BMB 212 Exam 2
1/72
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
73 Terms
What is a recombinant DNA molecule?
A circular DNA plasmid that carries essential elements that allow the DNA to function and be maintained as well as a gene of interest
What is a gene of interest?
DNA sequence that codes for a protein we want to make
What is a plasmid?
A small double stranded circular DNA found in bacteria in addition to the genome
Not generally required for basic survival of a bacteria but can provide a growth advantage like antibiotic resistance
Can replicate independently of the bacterial genome (using its own origin of replication) to make sure new bacteria inherit at least one copy
Explain how recombinant DNA technology is used to produce proteins of interest.
This technology allows us to produce large quantities of proteins in a humane way. Before this development, 10,000 pigs had to be killed for one tiny bottle of insulin. Now, a circular DNA plasmid can be inserted into a bacteria which will then use its own machinery to make the protein of interest.
Draw a basic outline of how an insert can be cloned into a plasmid using restriction enzymes and DNA ligase, and how that recombinant plasmid can then be transformed into bacteria.
*
What is an Ori?
This is the origin of replication that allows the plasmids to replicate independently of the bacterial genome.
What are the common plasmid elements?
Ori, antibiotic resistance gene, restriction enzyme sites, elements to aid gene expression, tags, and gene of interest
What is an antibiotic resistance gene?
This gene codes for proteins that provide resistance to certain antibiotics. This gene is often referred to as a selective marker because it allows the bacteria with the plasmid to grow under conditions that those without the plasmid could not.
Give two examples of antibiotic resistance genes
KanR gives resistance to kanamycin and AmpR gives resistance to ampicillin
What are restriction enzyme sites?
These are the sites recognized by restriction enzymes. Plasmids often contain a number of different enzyme recognition sequences clustered in an area called a multiple cloning site.
What are restriction enzymes?
These are enzymes that cut DNA at specific sequences
What are elements that aid in gene expression?
These include anything that help control or initiate transcription or translation. This can include promoters, terminators, etc
What are protein tags?
These sequences allow proteins that are expressed from a sequence cloned into a plasmid to have a tag fused to it. These tags help in detection and purification.
Give an example of protein tag.
His6- 6 histidine residues
This helps in purification because the residues will bind to nickel or cobalt beads.
Explain the technique used to purify plasmid DNA from bacterial cells.
The technique takes advantage of two differences between chromosomal E.coli DNA and extrachromosomal DNA like recombinant plasmids: chromosomal DNA is much larger and is linear where the extrachromosomal is closed and circular. Centrifugation separates the high MW E.coli DNA from the low MW plasmid DNA. Then the plasmid DNA is treated with a cell extract with mild alkali which breaks H bonds involved in base pairing. This irreversibly denatures chromosomal DNA and reversibly denatures plasmid DNA (returns to normal in normal pH).
Explain how absorbance is used to measure DNA concentration and estimate purity.
Nucleic acids in solution absorb UV light at a peak wavelength of 260nm while proteins have peak absorbance at 280nm. Concentration of molecules that absorb light can be determined based on the Beer Lambert Law (A= elc). You can begin by calculating the concentration if the absorbance as one, which is a standard conversion factor that is used to estimate the concentration of double-stranded DNA in a sample.
You have isolated 0.5ml of pure DNA. You take an absorbance measurement at 260nm of an undiluted sample. The reading is A260= .4. Find the concentration using both Beer's law and the standard conversion factor. Also find total yield.
Beer's Law:
0.4A260 = (.020 (ug/ml)-1(cm)-1(1cm)(C)
C= 20 ug/ml
Conversion Factor:
0.4A260/c = 1.0 A260/50 ug/ml
C- 20 ug/ml
To find total yield, you multiply concentration by total volume so 20 x .5ml= 10ug
Be able to analyze absorbance data to make conclusions about DNA concentration.
We can test a DNA solution for 260nm and 280nm and use the results to assess the purity of DNA. The A260/A280 ratio does this. For pure DNA, the ratio is between 1.8/2. A ratio lower than 1.8 (meaning higher A280 absorbance) indicates protein contamination and we cannot make accurate measurements of the actual DNA concentration. Above 2.0, there may be RNA contamination. This is because adenine and uracil have high 260/280 rations and uracil is only found in RNA.
If you take the absorbance of two different DNA samples and you get the following values, what does that suggest about the purity of each sample?
Sample 1:
A 260: 1.14
A280: 0.6
Sample 2:
A 260: 1.36
A280: 0.8
Sample 1 is pure and Sample 2 is likely contaminated with protein
Copy and paste the following templte into the answer box in order to record your data and answer the following questions (show your work).
Concentration of purified plasmid: 27.1
A260:A280 ratio: 1.76
Yield (ie how many total ng you purified):
What volume of the purified plasmid would you need to have 500 ng of DNA?
What does your A 260:A280 ratio suggest about your purification?
need 18.5 uL
Ratio suggests protein contamination
Assuming that we are talking about DNA molecules of the same size, but different shapes. Match the terms to indicate how the molecules would run on an agarose gel.
Supercoiled DNA
Linear DNA
Open Circle (nicked) DNA
Runs in between the other two
Runs fastest in gel
Runs slowest
Supercoiled DNA
Runs fastest in gel
Linear DNA Runs in between the other two
Open Circle (nicked) DNA Runs slowest in gel
Match the following terms to indicate how the following linear pieces of DNA would be expected to run on on an agarose gel.
Fastest, slowest, in between
1kb
4kb
2kb
1 fast, 4 slow, 2 in middle
Which of the following is used to visualize the DNA molecules on the agarose gel?
GFP fluorescence
Ethidium Bromide (EtBr)
no stain is necessary, the DNA molecules fluoresce on their own
None of the above
EtBr
Which of the following are true? Select all that apply.
A. If a plasmid has a single recognition site for a restriction enzyme, when you cut the DNA with restriction enzyme, you expect to end up with two DNA
B. If a plasmid has two recognition sites for a particular restriction enzyme, when you cut the DNA with that restriction enzyme, you expect to end up with two DNA fragments.
C. If a plasmid has a single recognition site for a restriction enzyme, when you cut the DNA with that restriction enzyme, you expect to end up with one DNA fragment.
D. If a plasmid has two recognition sites for a particular restriction enzyme, when you cut the DNA with that restriction enzyme, you expect to end up with three DNA fragments.
E. There is no way to determine the number of fragments that you will end up with after digesting a plamid with restriction enzymes.
B and C
Gel 1 is empty
Match the following"
Gel 1
Gel 2
Depicts analysis of pET30 GFP DNA
Depicts analysis of pET30 DNA (ie empty plasmid)
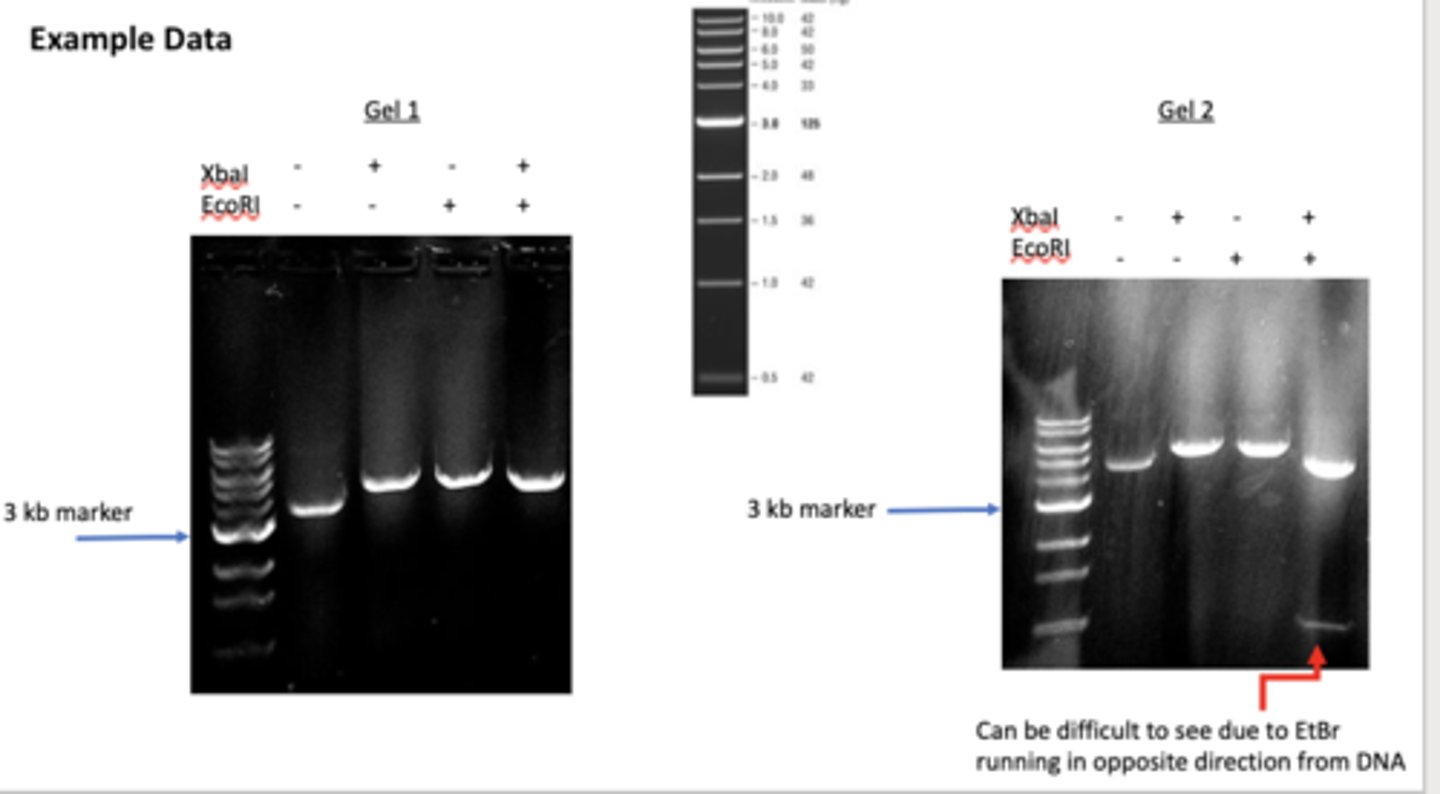
Describe what a restriction digest is and how a restriction digest can be used to determine identity of a mini prepped plasmid (ie empty vector or recombinant DNA molecule).
These are produced when restriction enzymes cut DNA. Since many restriction enzymes exist and each has a unique recognition site, analyzing the fragments produced when DNA is subjected to certain restriction enzymes can help identify different DNA. If a plasmid has one recognition site, the enzyme will cut once, creating linear DNA. If it has two sites, it cuts twice, cleaving the DNA into two DNA fragments.
Nicked DNA if there is one enzyme will travel the least, then linear travel faster and if there are two fragments that was not the empty plasmid
How does percentage of agarose gel affect separation?
Agarose gels range in concentration from .3-2%. Different concentrations optimize separation of different sizes/lengths of DNA. High percentage gels separate smaller/shorter fragments from each other whereas low percentage gels resolve larger/longer DNA fragments from each other. 0.5% is for 50-2 whereas 2 is for 2-.1
What does applied electrical current do when running the gel?
Voltage is applied at the ends of an agarose gel, generating an electric field with a strength defined by the length of the gel and the potential difference. DNA molecules exposed to this field migrate towards the anode (posiitve-red) due to the negatively charged phosphates along the DNA backbone. Migration velocity is limited by friction and the rate of migration depends on length and shape.
Why dye did we use to visualize DNA on the agarose gel?
The dye we used is ethidium bromide. It intercalates between stacked bases of nucleic acids and fluoesces rego orange when illuminated with UV light.
How does DNA shape/size affect gel results?
There are pores formed when the gel solidifies, and larger molecules move through them slower than smaller molecules. We can compare where the bands are to a known reference ladder to determine mass and kb. Circular plasmids purified from bacteria are ofte supercoiled. A single nick will release some supercoiling and result in a more open circle structure. NIcked DNA runs slower than a DNA molecule of the same size with linear form. Supercoiled molecules run faster than linear DNA. This is because different shaes can snake through pores differently.
review what was actually done in the procedure
Which of the following are true? Select all that apply.
When a protein is subjected to SDS PAGE, it is denatured and therefore loses its tertiary structure.
SDS PAGE gels separate proteins based on size (molecular weight) and shape.
SDS PAGE gels are made from an agarose matrix (identical to DNA gels) but unlike DNA gels, they also contain the detergent SDS.
None of the above are true.
A
According to the background material, presence does not equal ____________.
purity
How are proteins visualized after SDS-PAGE?
UV absorbance
Ethidium Bromide (EtBr) stain
Fluorescence
Coomassie stain
Coomassie stain
GFP is able to fluoresce because
it binds to a dye that fluoresces when exposed to UV light
the protein is mutated when exposed to UV light
the amino acids in the protein undergo a chemical reaction with each other to form an internal chromophore
None of the above
A
The his tag (select all that apply)
is used to help purify the protein
is a string of amino acids that is added to the end of the protein
is coded for by the DNA in the pET30 GFP plasmid
binds to nickel resin (beads)
All of the above
Put the following steps in order for an affinity purification
Bind
Wash
Elution
Denature
1, 2, 3, not used
What is lacO?
An operator where the lac repressor binds (lacI)
Where does IPTG bind?
It binds to the lacI repressor and turns on expression from the promoter
What binds the promoter?
region of DNA where RNA polymerase binds
Where does lacI bind?
Binds to lacO sequence and blocks transcription
Where does RNA polymerase bind?
Binds to the promoter sequence and is responsible for transcription
add last two post labs when they are graded
Describe the steps in gene expression (transcription and translation) and how promoter sequences play a role in the expression process. Specifically discuss the pET30-GFP plasmid.
Transcription
RNA polymerase assembles at promoter sequences along with transcription factors. Then, it moves forward until it finds a transcription start site, where it begins to read DNA to make RNA. In the recombinant plasmid (pET30-GFP), the GFP DNA sequence was placed after the promoter in the pET30 plasmid. This promoter is called the T7 promoter. So the RNA binds and reads until it reaches the T7 promoter sequence, it will read the GFP gene and make the mRNA for the GFP sequence.
Translation
Ribosomes and translation factors will then read the GFP mRNA and use it to make the GFP protein, which fluoresces green.
Describe how E. coli cells are genetically engineered to allow for expression from the T7 promoter, and why genetic engineering of the E.coli is necessary to get expression of genes that use the T7 promoter.
The expression of GFP is controlled by the T7 promoter, but this is not the natural E.coli promoter. Since this promoter is from a bacterial phage or virus, the native bacterial RNA polymerase cannot recognize and bind to the T7 promoter to start GFP expression. In order to have bacteria express the GFP gene, we have to put the plasmid in a specific strain of bacteria that have been genetically modified to make the T7 RNA polymerase. Expression of that T7 RNA polymerase can be controlled so we can decide when the bacteria makes that polymerase and thus any genes that require that polymerase.
Why is it beneficial to be able to control expression in bacteria?
The bacteria could produce lots of foreign proteins too early and it can kill them or harm them before we can get any proteins.
The protein can be toxic based on the function of the cell.
We can use control of expression to study the effects of a protein expressed.
Sometimes we want to use bacteria to express proteins that don't fold well at an optimal temperature for the bacteria. So first we grow it then shift the temperature and start expressing the protein of interest.
What is the lac promoter an example of in terms of expression control?
inducible promoter- turned on/off in the presence of certain chemicals
Explain how the lac operon sequences (lac promoter, operatore, and lacI gener) are used to control gene expression.
The lac operon in E.coli, which is a native cluster of genes that are required for producing the enzyme lactase to breakdown lactose for nutrient acquisition. The E.coli only use energy to make the enzyme to breakdown lactose when there is lactose around and needed as a food source. When lactose is not around, the lacI (lac repressor) binds to the operator (lacO), which is between the promoter and lactase gene. It acts as a roadblock for the RNA polymerase. When lactose is available, it binds to the lac repressor protein and makes it change its shape and it leaves the operator.
In some bacterial strains, the native lac operon has been genetically modified in such a way that a viral T7 RNA polymerase is inserted into the genome under the control of a lac promoter. So, it can produce the RNA polymerase and any other genes that use the T7 promoter. Therefore, we can control the product of GFP by deciding when to supplement lactose or similar genetically modified molecules. Since bacteria quickly break down lactose, this induction is temporary.
Describe the role of IPTG in gene expression.
We use IPTG because bacteria breaks down lactose quickly so induction is temporary. We circumvent this by using a lactose analog called IPTG, which looks like a lactose molecule but cannot be broken down. So IPTG continuously induces GFP production, leading to the higher yield.
Be able to make predictions about when genes would be expressed to significant levels if they are controlled by sequences such as T7 promoters and lac operators.
**
Explain what each component does in the E.coli expression system.
Bacterial Genome
Plasmid
Bacterial genome: This provides the genes for the machinery that is needed for gene expression (RNA poly, transcription factors, etc)
Plasmid: This contains genes for the protein we want to make (GFP). Some plasmids contain DNA sequences tha help control gene expression. For example, the lac operator is between the T7 promoter and GFP genes. Also, the pET plasmid contains the sequence for the lacI protein which is an additional copy to help control expression. IN summary, the lac operator/repressor system is being used to control expression of both T7 polymerase from the genome and the GFP gene from the plasmid.
What is leaky expression?
Sometimes the lac I protein releases from lacO which allows a RNA poly to sneak through and transcribe the gene. By putting a lac operator sequence between the lac promoter and T7 poly genee and between lac promoter and GFP, that prevents the majority of it.
What was done to the E.coli before we came into lab?
A specialized strain of E coli called BL21-DE3, which uses the lac promoter to control expression of the T7 RNA polymerase, was chosen. These cells were transformed with the pET30-GFP plasmid and grown. Before the bacteria reached stationary phasem IPTG was added to induce GFP production. In the presence of IPTG or lactose, IPTG binds to the lac operon repressor, which is inhibiting expression of genes in lac person. The binding of IPTG releases the repressor, leading to product of T7 RNA polymerase. This recognizes and bines the T7 promoter in the pET plasmid, turning on expression of GFP.
Describe the steps necessary to purify a protein from an organism like bacteria using an affinity purification approach.
Make a protein extract
Purify protein, which means separating the POI from other proteins. We focus on affinity purification
Bind: protein of interest binds to an affinity matrix
Wash to get rid of unbound or loosely bound contaminating proteins
Elute to release your protein from the affinity matrix
How do we get access to protein extracts for purification?
Sometimes we can collect growth media if the cells secrete the protein. MOre often, the protein is inside the cell, so we have to break open the cells to get access. We used BugBuster which is designed to be gentle and efficient and releases soluble proteins from bacteria, yeast, plant, mammalian, and insect cells.
How was the GFP protein purified?
Once we have the protein extract, we need to purify the GFP protein from other bacterial proteins. To do this, we take advantage of the his tags. They have a high affinity to the Ni2+ ion.
When we apply the entire cell lysate to nickel resin, the his tagged proteins will preferably bind to the resin. The native proteins will not and are then washed off with buffer. After washing, the his tagged GFP wtll be eluted with buffer that has imidazole. This is a histidine analogue, so it competes with the his-tag for binding leading to elution of the his-tagged proteins.
Describe the levels of structures in proteins.
Amino acids are made of a backbone (amino, alpha carbon, carboxylic acid) and an R group or side chain connected to the alpha carbon. Proteins (polypeptides) are simply a long chain of amino acids that adopt a very specific structures that allows the protein to function.
Primary structure is defined as the sequence of amino acids in a protein. Secondary structure is defined by interactions between backbone molecules in each amino acids. Common structures are the alpha helix and beta sheets. Tertiary structure is defined by the interactions between amino acid side chains, and quaternary structures defines structure that results from interactions between multiple protein subunits.
Describe the structure of the GFP protein and how it allows for the protein to fluoresce when exposed to UV.
The GFP protein has a primary structure that adopts a secondary structure full of beta sheets. The funal arranged result in a tertiary structure that has certain amino acids (serine, tyrosine, glycine) that are close enough and in the environment to form an internal chromophore. This chromophore allows the GFP protein to fluoresce. This is because GFP absorbs the UV light and emits a lower energy green light.
Explain the theory behind SDS-PAGE and how proteins are separated.
SDS-PAGE allows separation of proteins according to size through a polyacrylamide matrix. The goal is to separate by molecular weight rather than by shape. It does this because it includes the SDS detergent which denatures proteins prior to and during the electrophoresis run. Denatured proteins unfolded int linear-like polypeptides so they lose their secondary and tertiary structural components. This means the primary structure mostly determines where a protein will end up on SDS PAGE.
What is the purpose of 4X protein sample buffer?
- Contains SDS and 2-mercaptoethanol
-SDS is an anionic detergent that ha an anionic head and hydrophobic tail. The tail interacts with the backbone and the head gives the protein a negative charge. It also denatures the protein. The 2-mercaptoethanol is a reducing agent that breaks disulfide bonds and helps denature.
What are the 12% Mini-PROTEAN gels?
These allow for separation of proteins. Since it is 12% it allows for separation int e 12-200 kD range of molecular weight.
What are the protein standards?
This is a mixture of ten recombinant proteins that give references to estimate protein size. It is like the DNA ladder from last time.
What is the SDS running buffer?
This includes Tris which is a buffering reagent and glycine whose ionic state is critical to gel separation.
What is the coomassie stain?
This is used to visualize proteins on the gel. The dye binds to hydrophobic and basic amino acids within proteins in acidic conditions.
What does the destain do?
The destain contains methanol and acetic acid which cause dye that isn't attached to protein to come out of the gel.
Explain the relationship of the SDS-PAGE portion of this module with the previous module on DNA and previous parts of the protein module.
In last week's lab, we were analyzing DNA on an agarose gel. That gel has large pore sizes to accommodate different shapes whereas the polyacrylamide has a gradient in pore size. The DNA gel separates based on size and shape and the protein gel separates by MW and can help us tell purity.
Last week we tested pure DNA and this week sis protein purity.
Explain how to access information on protein sequence and analyze that sequence for molecular weight.
First we search the NCBI database to find the GFP protein sequence that was cloned into the pET30-GFP vector that we used to express GFP in the E.coli cells.
Go to NCBI homepage, choose protein database
Search for protein name
Click protein title and copy the protein sequence from FATSA link
Copy the amino acid sequence of the GFP protein and put it in the protparam tool to find the MW
What did we do in the protein purification lab?
We ran the collected samples from GFP purification on the gel, which included uninduced soluble cell lysate, induced soluble cell lysate, column flow through, washes 1-3, and elutions ½. By running the samples, we can get an idea as to the efficiency and effectiveness of the purification scheme.
What did we expect to see on the SDS-PAGE gel when comparing induced and uninduced? Also discuss flowthrough, washes, and elution
We expect to see an increase in a protein that is the size of the GFP molecules when comparing the induced to uninduced. When we run a sample of the flow through on the gel, we can get an idea of the proteins in the cell that we were able to get rid of during the purification scheme. These proteins are ones that do not have an affinity for the nickel column. By running washes, we can determine if we are losing any of the GFP and an idea of the other proteins that have a weak binding affinity for the nickel resin in the column we used for purification. We can look at the elution step to see if there were proteins different than GFP bound to nickel. The take home message is presence does not equal purity (like with last week).
What is the process of protein expression and purification?
DNA isolation, transformation, protein expression, obtain cell lysate, sample (cell lysate) loaded onto column, column wash and POI elution
Draw the diagram
*
How does affinity column chromatography work?
Proteins of interest that have his tags will be attracted to nickel beads in the column and bind to them. Note that different tags will use different type of beads or columns. The proteins that were bound on the will be elute out imidazole
What is SDS PAGE and why do we run it?
Sodium dodecyl-sulfate polyacrylamide gel electrophoresis
We run it to assess purification method and sample purity