. The pictures below show the formation of the peptidoglycan monomer until its maturation in the layer.
This drug target the cross-linking (transpeptidase) - D-Alanyl-D-Alanine
PENICILLINS
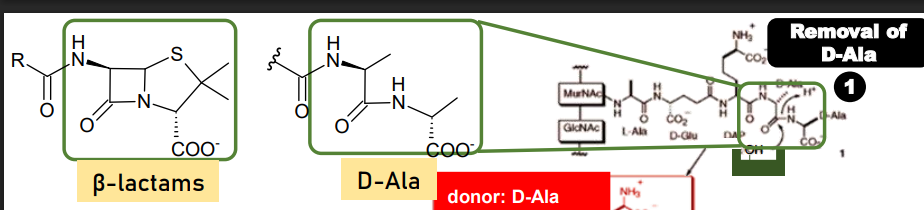
We can see the resemblance of the beta-lactam ring to the D-Ala transition state (D-Ala is in the middle of breaking and forming new bonds) and drugs that look like the transition state have higher affinity towards the enzyme because they resemble the active conformation of the enzyme, meaning less energy is needed to change into the active form. Therefore, there is higher affinity of beta-lactams compared to the D-alanyl-D-alanine peptide.
Consequence: high affinity in exchange for stability.
Beta lactams kinemerut mej MOA
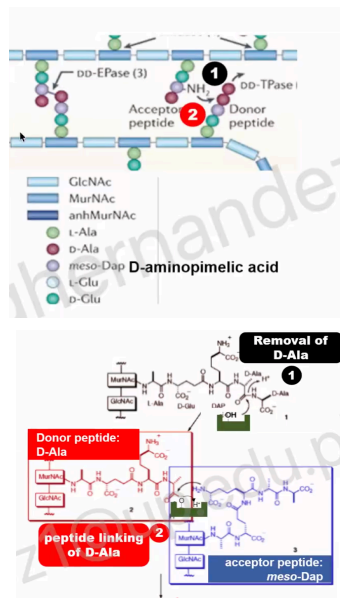
This cross-linking involves three major steps:
First step
Removal of D-Ala - the one at the C-terminus will be removed.
The main substrate is WHAT, which is a terminal peptide of the donor peptides in cross-linking
D-Alanyl-D-Alanine
which indicates that this particular enzyme only accepts peptide bonds belonging to both D amino acids
DD-TPase = DD-transpeptidase
Explain penicillin’s selectivity towards bacterial transpeptidases.
Implication: In terms of selectivity, in terms of ease of designing drugs against bacteria more than in human protein targets, the transpeptidase, although we also have transpeptidases in our body, there’s no chance that penicillins will attack them because our transpeptidase only accommodates L-amino acids.
The bacteria's cell wall is built using D-amino acids (specifically the D-Ala-D-Ala terminus). The enzyme responsible for cross-linking these building blocks is DD-transpeptidase. The "DD" prefix is critical; it signifies that the enzyme's active site is stereospecifically designed to only recognize and bind to D-configuration peptide bonds. While humans do have transpeptidases, our enzymes are evolved to process L-amino acids (the standard building blocks of human proteins). Because Penicillins are designed to mimic the D-Ala-D-Ala transition state, they fit perfectly into the bacterial "DD" enzyme but are physically ignored by human "LL" enzymes. This stereochemical mismatch provides a natural safety barrier, ensuring the drug only targets the pathogen.
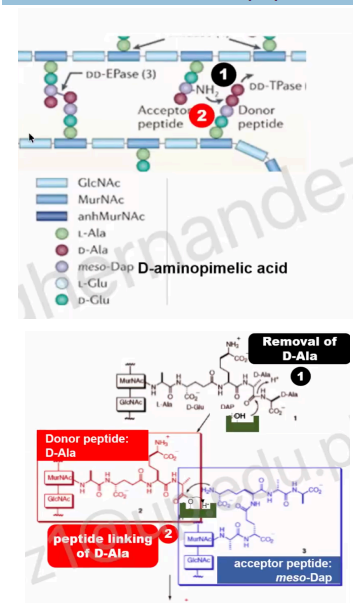
This cross-linking involves three major steps:
Second step
Peptide linking of D-Ala (that was left, which is the new C-terminus) to the meso-Dap.
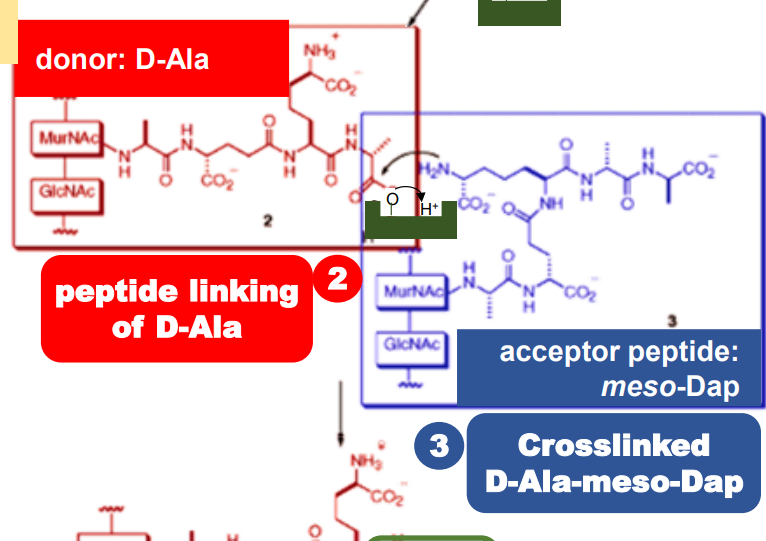
This cross-linking involves three major steps:
Second step
The remaining D-Ala binds to the enzyme when
the D-alanine residue is removed in the first
step. This is now ready to link with the
meso-Diaminopimelic acid (meso-DAP).
Acceptor peptide; already attached to the existing peptidoglycan layer. The one to cross-link with newly made peptidoglycan units.
meso-DAP
The donor peptide to be attached; currently bound to the enzyme.
D-alanine
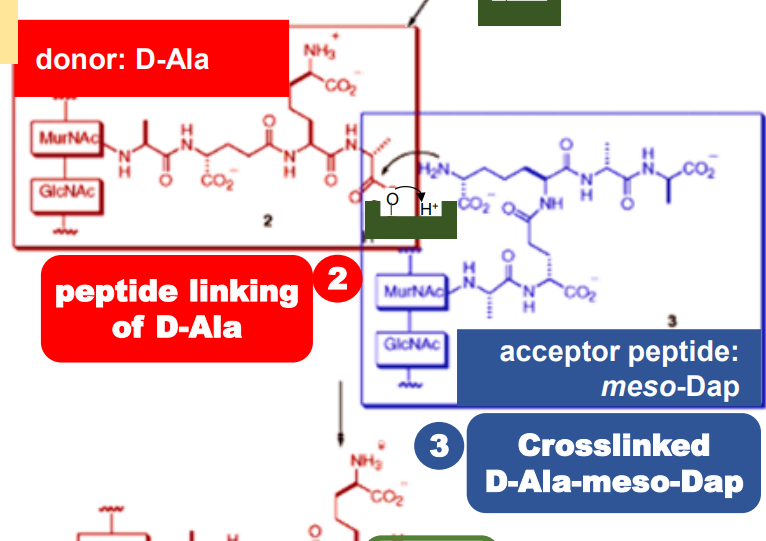
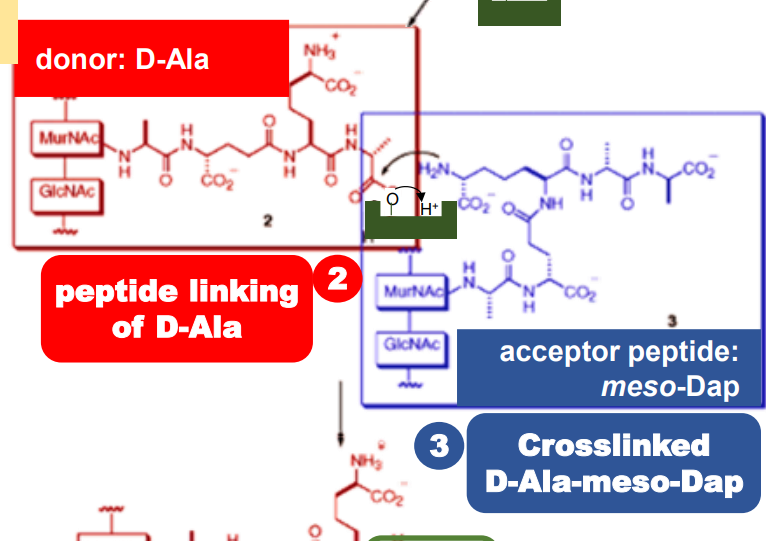
What happens when there is a formation of peptide bond
Once there is formation of a peptide bond (condensation reaction) between the carbonyl carbon and the amino residue of the meso-DAP, when the peptide linkage is formed, it will eventually return to the peptide form itsel
This cross-linking involves three major steps:
3rd step
(3) Crosslinked D-Ala-meso-Dap
■ It looks like it’s still in the transition state but eventually it will adapt to a form such that it will be an ordinary straight chain like other peptide bonds. It will be liberated from the enzyme binding site.
Penicillin
MECHANISM: COMPETITIVE INHIBITION
mimic the transition state, which is the active state—in the middle of bond formation and bond breaking.
Once the penicillin binds at the same site as the peptide linkage, and due to steric hindrance, the bond formed between the penicillin and the seryl residue of the penicillin-binding protein cannot be hydrolyzed. Due to this, the substrate (D-Ala-D-Ala) cannot bind, keeping it away from the active site.
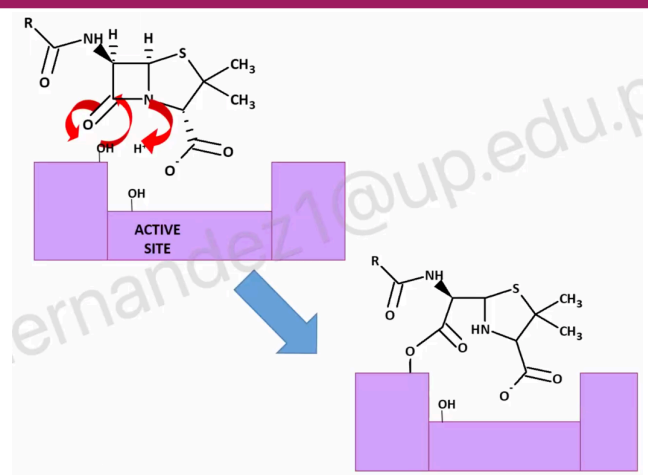
Penicillin
MECHANISM: UMBRELLA EFFECT
Non-competitive, allosteric, but because of the size of the molecule, it blocks the substrate from binding to the active site. ● Instead of the seryl residue found directly at the active site, it binds to the seryl residue in the allosteric site.
INTRINSIC RESISTANCE OF GRAM (-) BACTERIA against Penicillins
Gram (-) bacteria are intrinsically resistant against penicillins, because, unlike Gram (+) bacteria, Gram (-) bacteria, to begin with, are already producing beta-lactamases, and the more they are exposed to penicillins, the production of beta-lactamases is promoted.
Another reason for the intrinsic resistance of Gram (-) bacteria is that their porin channels do not allow the entry of large, hydrophobic, and negatively charged molecules. ○ Penicillins are acidic (free carboxylate form) and are relatively large. So it can’t just enter the porin channels.
RESISTANCE TO PENICILLINS: β-LACTAMASE ENZYMES
Beta-lactamases target the hydrolysis of beta-lactam rings. Once it opens, it will no longer bind to the penicillin binding protein.
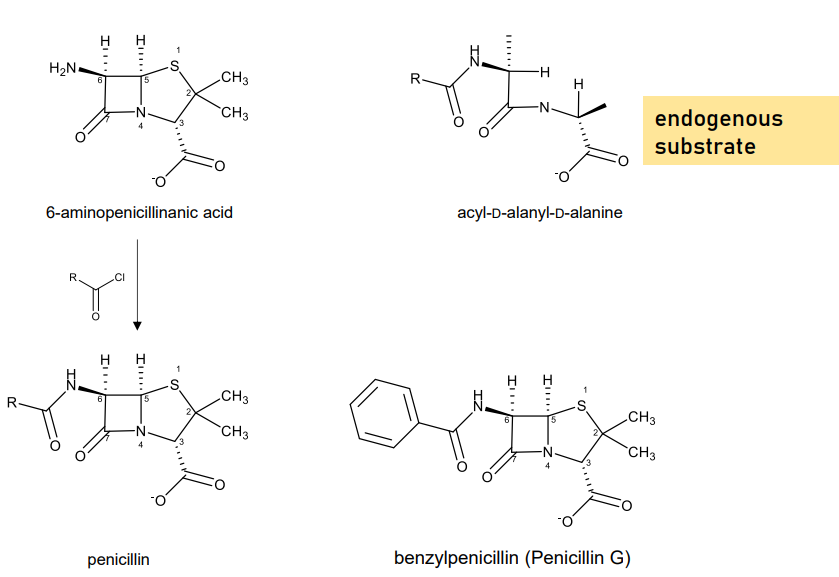
The penicillins we know today, are actually, more likely (especially benzylpenicillins and other penicillins), produced semi-synthetically and are synthesized from
6-aminopenicillanic acid.
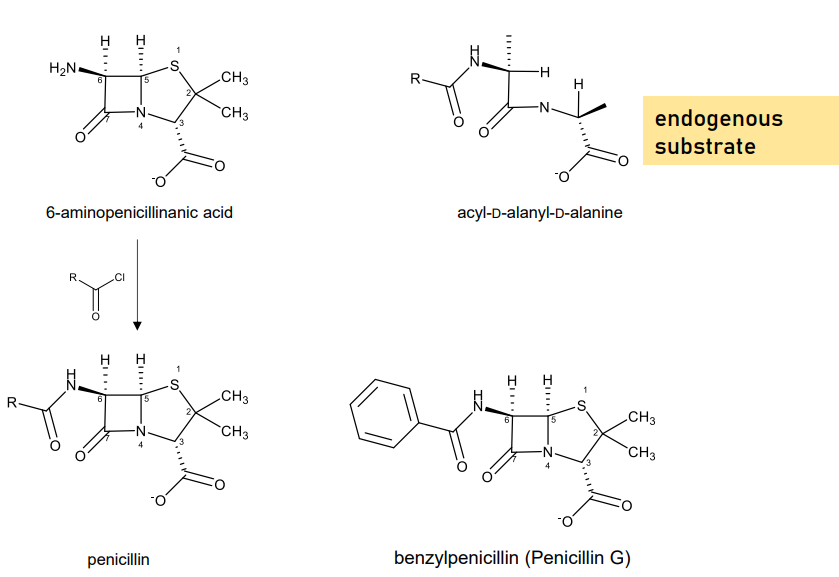
The fermentation product of molds that produce penicillins.
Much more abundant than penicillin itself.
More stable so it is isolated easily.
6-aminopenicillanic acid
Resitance to Penicillins
Production of high levels of transpeptidase
Change in affinity of the transpeptidase to penicillin
Efflux Mechanism
Mutations and genetic transfers
Bacterial tolerance
Resitance to Penicillins
Inherent to Gram-negative bacteria
Production of high levels of transpeptidase
Resitance to Penicillins
relative proportions varies across bacterial species
variable susceptibility to different penicillins
Change in affinity of the transpeptidase to penicillin
Resitance to Penicillins
Over-expression of P-glycoprotein or ATP-binding cassette proteins. ■ Pronounced in Gram-negative bacteria.
Efflux Mechanism
Resitance to Penicillins
Plasmids: genetic carrier of resistance genes
“Memory”
Mutations and genetic transfers
Resitance to Penicillins
Bactericidal to bacteriostatic
Bacterial tolerance
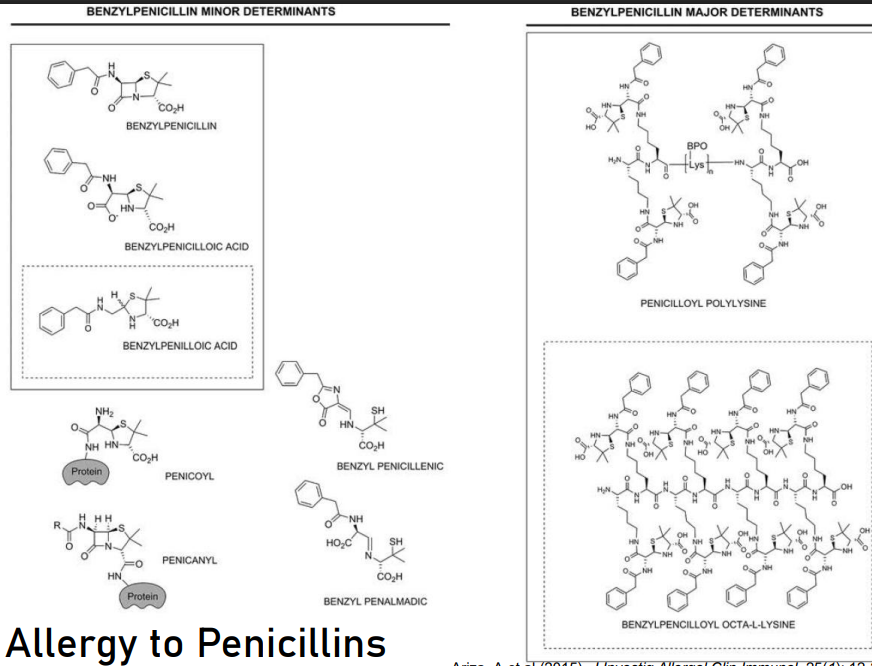
ALLERGY TO PENICILLINS
It's better to know if you are allergic to penicillin.
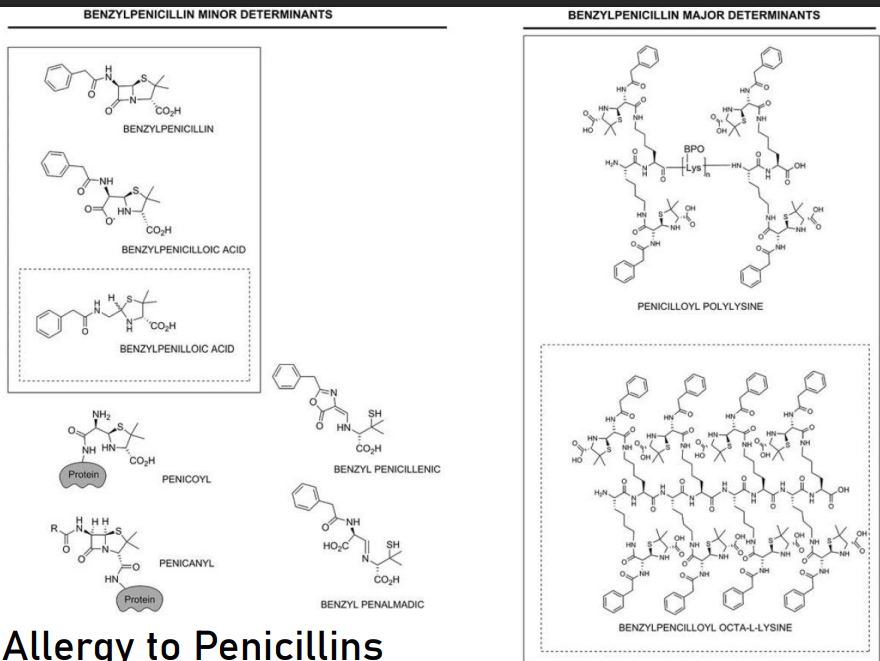
They resemble the structure of peptides; these penicillins are amino acid derivative products. The enzyme involved in their synthesis is that they are produced by non-ribosomal peptide synthetases.
Because of their resemblance, there is a higher chance that they can be immunogenic.
Determinants of allergic reaction
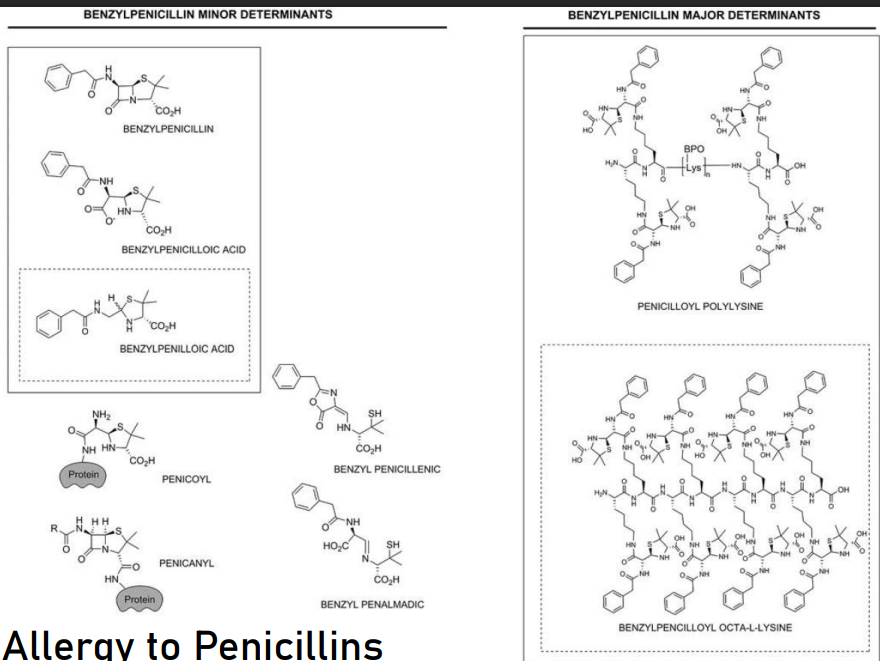
Explain Allergy to Penicillins
These are just some of the examples of the formation of an allergy towards penicillins:
○ In the form of penicillin itself.
○ Penicilloic acid (hydrolyzed form)
○ Benzylpenicilloic acid
It can form adducts with endogenous proteins (they resemble peptides); the body will consider it foreign, leading to an immune response. In certain cases, there can be formation of penicilloyl polylysine and penicilloyl octa-L-lysine. These are the determined (so far) forms of penicillin that cause allergic reactions.
The immunogenicity of Penicillins stems from their nature as dipeptide mimics. Because they are synthesized by non-ribosomal peptide synthetases in fungi, they possess a structure that closely resembles human peptide bonds.
The actual allergic trigger is rarely the "free" drug. Instead, it’s the formation of hapten-protein adducts. Penicillin (or its hydrolyzed form, penicilloic acid) is a highly reactive electrophile. It reacts with the amino groups (like lysine residues) of your own endogenous proteins (self-proteins). This "labels" your protein with a penicillin molecule. The immune system no longer recognizes the protein as "self" and attacks the penicilloyl-protein complex. Specific diagnostic markers for this include penicilloyl polylysine, which are used in skin testing to confirm a Type I hypersensitivity.
Penicilln SAR: Beta Lactam Ring and Carboxylate Residue are important for
binding
If this is modified (e.g beta-lactam ring is opened), it will not bind to transpeptidase or penicillin-binding proteins.
Carboxylate residue binds to the lysine residue of the target enzyme.
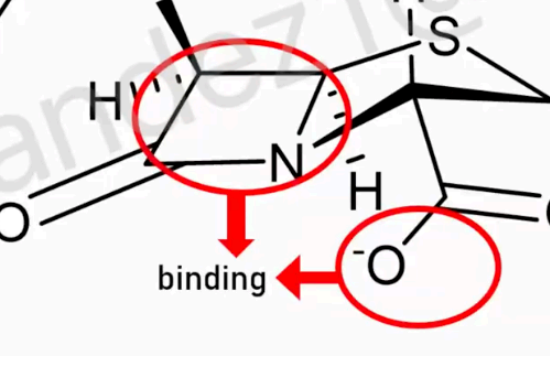
whole beta lactam ring including thiazole structure
The whole beta-lactam ring which includes thiazole introduces greater strain on the ring alone. This is the reason why it is unstable.
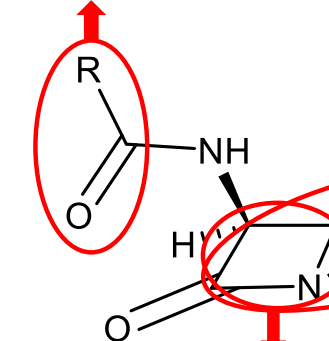
Acyl connected amino (which is also attached to the beta-lactam ring:
this is the part that can be modified”:
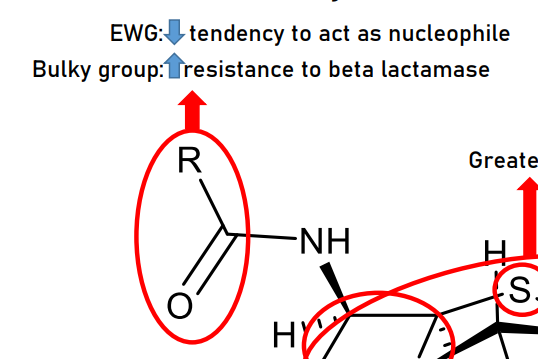
Acyl connected amino (which is also attached to the beta-lactam ring ATTACHED WITH BULKY GROUP
more resistant to beta lactamases
If bulky groups are attached in this portion → increase tendency to be effective against gram-negative bacteria
Acyl connected amino (which is also attached to the beta-lactam ring ATTACHED WITH electron-withdrawing group
(EWG)
If changed to an electron-withdrawing group (EWG) → lower tendency to act as nucleophile
Acting as a nucleophile has added benefits for the molecule since it has added binding
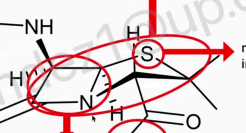
T/F sulfur is has the most important role (based on SAR studies)
F. All penicillins have sulfur but have no important role (based on SAR studies).
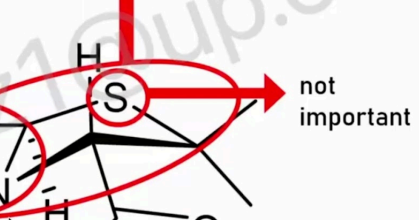
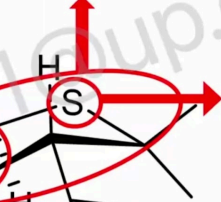
If this will be changed with an atom that has almost similar electronic properties or if changed to Carbon
not affect the activity of Penicillins
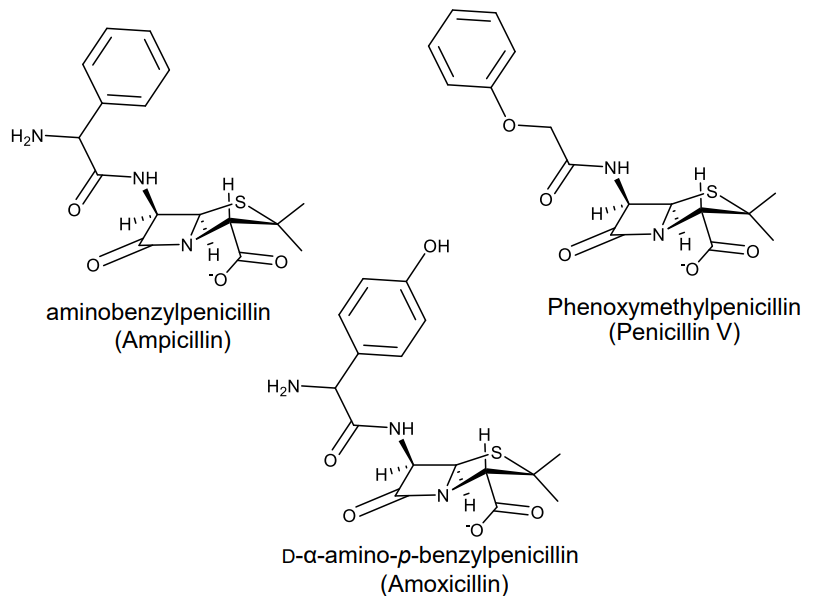
Aminobenzylpenicillin (Ampicillin)
Phenoxymethylpenicillin (Penicillin V)
D-α-amino-p-benzylpenicillin (Amoxicillin)
ACID-RESISTANT PENICILLINS
Bulky groups attached to acyl residue attached to amide (also attached to beta-lactam ring).