Ultrasound Findings / Birth Defects, Prenatal Diagnosis
1/136
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
137 Terms
spina bifida cause
Incomplete closer of spinal cord and vertebral column, typically due to failure of the neural tube to close during early embryonic development. Most commonly related to folate deficiency.
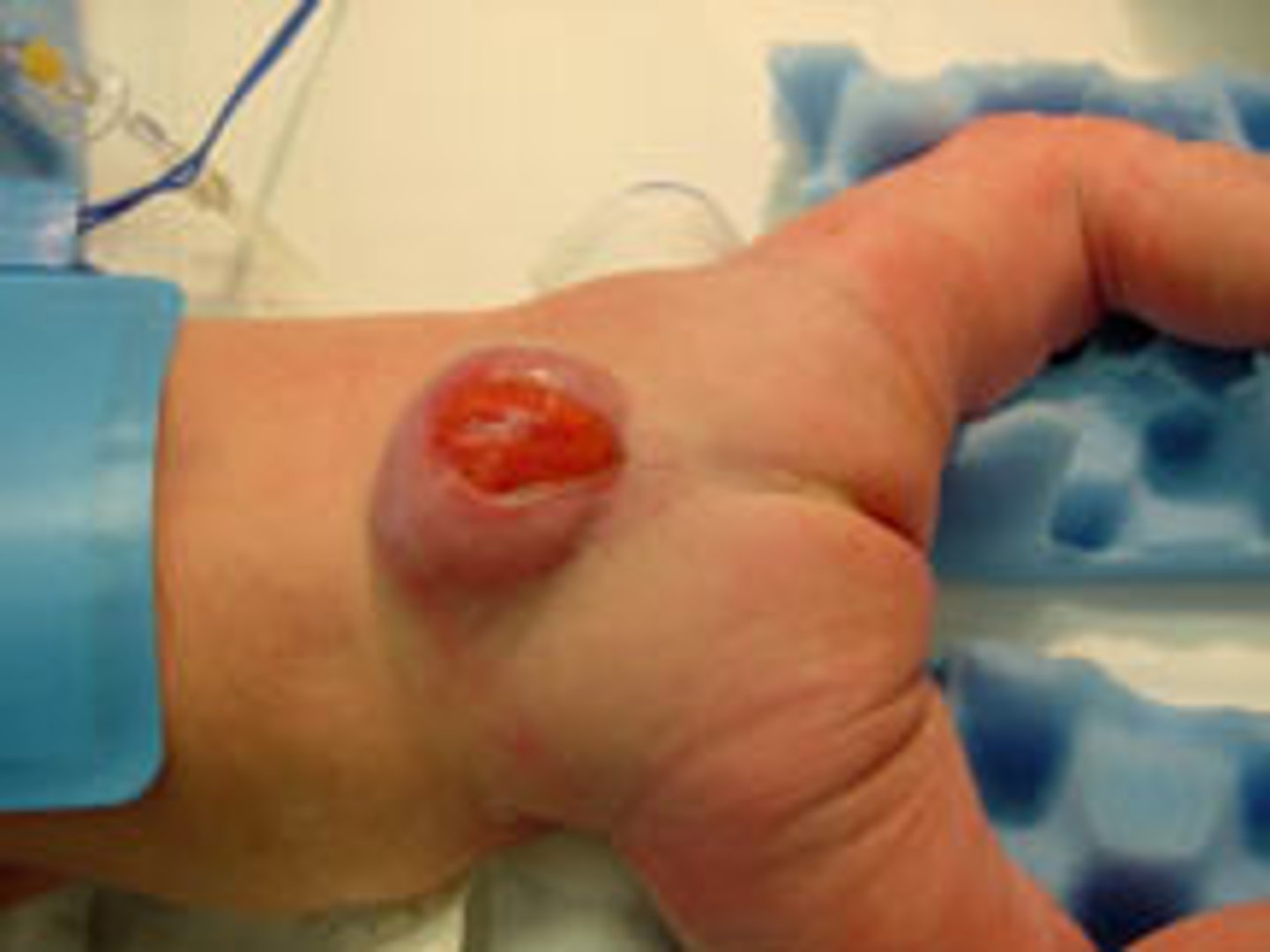
myelomeningocele
most severe form of spina bifida in which the spinal cord and meninges protrude through the spine
meningocele
the congenital herniation of the meninges ONLY, a milder form of spina bifida
spina bifida occulta
most common and least severe form of spina bifida without protrusion of the spinal cord or meninges
Anencephaly cause
defect in closure of the cephalic portion of the neural tube that results in incomplete development of the brain and bones of the skull
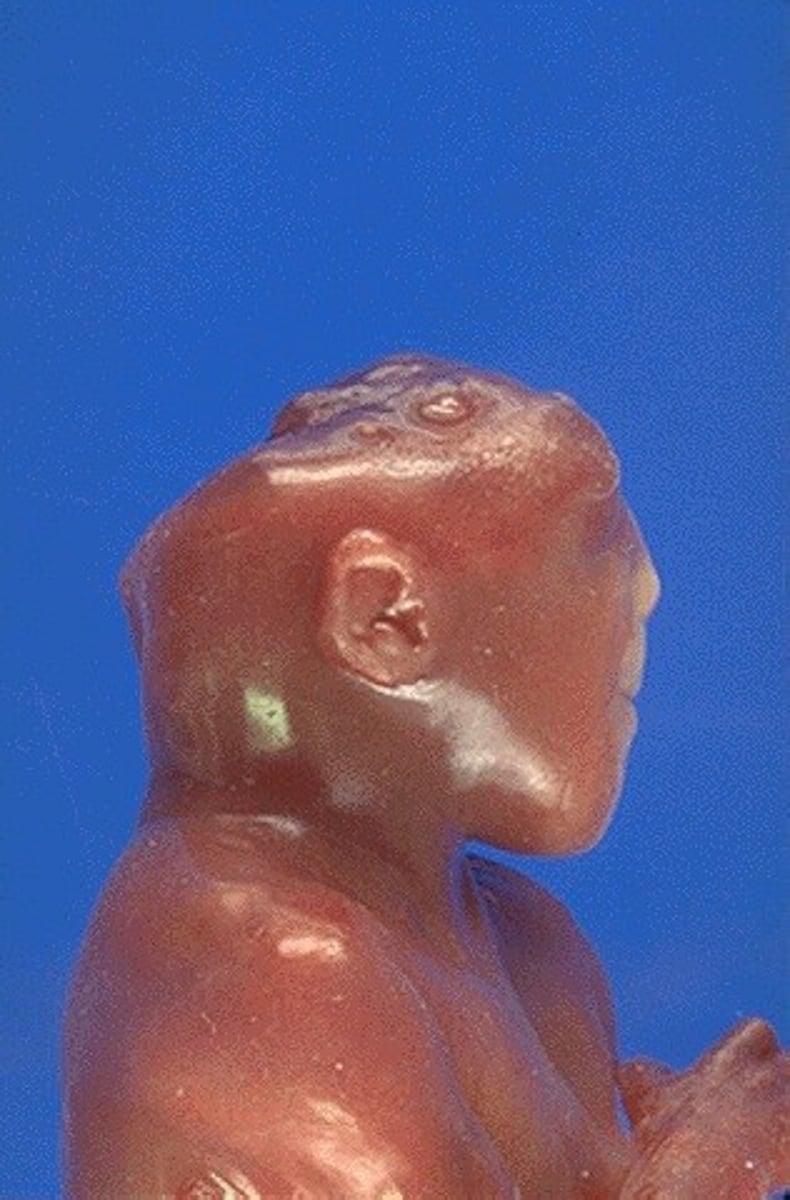
anencephaly prognosis
Most die shortly after birth, have premature reflexes only.
encephalocele
a congenital herniation of brain tissue through a gap in the skull. Similar to spina bifida, but due to a failure of the cranial end of the neural tube to close.
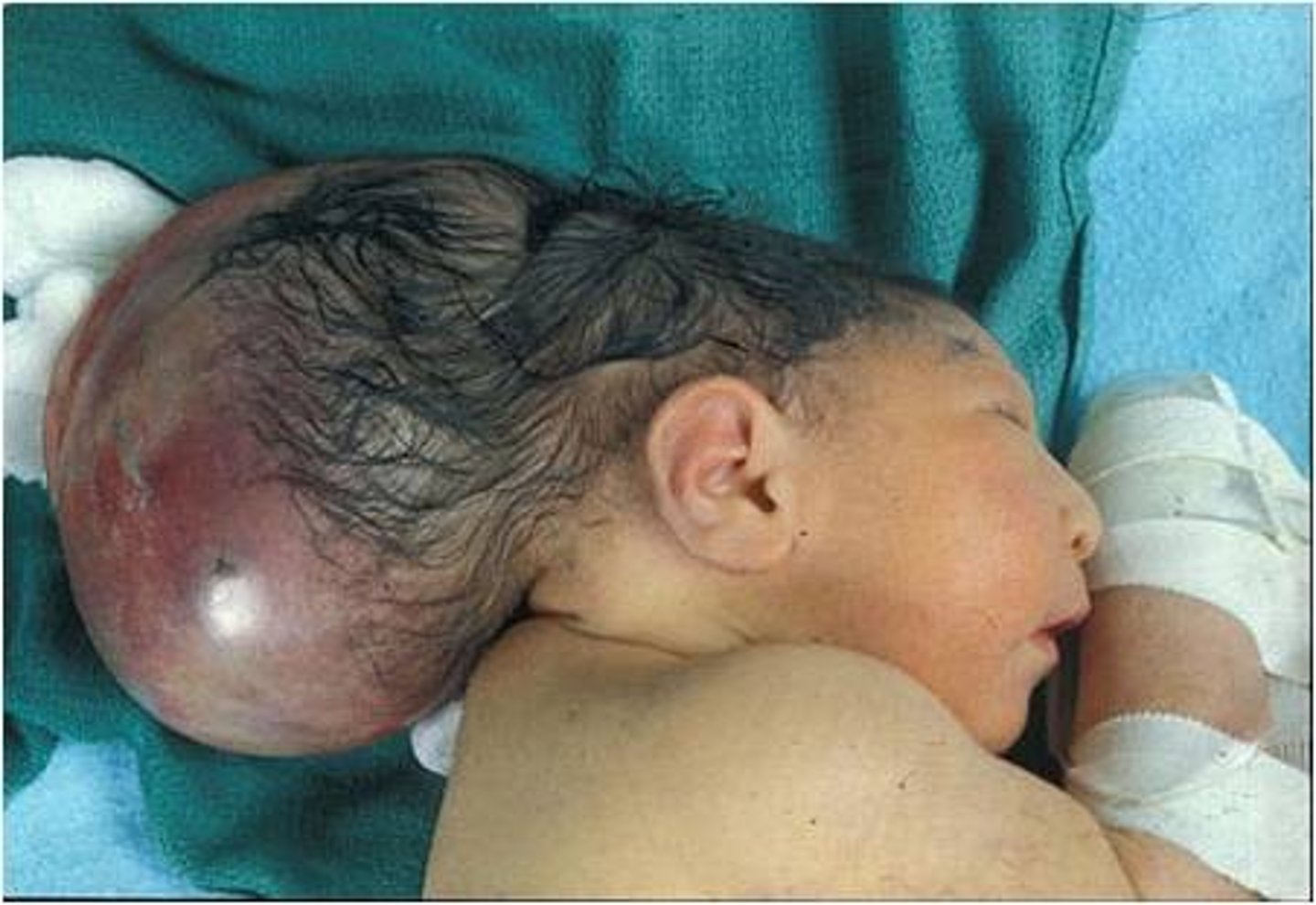
encephalocele prognosis
frontal encephaloceles have generally good prognosis since brain tissue generally does not herniate out. A covering of skin over the sac also improves prognosis. General prognosis of occipital encephaloceles = 55% survival. Surgery generally required within first few days - first year of life
Meckle-Gruber Triad
occipital encephalocele, multicystic dysplastic renal disease, post-axial polydactyly.
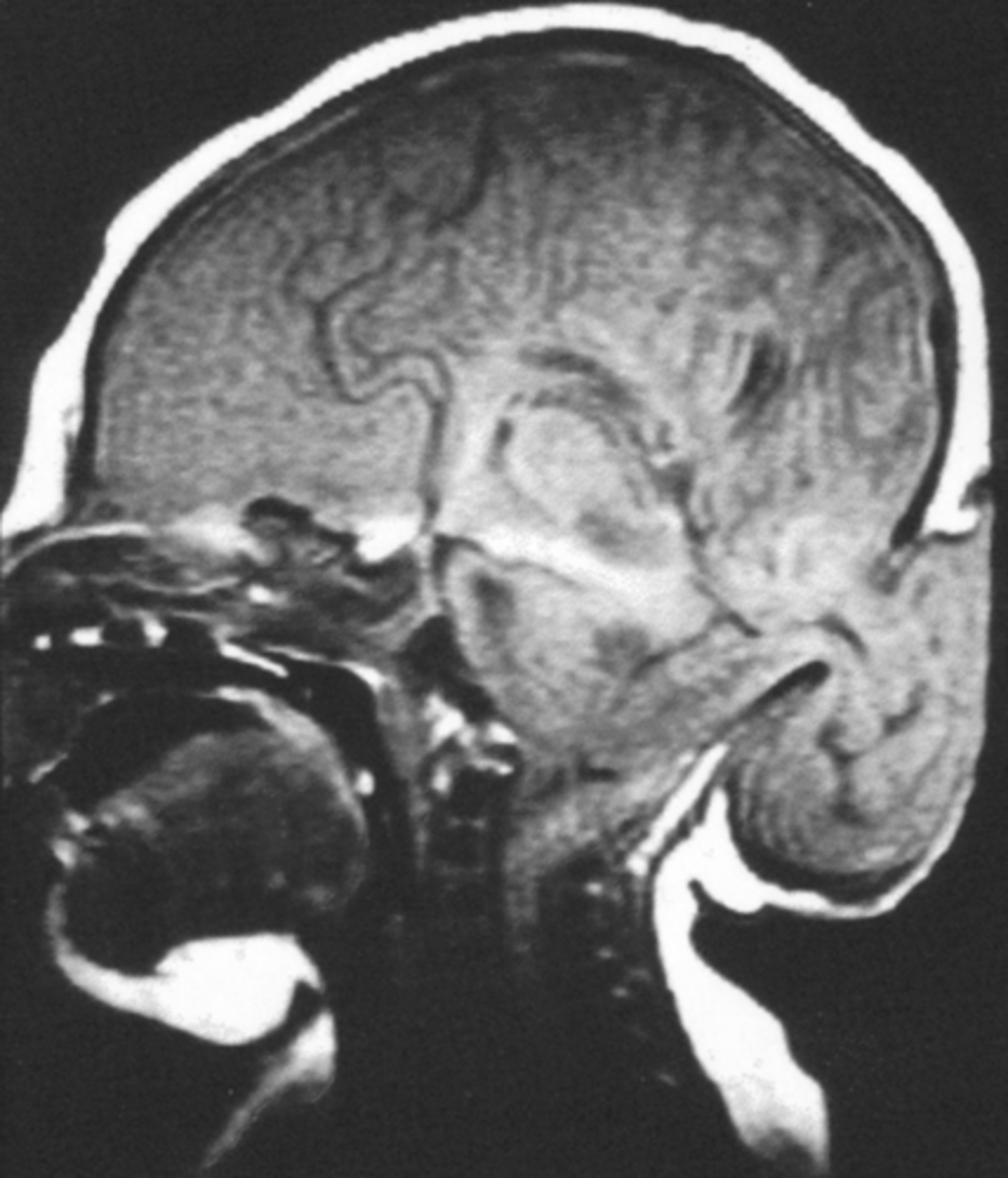
Meckel-Gruber etiology
The MKS gene family encodes cilia proteins, which act as chemosensory and mechanosensory signalling proteins on cells. Most MKS proteins localize to the transition zone (TZ), which regulates the trafficking of cargo proteins or lipids.
Meckel-Gruber genetics
autosomal recessive with 13 known genetic causes. No digenic inheritance
Ciliopathies features
Syndromes linked to defects in cilia, which are involved in cellular signaling and transport. Three well-known ciliopathies (Bardet-Biedl, Joubert, Meckel Gruber) share similar features (on a spectrum of severity) including: polycystic kidney disease, limb defects, retinal disease, intellectual disability, and male infertility
Ciliopathies Examples
Meckel-Gruber, Bardet-Biedl, Kartagener, Joubert
Heterotaxy
A condition in which the internal organs are abnormally arranged in the chest and abdomen, typically somewhere between situs solitus and situs inversus. Individuals usually have complex birth defects affecting the heart, lungs, liver, spleen, intestines, and/or other organs.
situs inversus
complete reversed position of organs, which tends to be asymptomatic. Compare with heterotaxy, in which organs have mixed L-R orientation, which results in complex birth defects
Pierre Robin Sequence
A series of birth defects that result from underdevelopment of the lower jaw. Micrognathia, glossoptosis, cleft palate, airway obstruction. Requires emergent management in the NICU to ensure tongue will not fall backwards, occluding the airway.
isolated CL/P genetic testing
Generally genetic testing is not indicated for isolated CL/P. However, a geneticist should perform a thorough exam to check for other subtle syndromic features. Parental exams may also be indicated, due to subtle features of dominant disorders such as Stickler
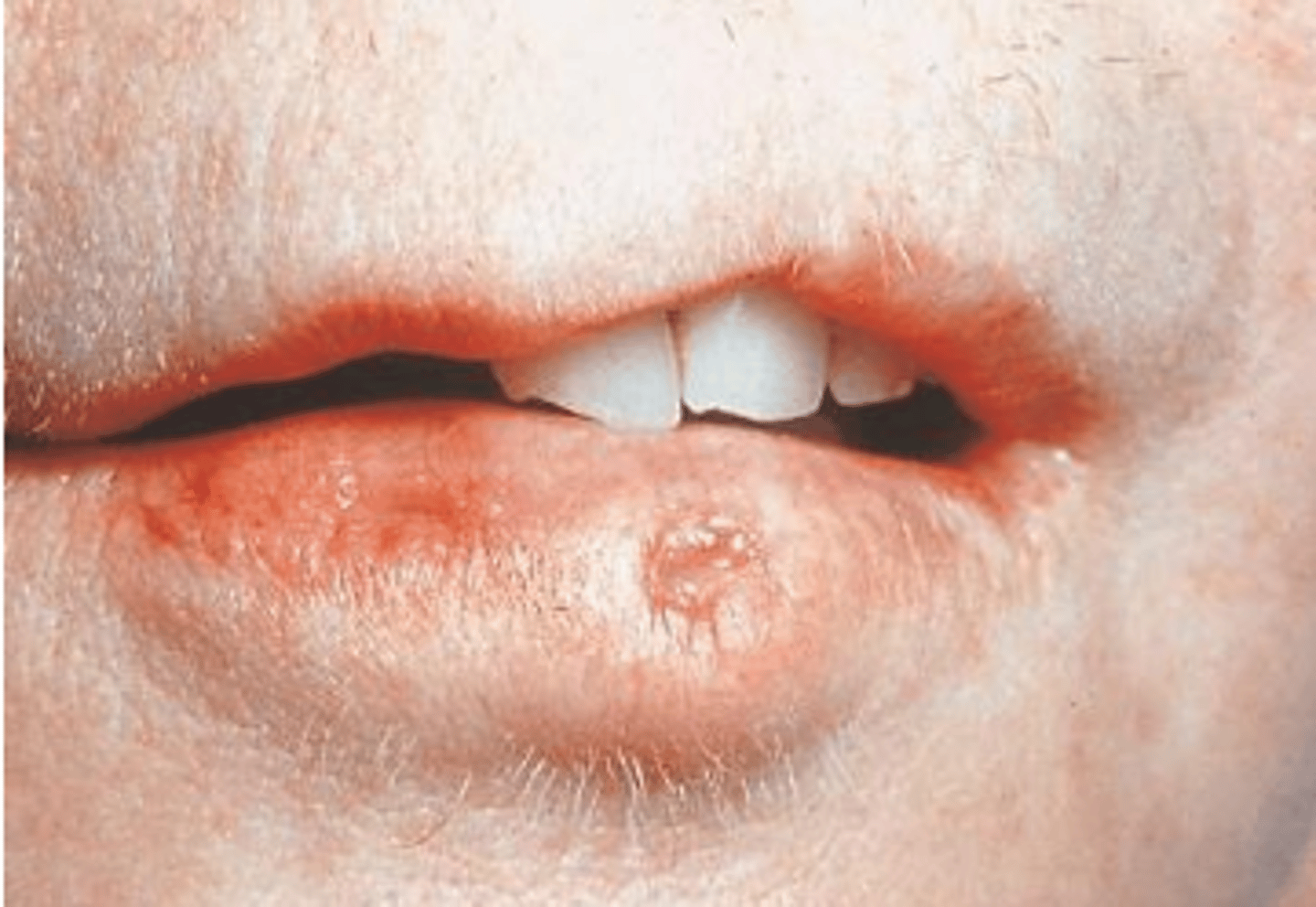
Van der Woude Syndrome
Dominant non-syndromic CL/P, lip pits. Normal intelligence but may struggle with language acquisition, even after repair.
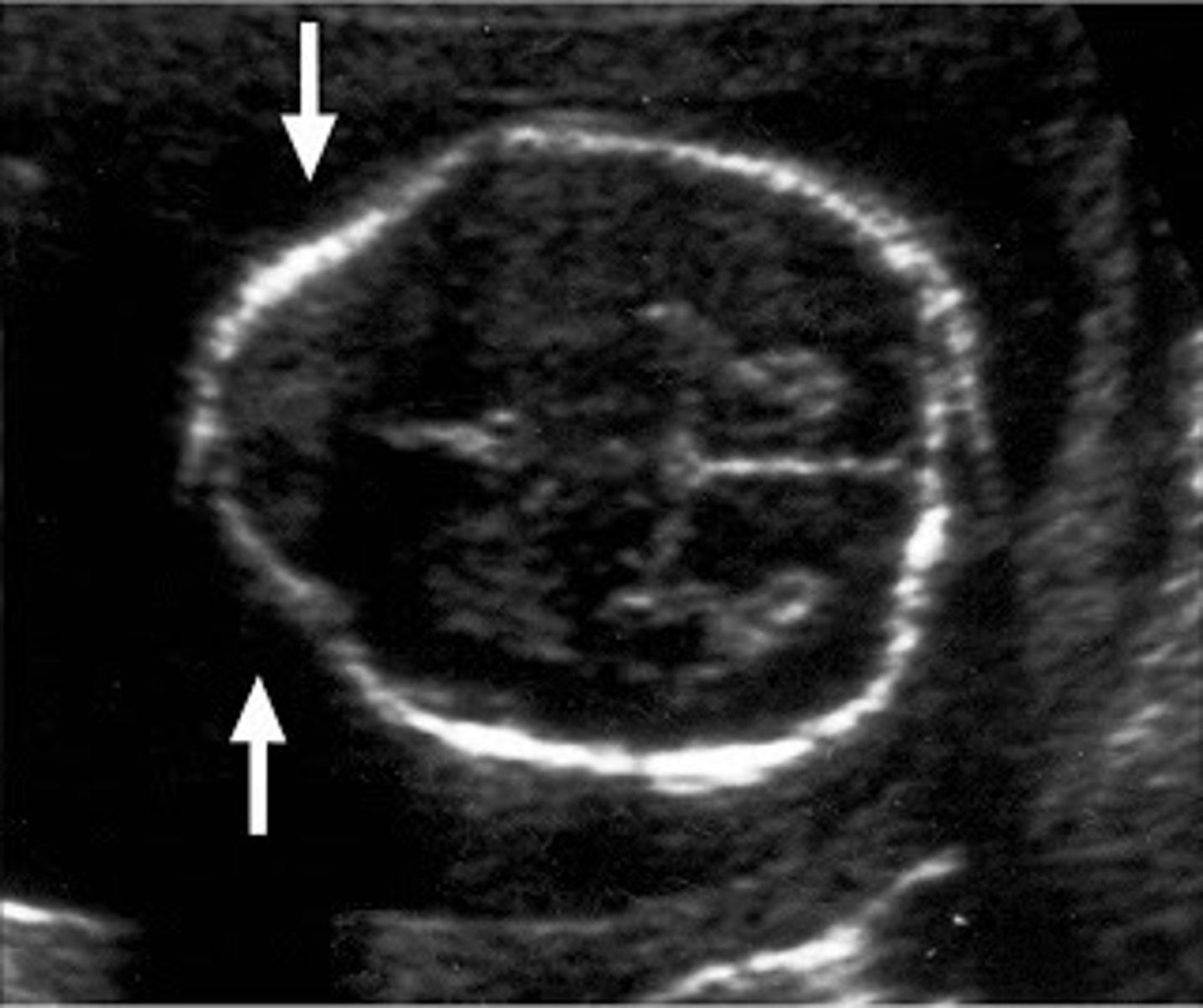
Lemon sign
frontal bone scalloping or indentation, presenting as flat or concave rather than the normal convex shape, most often found in the fetus with spina bifida
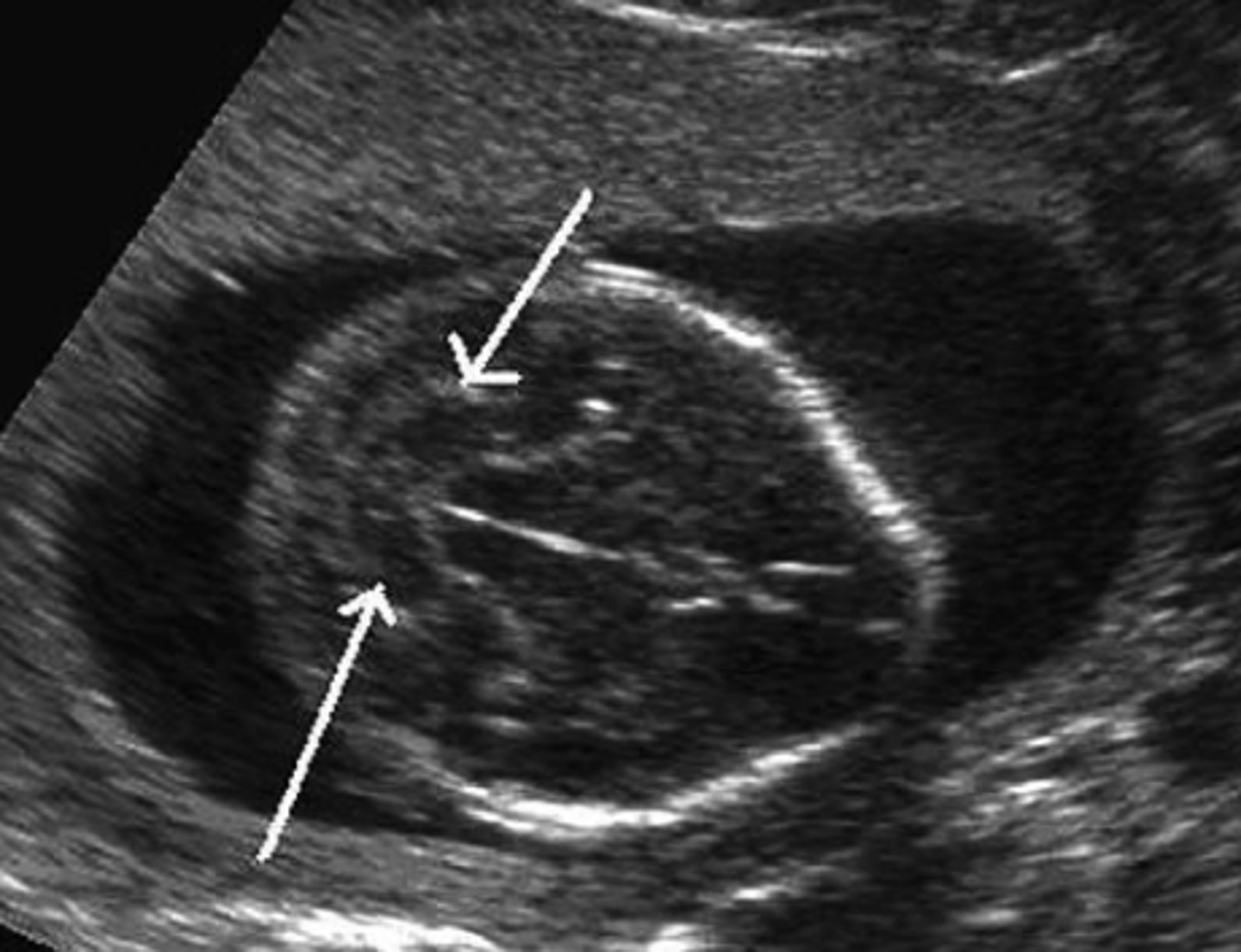
banana sign
elongation / squishing of the cerebellum showing a posterior fossa abnormality and obliteration of the cisterns magna.
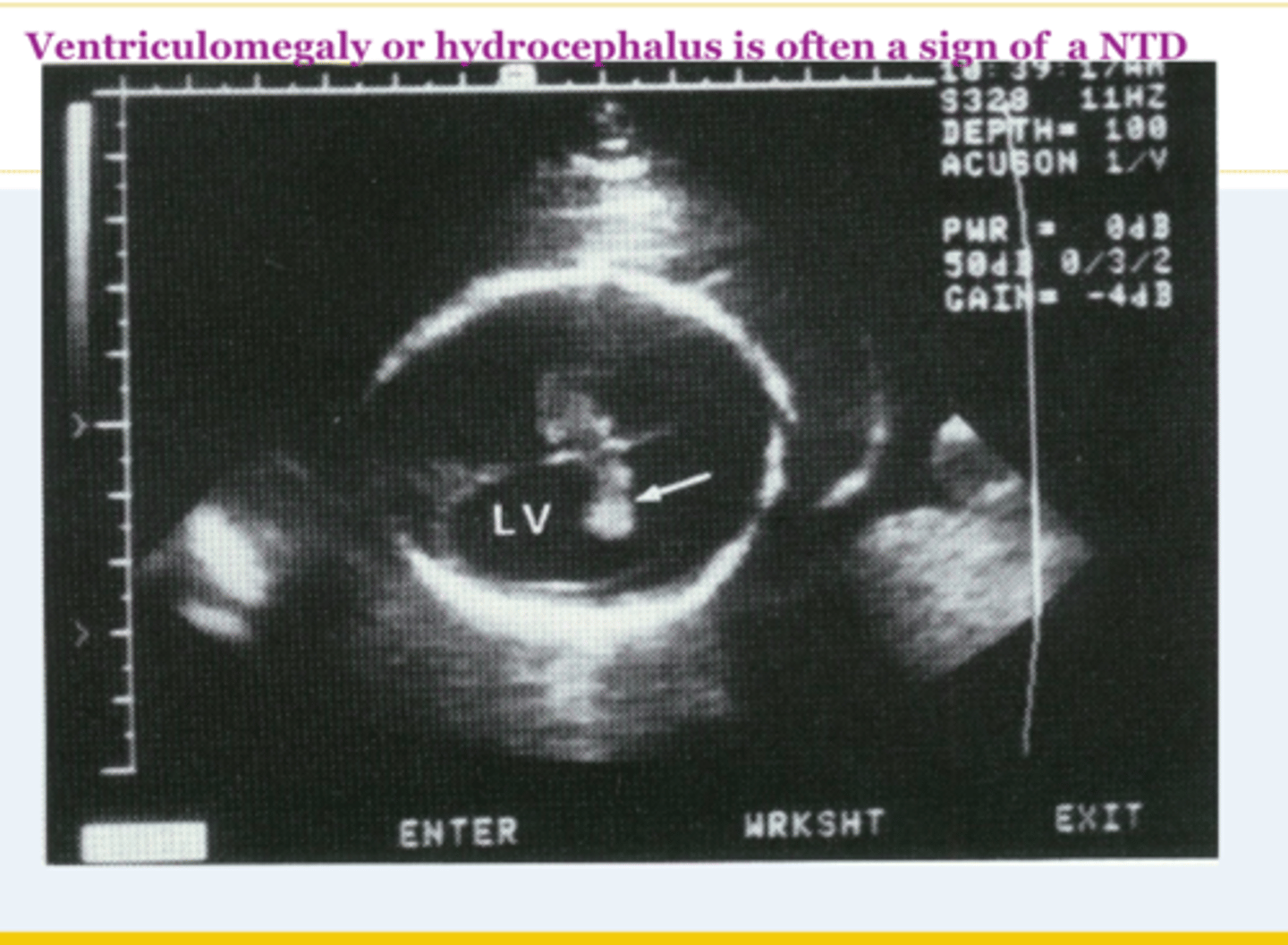
Ventriculomegaly
Dilation of the cerebral ventricles without enlargement of the cranium. Occurs because of obstruction of outflow of CSF from the ventricles. Often sporadic and benign. Non-specific, but can indicate Dandy-Walker malformation, possible aneuploidy, variable future cognitive deficits
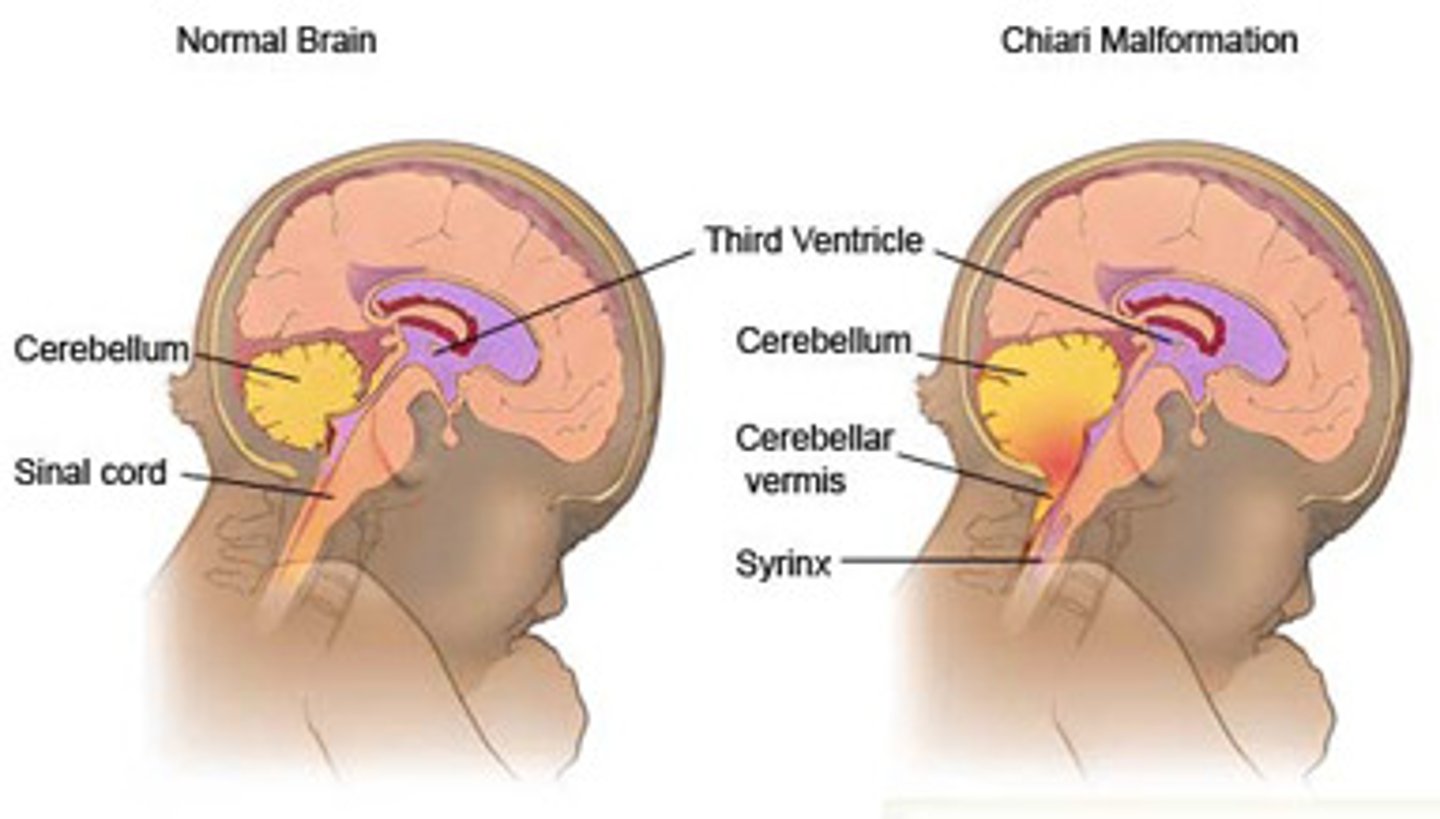
Chiari 2 malformation
Herniation of low-lying cerebellar vermis and tonsils through foramen magnum. Causes hydrocephalus / ventriculomegaly. Associated with lumbosacral meningomyelocele.
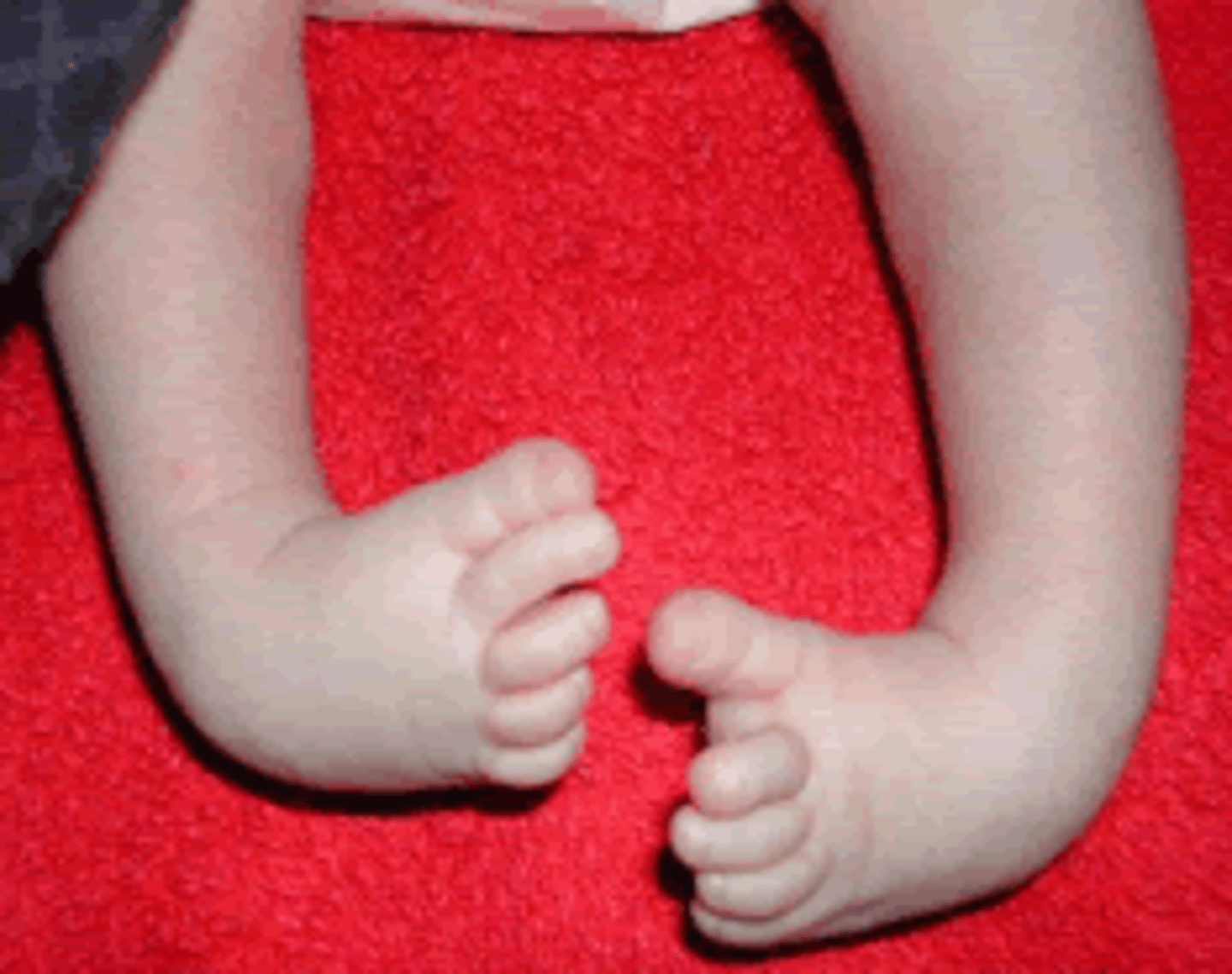
Clubfoot causes
low amniotic fluid or congenital contractures
Clubfoot (Talipes Equinovarus)
A birth defect in which one or both feet are interiorly rotated. Often isolated. May be related to spina bifida, myotonic dystrophy, Potter's sequence, or fetal positioning.
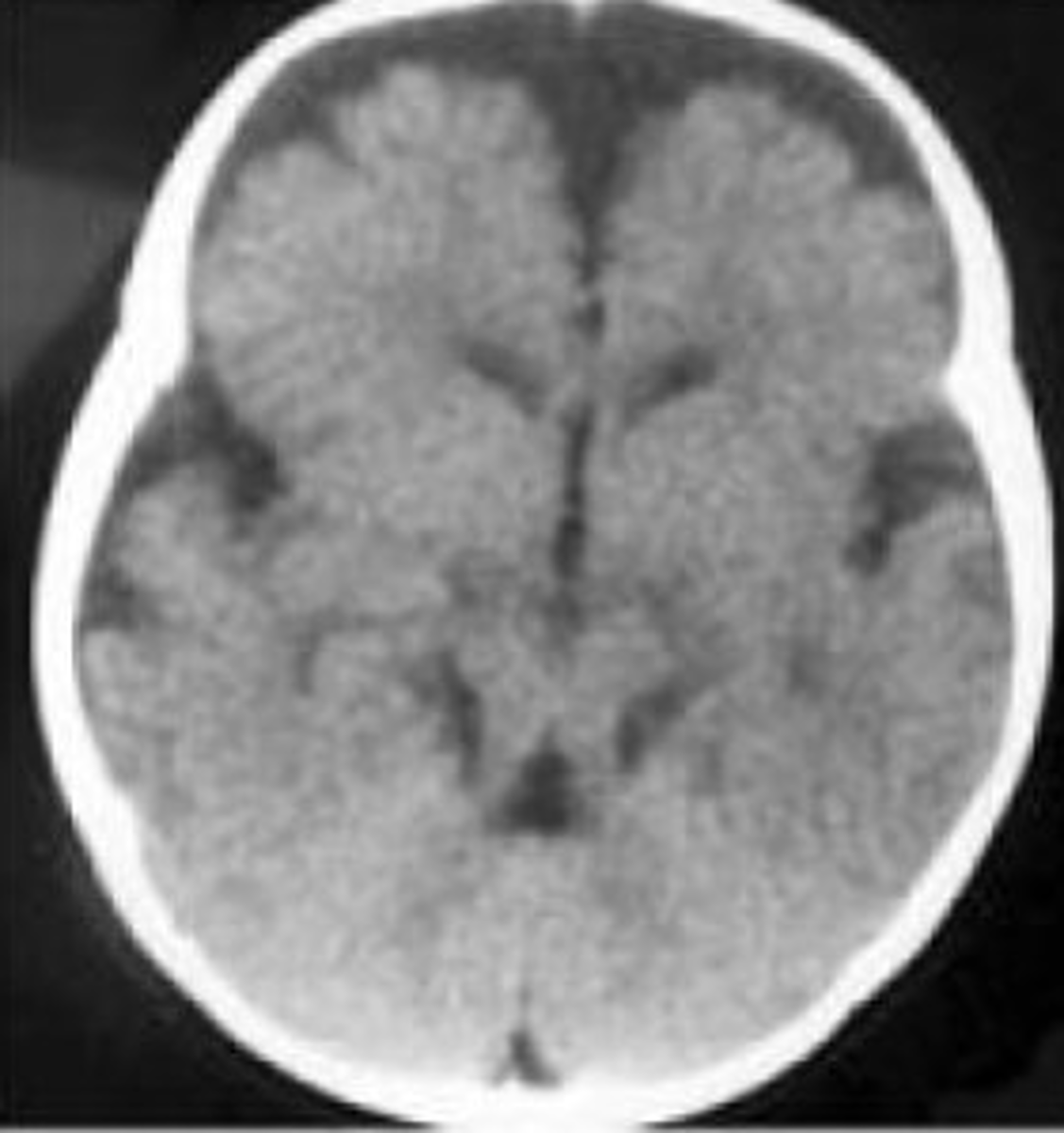
Molar tooth sign
Brain finding which may be suspected on prenatal ultrasound & confirmed by MRI. Occurs due to several related brain changes: abnormally deep interpeduncular fossa; elongated, thick, and maloriented superior cerebellar peduncles; and absent or hypoplastic cerebellar vermis. Highly associated with Joubert but can rarely have other causes.
malformation
type of birth defect in which a tissue never formed properly-- an intrinsic error. Examples: endocardial cushion defect, renal agenesis, duodenal atresia
deformation
birth defect in which a tissue develops differently or becomes bent into a different shape due to external force. Examples: flattened face in Potter's Sequence, clubfoot as a result of joint contracture in spina bifida
disruption
birth defect caused by restricted oxygen flow to tissue, result in tissue death. Examples: amniotic bands / constriction ring
Duodenal atresia
Occurs due to failure of recanalization of the duodenum. Results in polyhydramnios, bile-containing vomit, distended stomach. Associated with Down Syndrome.
Double bubble sign
Overfilled stomach and duodenum caused by duodenal atresia, most often related to trisomy 21
atrial septal defect
Failure of closure of the septum between the left and right atrium. Relatively common birth defect. Genetic/Teratogenic causes include: T21, FASD, holt-oram, lithium (resultant of ebstein's anomaly)
ventricular septal defect
an opening in the septum separating the ventricles, most commonly the membranous portion. Most common congenital heart defect. Genetic causes include Down Syndrome and rare single gene disorders. However, VSDs are most often sporadic.
Dysplasia
abnormal maintenance of cells in a tissue as it develops. Example: skeletal dysplasias are due to dysplasia in the developing bone and cartilage.
4-6th week of development
time period in which most internal organs form in the fetus
4-10th week of development
time period in which limbs and digits form
VACTERL
V-vertebral anomalies
A-anal atresia
C-cardio anomalies
TE-Tracheo-Esophageal fistula
R-renal anomalies
L-limb anomalies
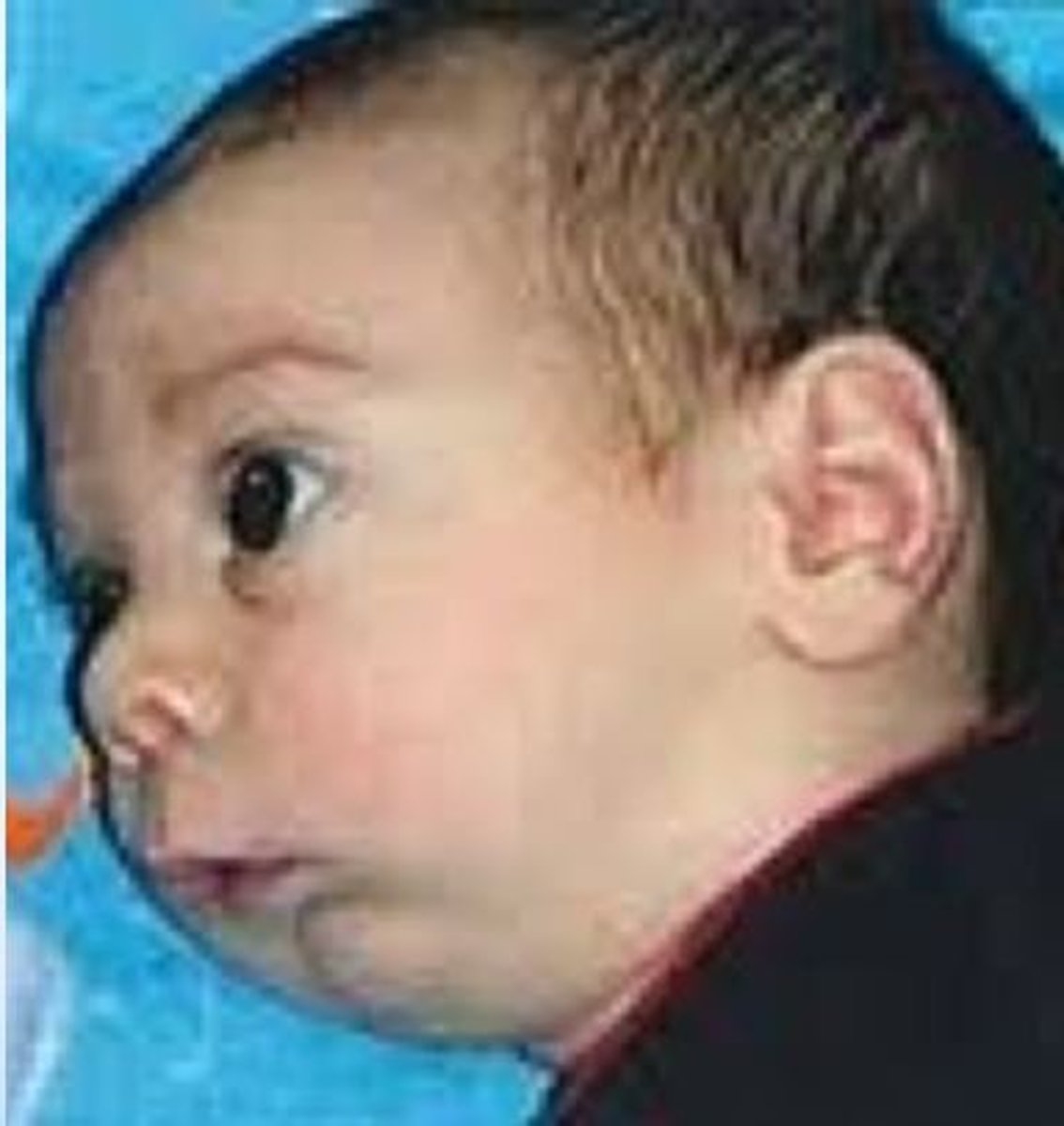
Goldenhar Syndrome
Hemifacial microsomia which is believed to be a disorder of the dysfunction of the stapeidal artery during embryonic development. Etiology is unknown but not believed to be genetic. In addition to facial asymmetry: facial nerve palsy, ear tags, macrostomia, CL/P, benign eye tumor, cardiac / renal / radial ray anomalies.
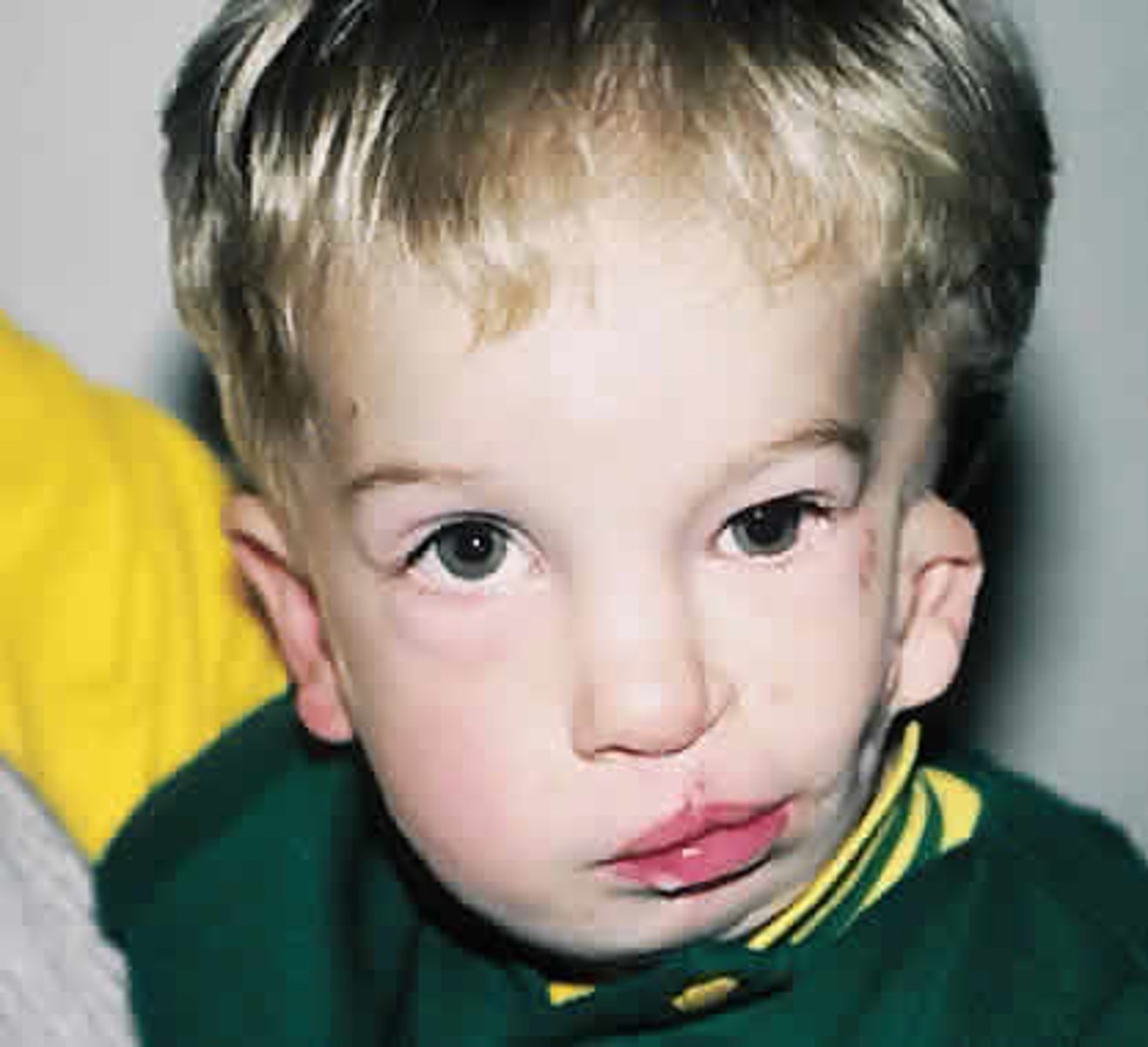
Holt-Oram Syndrome
AD at TBX5. Congenital heart defect (ASD, VSD), skeletal defects. upper limb and shoulder girdle malformation, thumb defects: triphalangeal (finger-like) thumb, absent thumb, extra carpal bones, bone protrusions
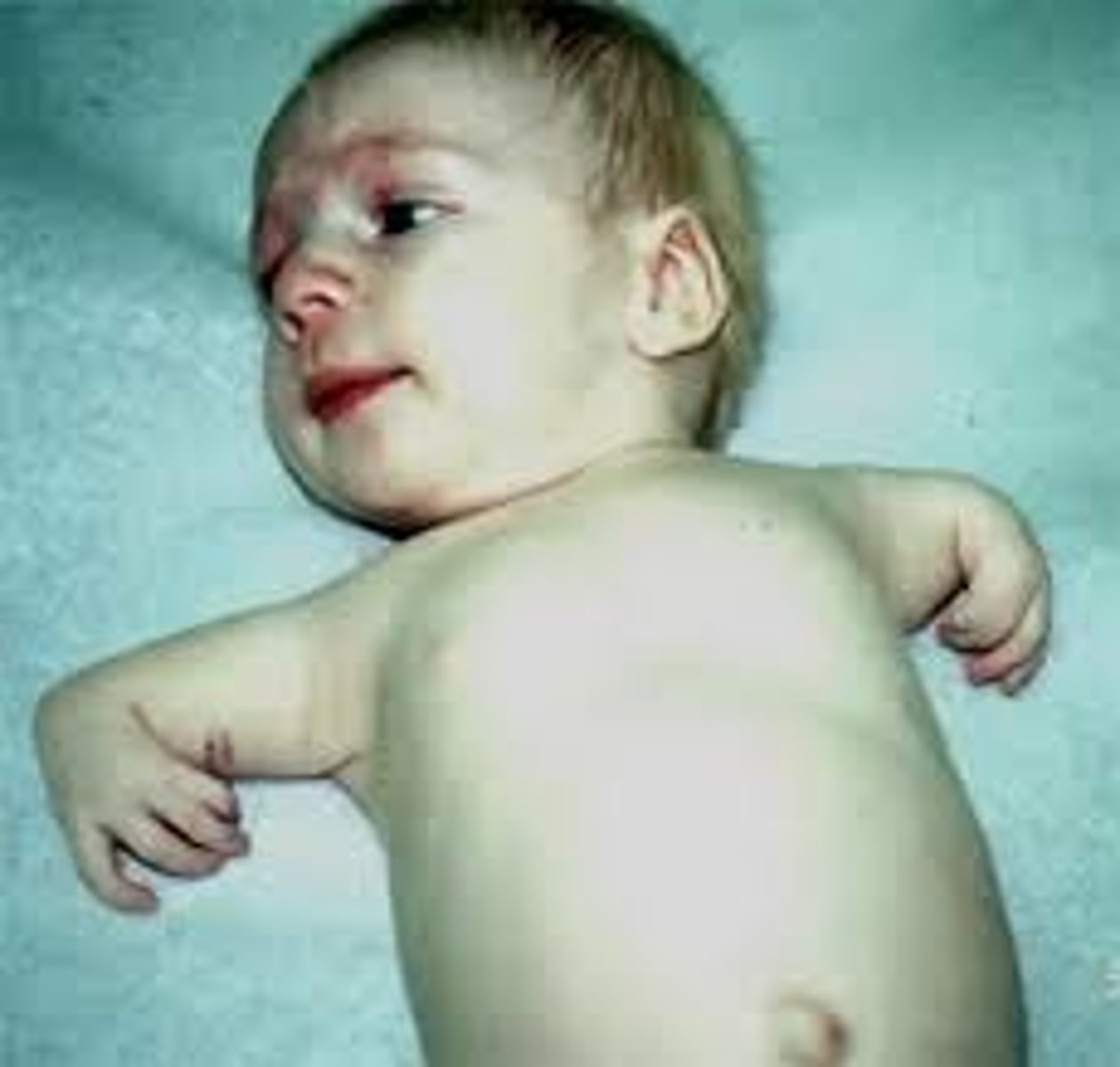
Nail-Patella Syndrome
AD at LMX1B. Eye issues (glaucoma, nystagmus), absent or deformed fingernails/toenails, patellar absence / malformation / frequent dislocation, abnormalities of the hip and pelvis bones. Pathognomic features: iliac horns (asymptomatic), triangular lunulae
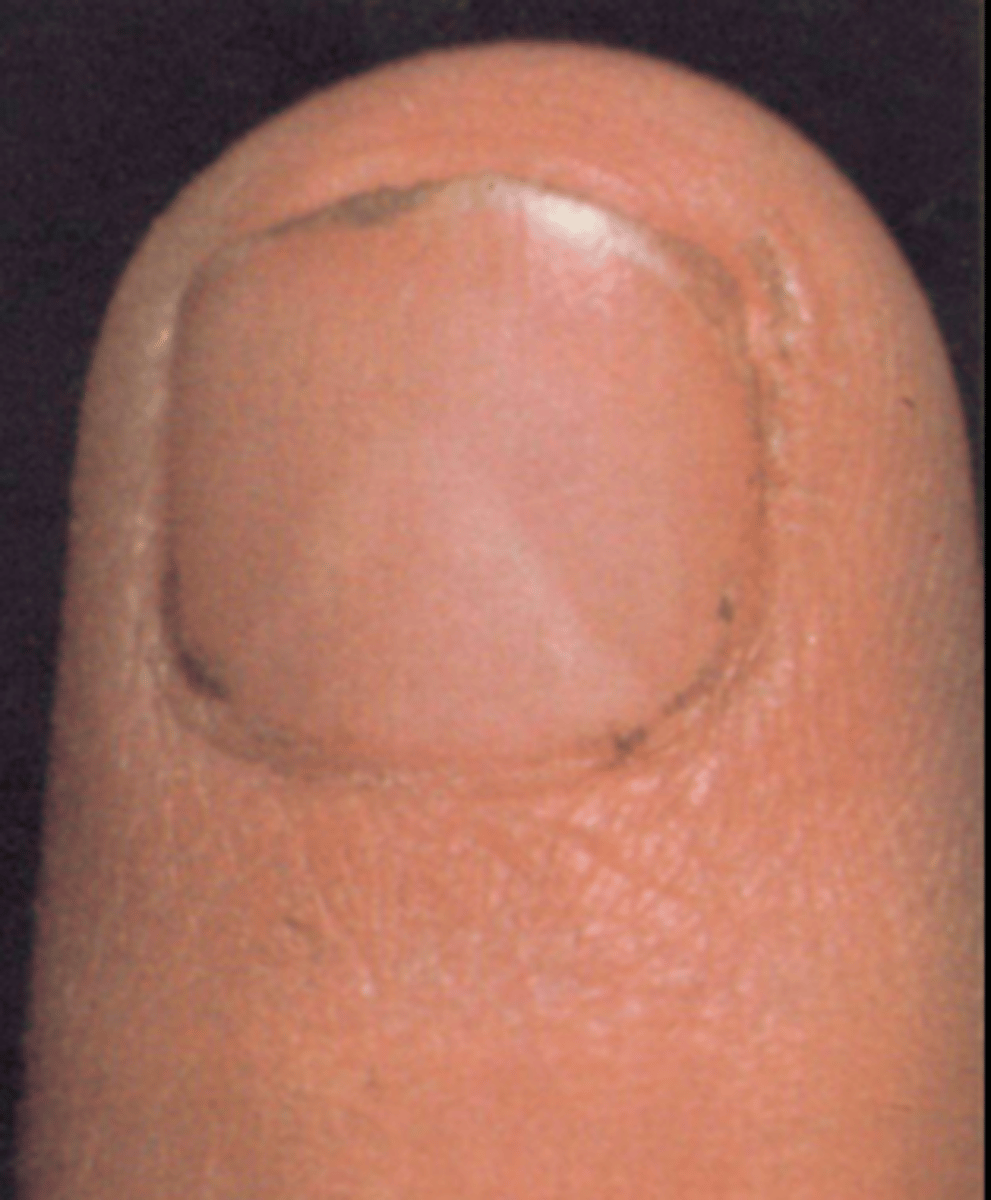
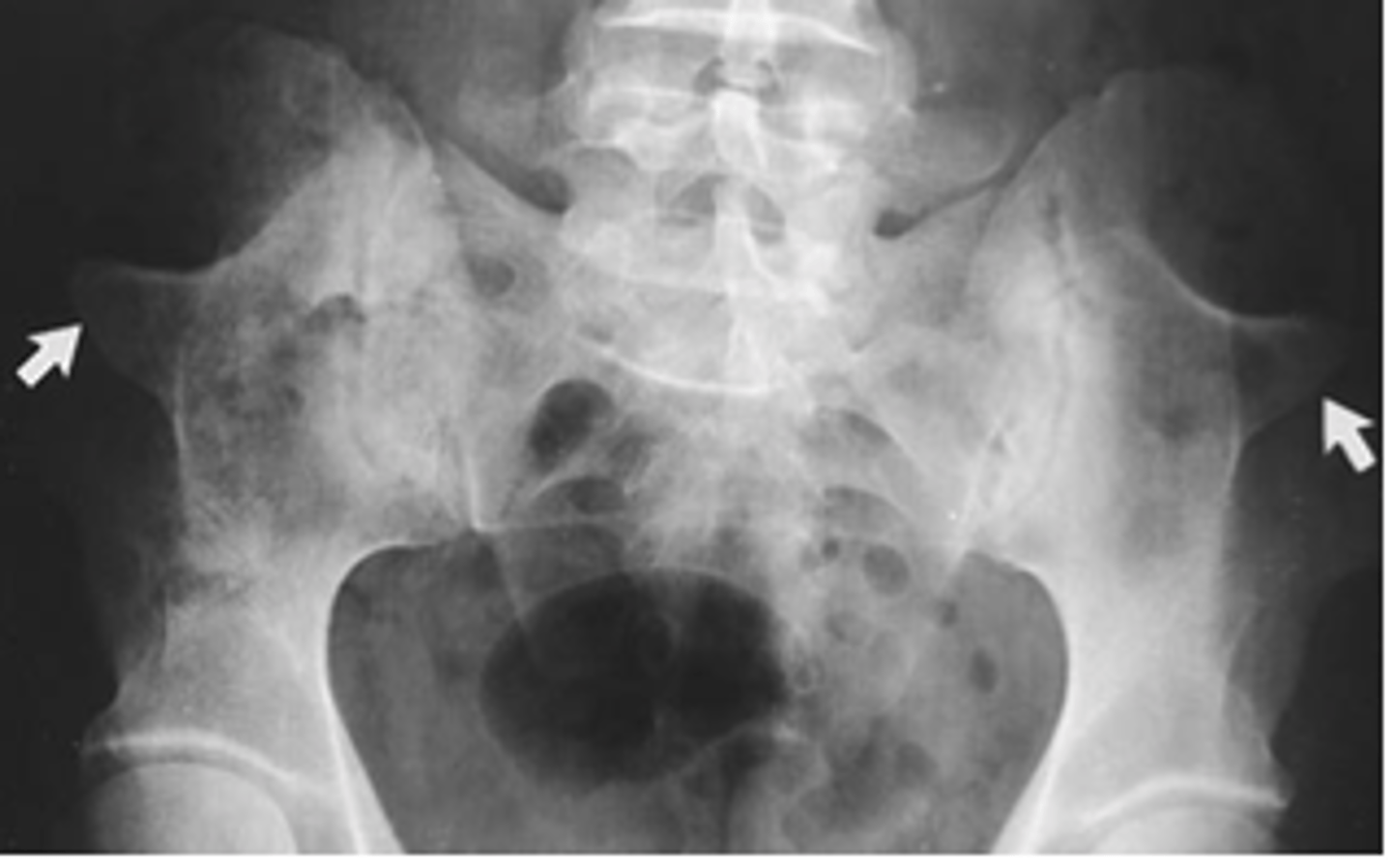
Iliac horns
post-natal radiographic finding of bone protrusions from the iliac crest of the hips. Asymptomatic but essentially pathognomic of Nail-Patella Syndrome.
Blepharophimosis, Ptosis, Epicanthus Inversus Syndrome
AD at FOXL2. narrow eyes, droopy eyelids, an upward fold of skin of the inner lower eyelids and widely set eyes. BPES type I is also associated with loss of ovarian function or premature ovarian insufficiency (POI)

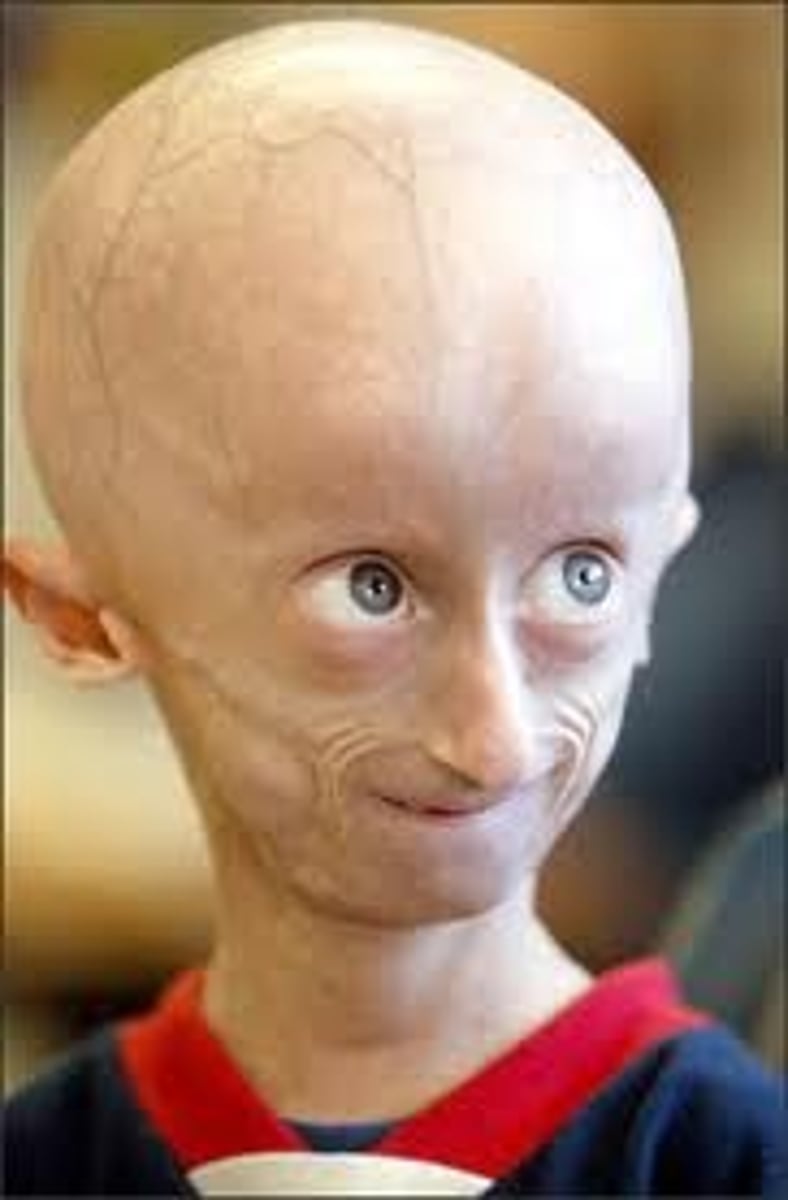
Hutchinson Gilford Progeria
AD (de novo) at LMNA. Limited growth, full-body alopecia, small face with a shallow / recessed jaw and a pinched nose). Progressive wrinkled skin, kidney failure, loss of eyesight, stiff ear canals resulting in conductive hearing loss, and atherosclerosis and other cardiovascular problems, Scleroderma (a hardening and tightening of the skin on trunk and extremities of the body). Small, fragile bodies, like those of older adults. Average age of death 13 years
mildly increased NT (3.0-3.5 mm)
93% likelihood of A&W birth
severely increased NT (4.5+ mm)
50% or less chance of A&W birth
Increased NT Differential
Down syndrome, Turner syndrome, Noonan syndrome, other aneuploidy, Congenital Heart defect, skeletal dysplasias
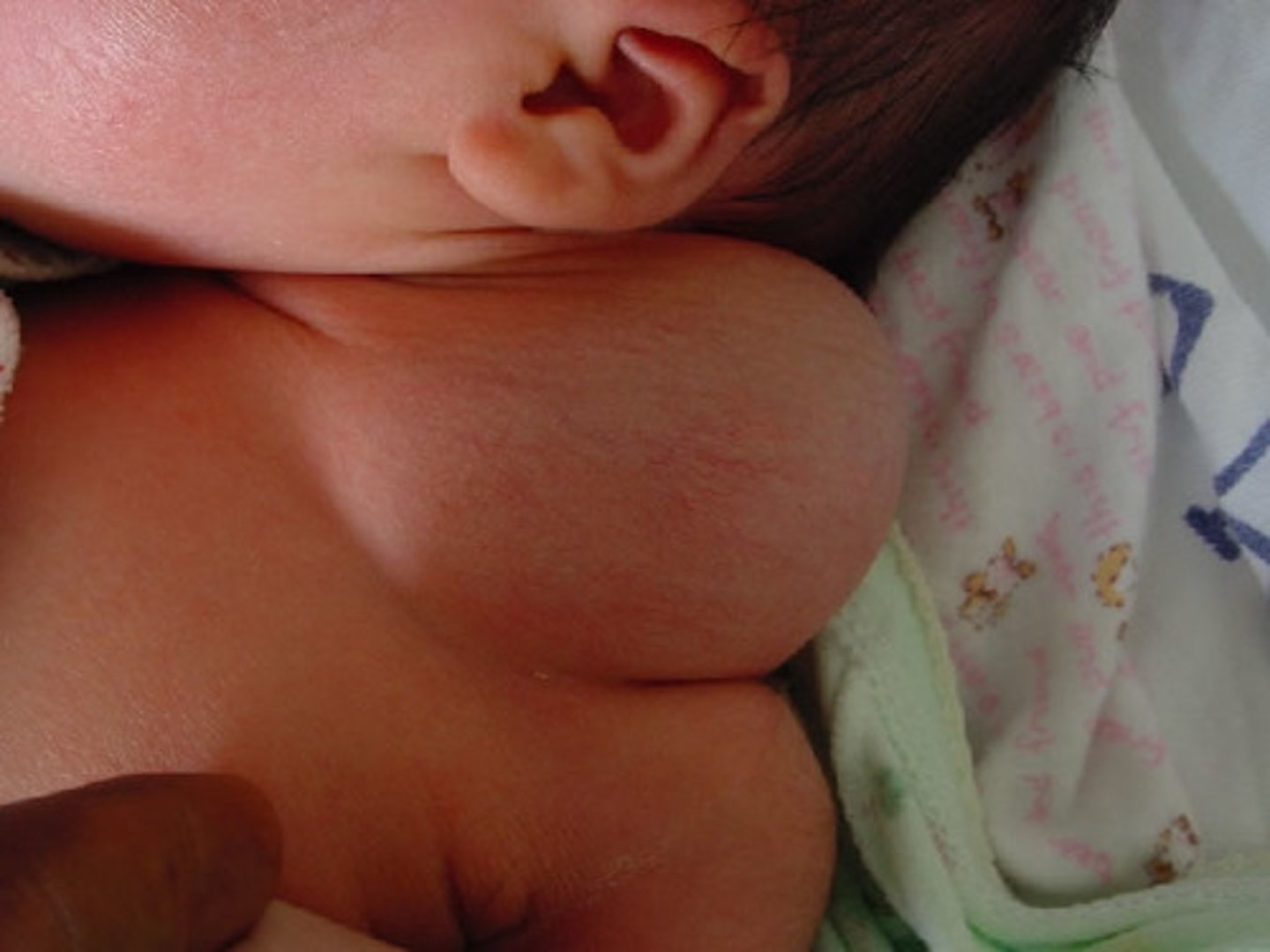
nuchal fold
The accumulation of fluid between the posterior cervical spine and the overlying skin in the fetal neck identified during an ultrasound examination. 2nd trimester finding, compared to NT which is measured in the first trimester
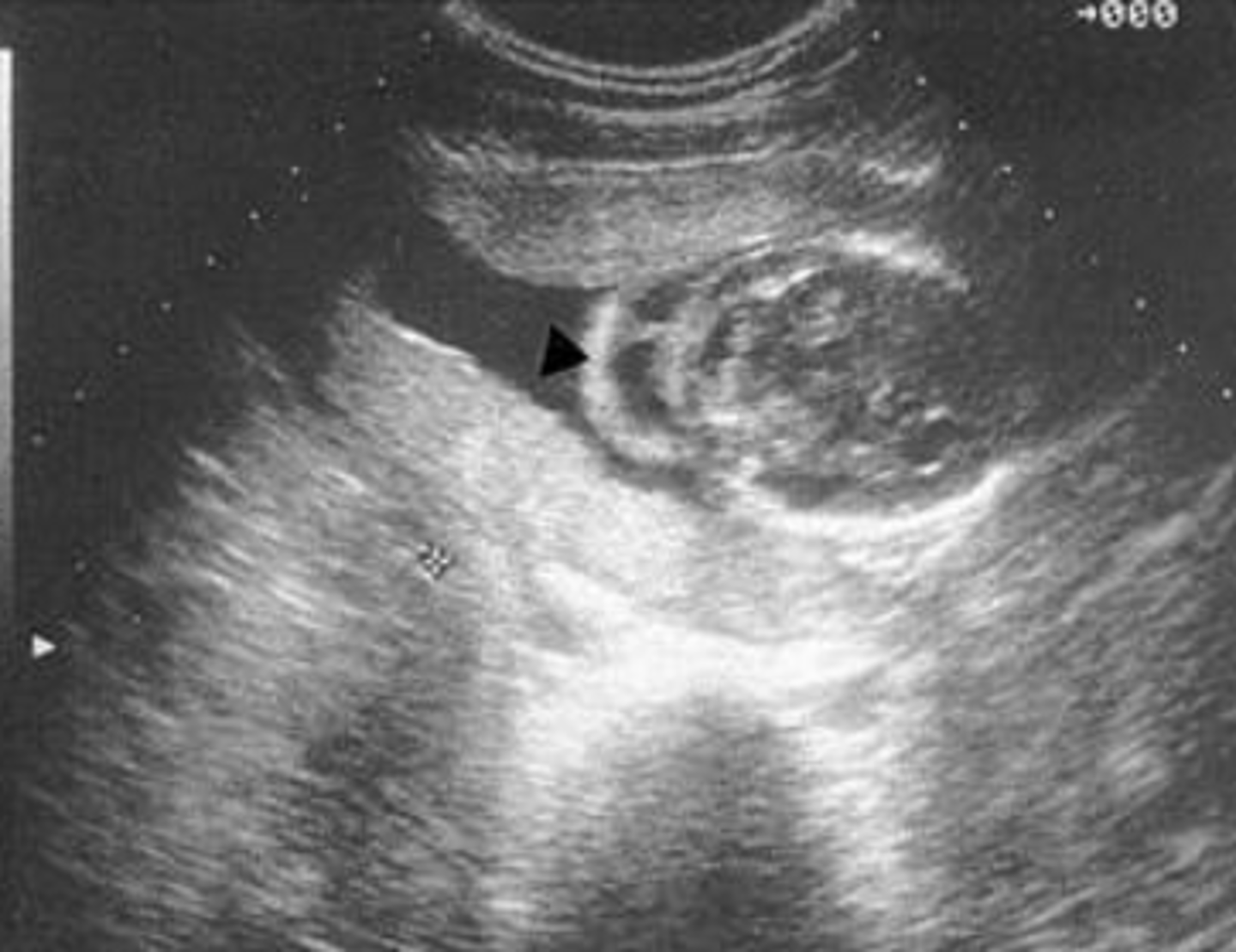
cystic hygroma
A cystic hygroma is similar to increased NT or NF in that it is a build up of fluid at the back of the fetal neck. Cystic hygromas are more severe and contain cysts within the fluid. The majority of cystic hygromas eventually grow larger than the fetal head.
choroid plexus cysts
Small cysts in the choroid plexus, caused by trapped CSF. The choroid plexus is a tissue found in each ventricle of the brain that produces cerebrospinal fluid. Often sporadic but is associated with T18
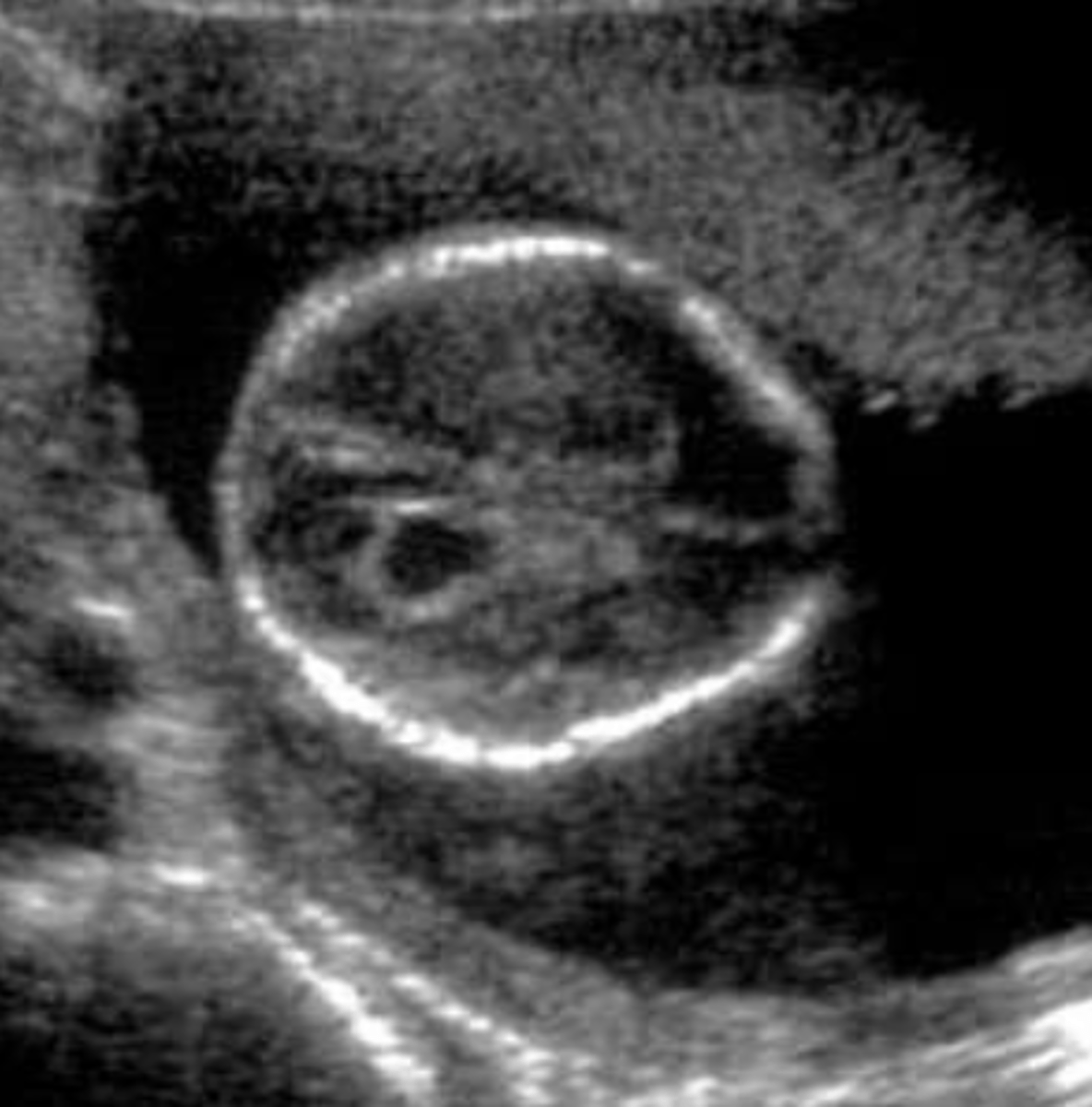
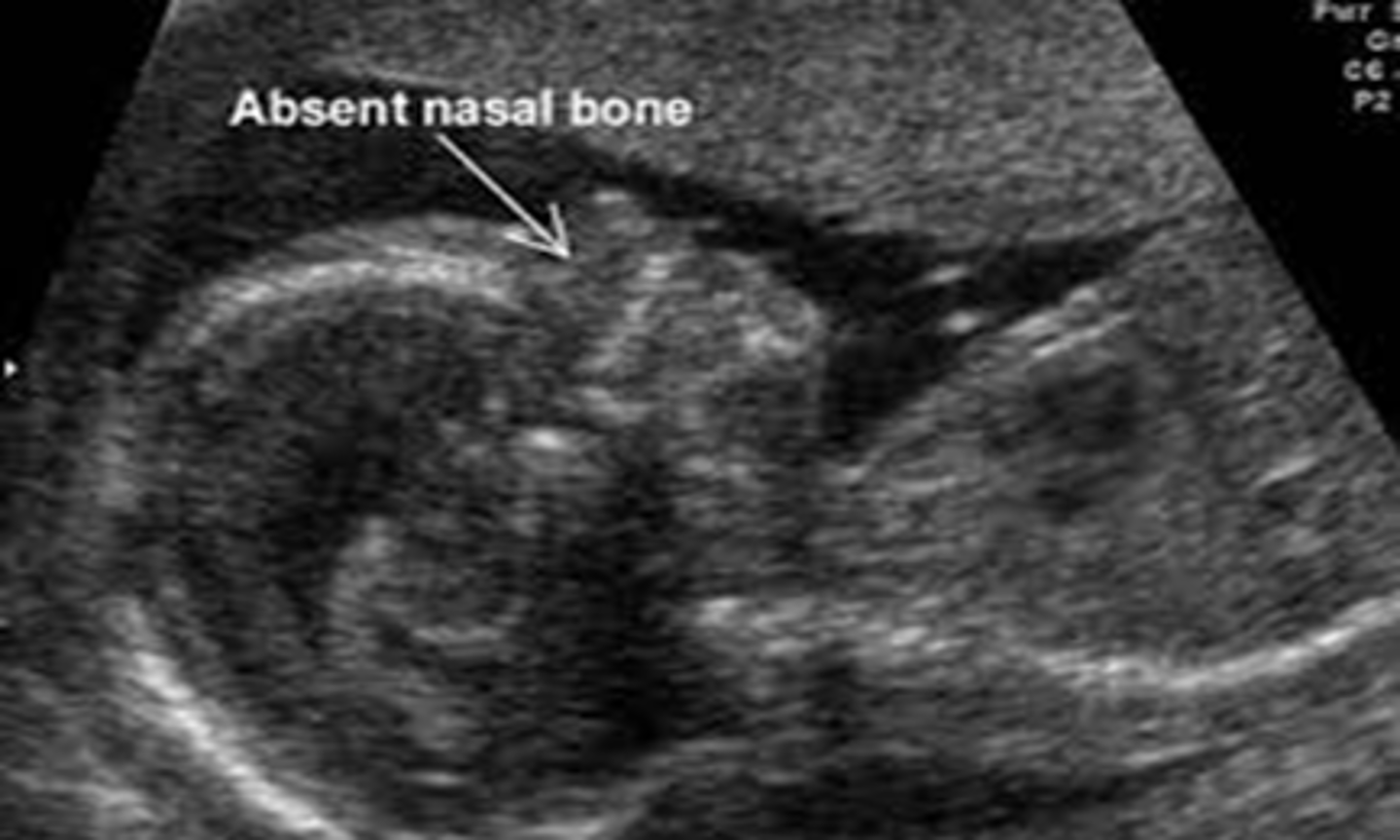
Absent/hypoplastic nasal bone
Nasal bone is either absent or underdeveloped, per visualization on 1st or 2nd trimester ultrasound. Is a common finding in certain ethnic groups, but is also a soft marker for aneuploidy especially T21 and a part of Warfarin embyropathy
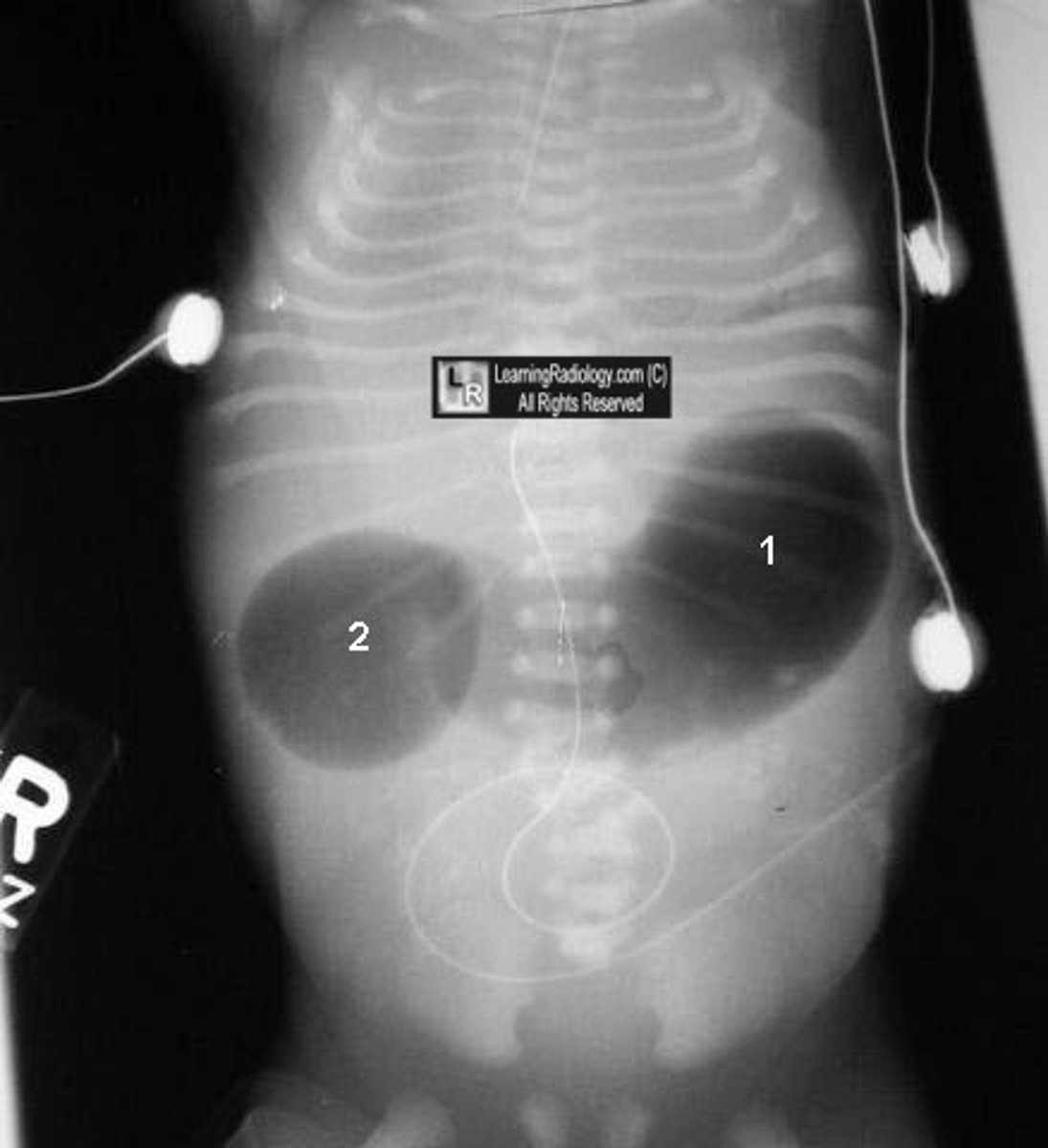
diaphragmatic hernia
Birth defect where there is a hole in the diaphragm. Organs in the abdomen (such as intestines, stomach, and liver) can move through the hole in the diaphragm and upwards into a baby's chest. A diaphragmatic hernia can prevent the baby's lungs from developing completely, causing breathing difficulties for the baby at birth. Often sporadic but can be associated with aneuploidy
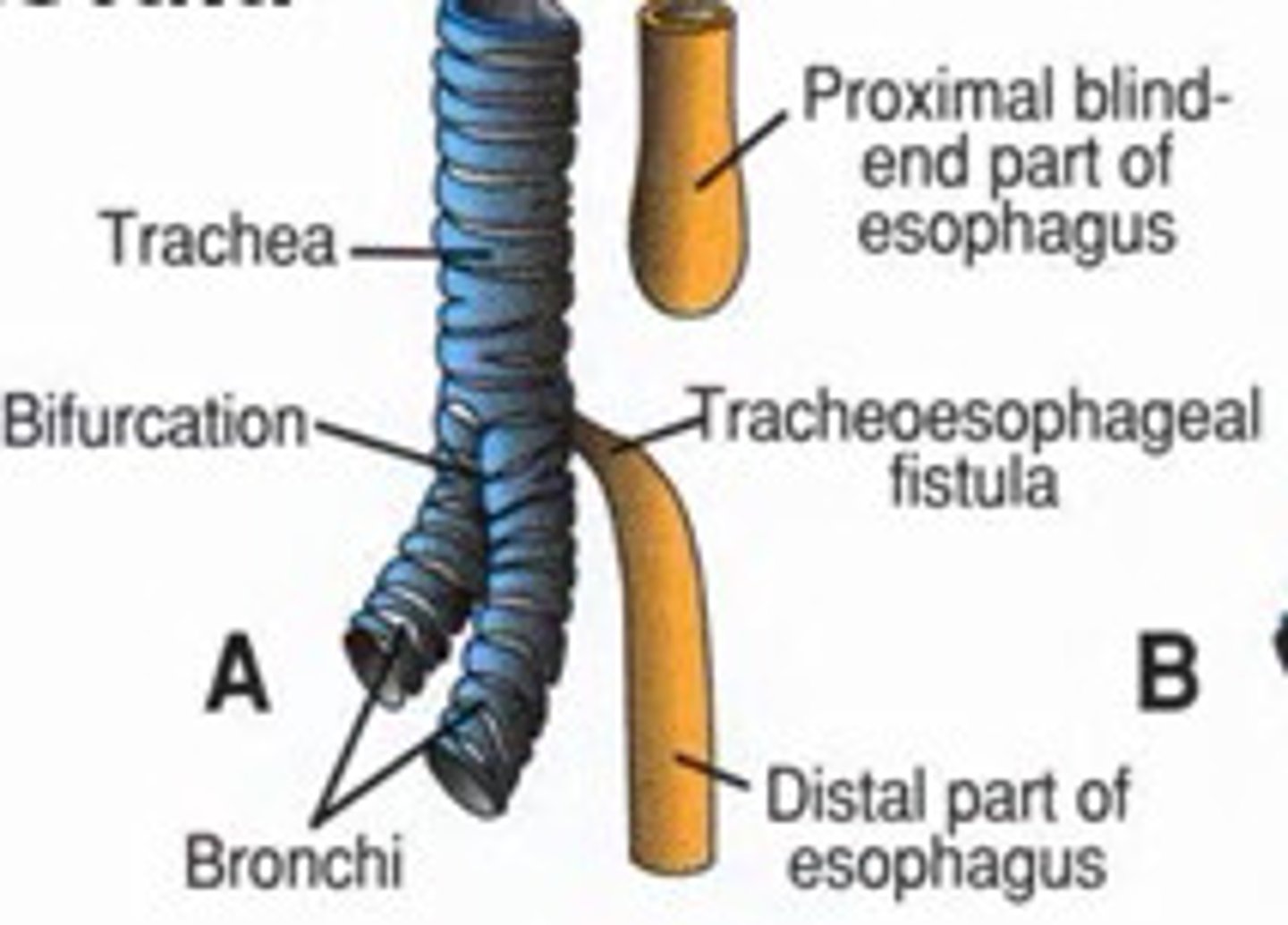
Tracheoesophageal fistula (TEF)
The trachea and the esophagus start forming as 1 tube. At 4 to 8 weeks of pregnancy, a wall forms between the esophagus and trachea. This separates them into 2 tubes. TE fistula and esophageal atresia happen when this wall doesn't form as it should. Usually sporadic but also associated with aneuploidy and VACTERL
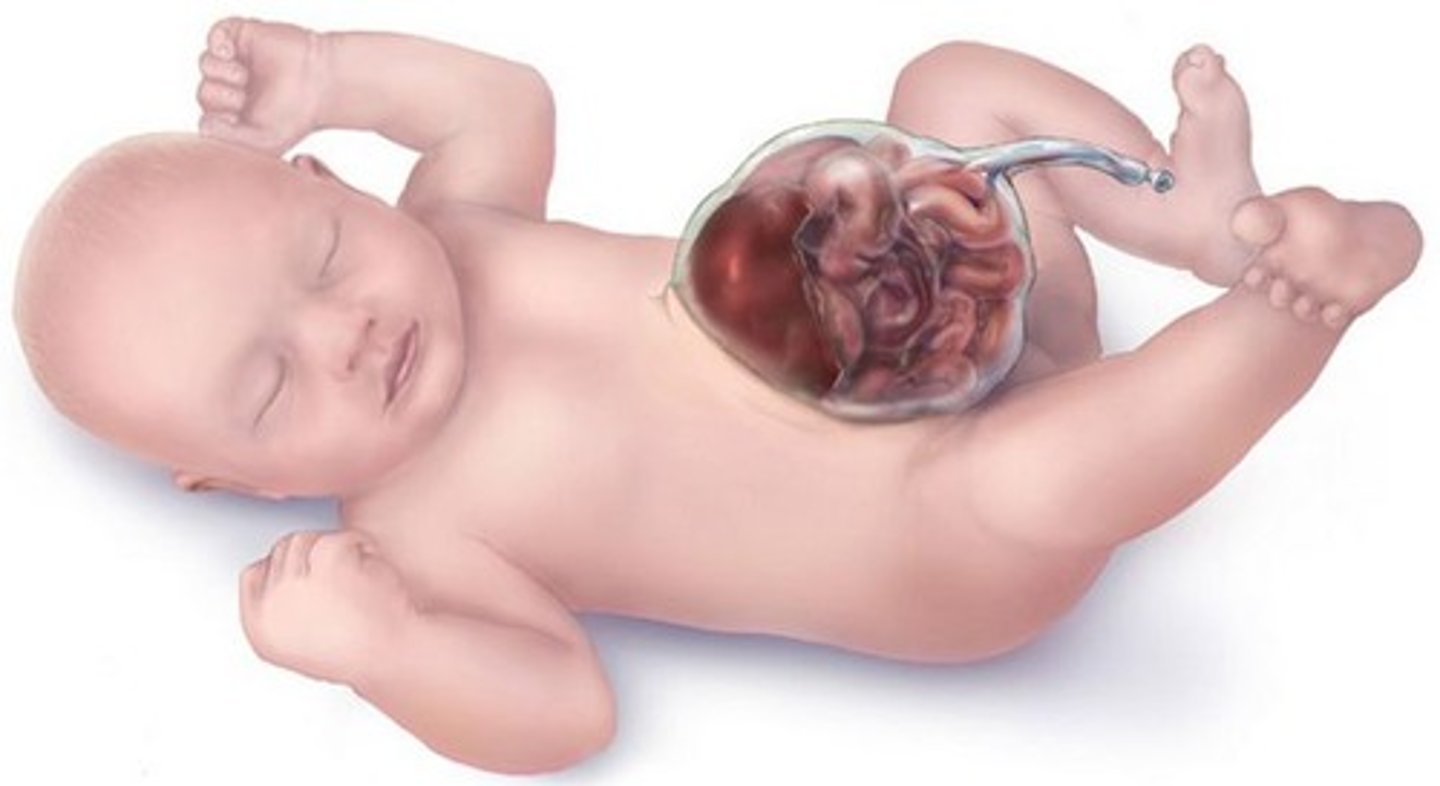
omphalocele
At six to ten weeks of embryonic development, a fetus's intestines will herniate into the umbilical cord. The fetus's abdomen will grow, and then the intestines will return to the abdomen at ten or eleven weeks. An omphalocele occurs when the intestines do not return and instead remain outside of the abdomen. In contrast to gastroschisis, omphaloceles are covered with a membrane.
omphalocele differentials
Beckwith-Wiedemann, anueploidy, CHARGE, Meckle-Gruber, Pentalogy of Cantrell
Gastroschisis
At six to ten weeks of embryonic development, a fetus's intestines will herniate into the umbilical cord. The fetus's abdomen will grow, and then the intestines will return to the abdomen at ten or eleven weeks. Gastroschisis occurs when the intestines do not return and instead remain outside of the abdomen. Unlike in omphalocele, the intestinal contents are uncovered in gastroschisis.
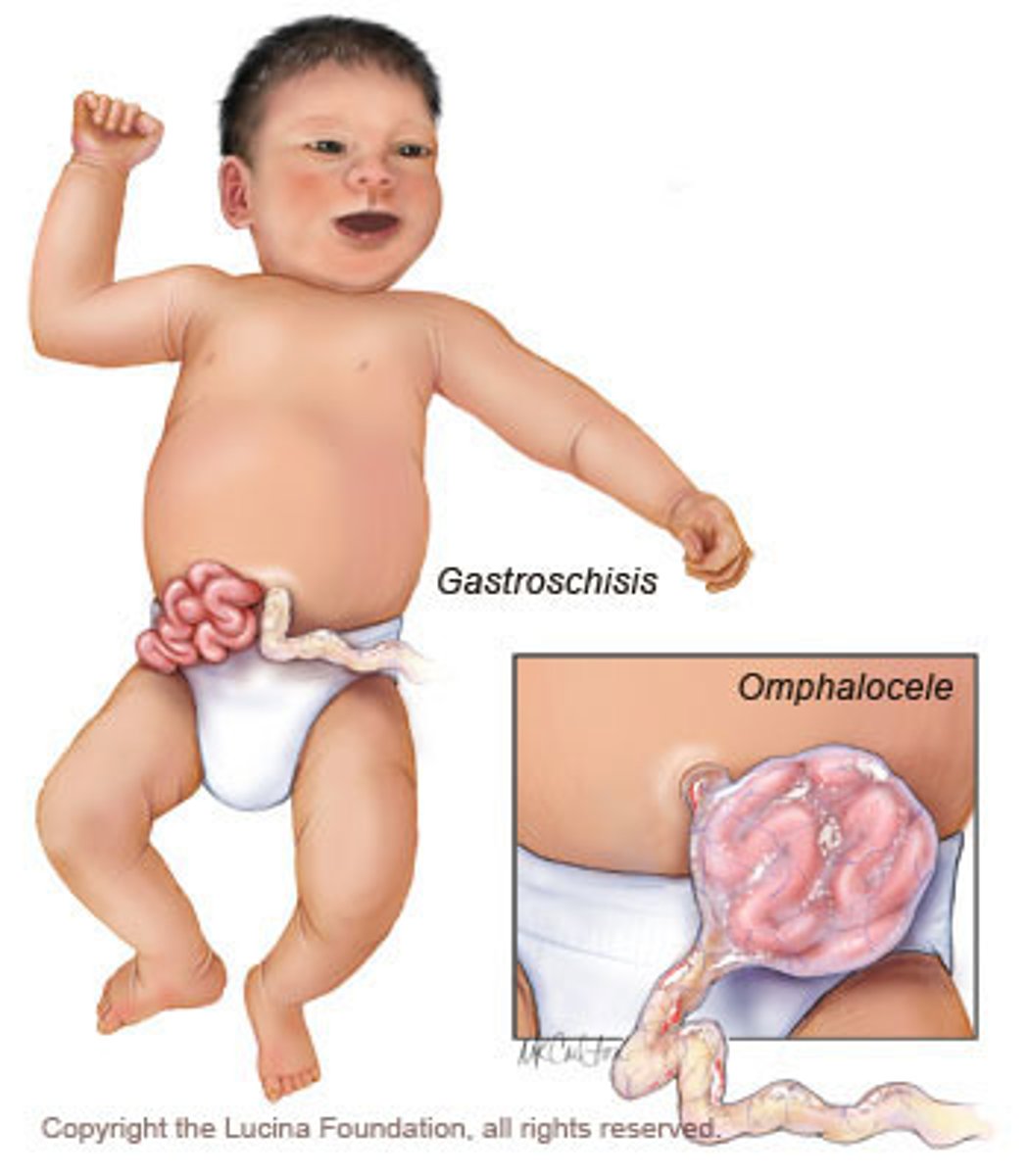
Gastroschisis risk factors
young maternal age, cigarette smoking
Echogenic bowel
Bright bowel caused by a build-up of meconium, gastrointestinal pathology, or fetal ingestion of blood. The diagnosis is made if the bowel is as echogenic or more echogenic than bone.
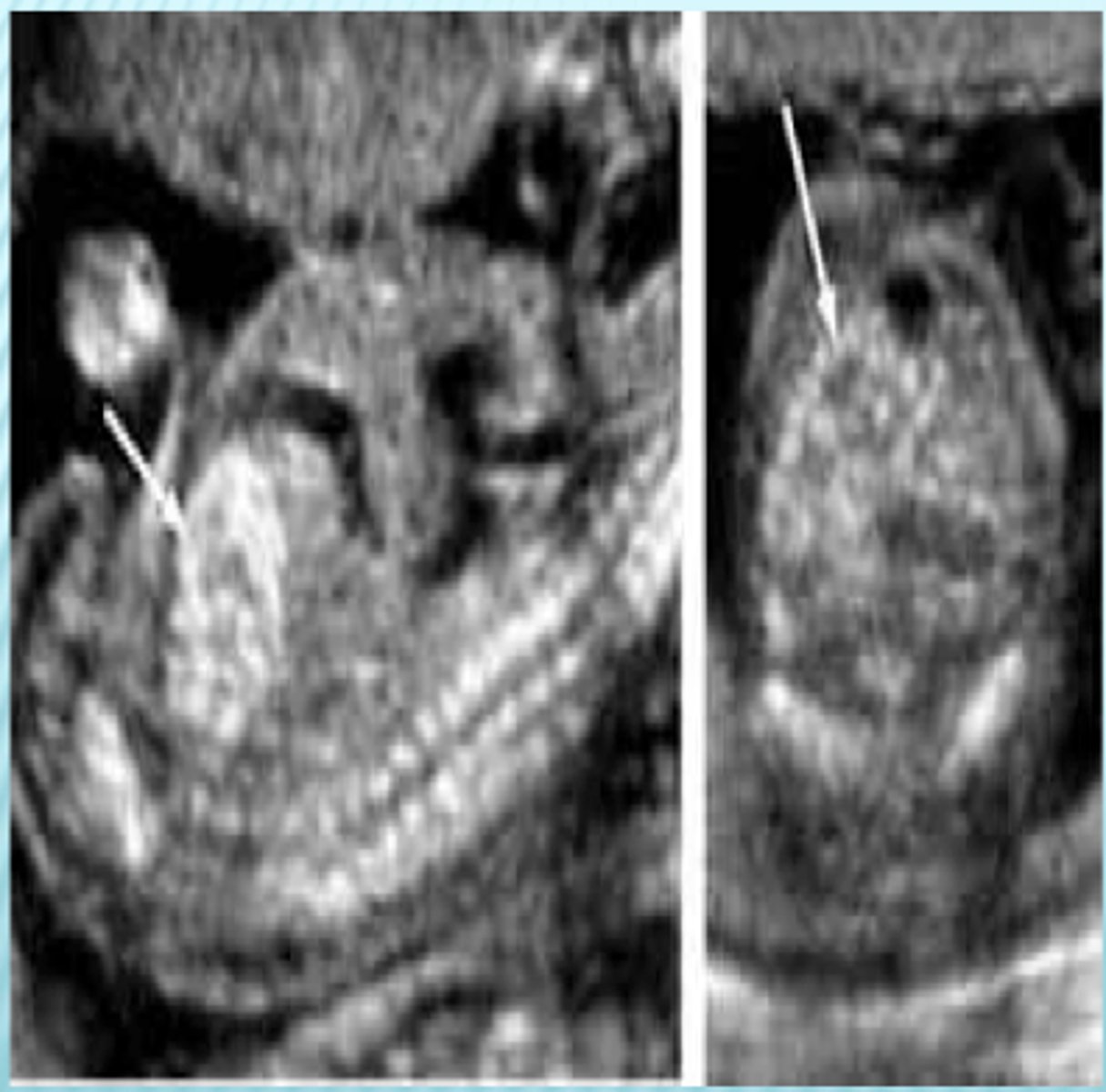
echogenic bowel differentials
Aneuploidy (27%) especially Trisomy 21 (15%), intraamniotic bleeding, cystic fibrosis (1-13%), congenital infection (1-10%).
ventricular septal defect differentials
Most often sporadic. Genetic / Teratogenic causes include aneuploidy (especially T21), DiGeorge, VACTERL, Noonan, maternal illness or exposures
Tetralogy of Fallot
Combination four congenital heart defects: ventricular septal defect (VSD), overriding aorta, pulmonary stenosis, and right ventricular hypertrophy (RVH), which together limit the oxygen in the blood that gets pumped to the body. Severe pulmonary stenosis associated with TOF can progress during fetal life ultimately leading to pulmonary atresia.
Tetralogy of Fallot differentials
22q, Down Syndrome, T13, T18, Alagille, CHARGE, VACTERL, maternal exposures
Echogenic Intracardiac Focus (EIF)
One or more bright spots in the heart. Most frequently seen unilaterally in the left ventricle. Parts of the heart muscle with more calcium could result in a bright spot.
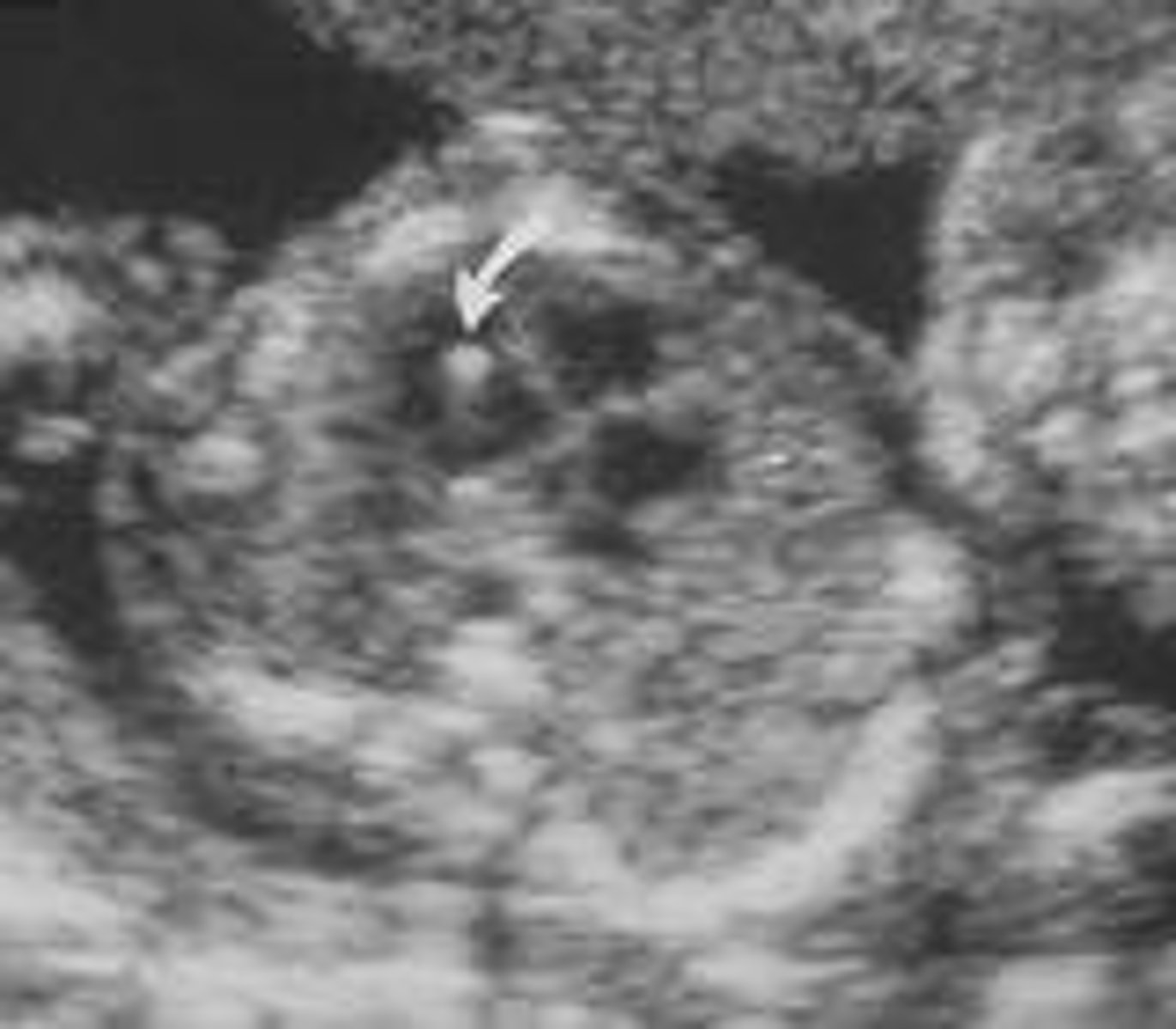
EIF differentials
Very common (1/20-30) pregnancies, usually sporadic and not associated with other findings. Soft marker for Down Syndrome, but one of the weakest soft markers. Also weakly associated with T13, and general congenital heart disease.
Single umbilical artery (SUA)
Single umbilical artery (SUA) = one artery in the umbilical cord is missing (could be either the left or right artery), more frequently the left.
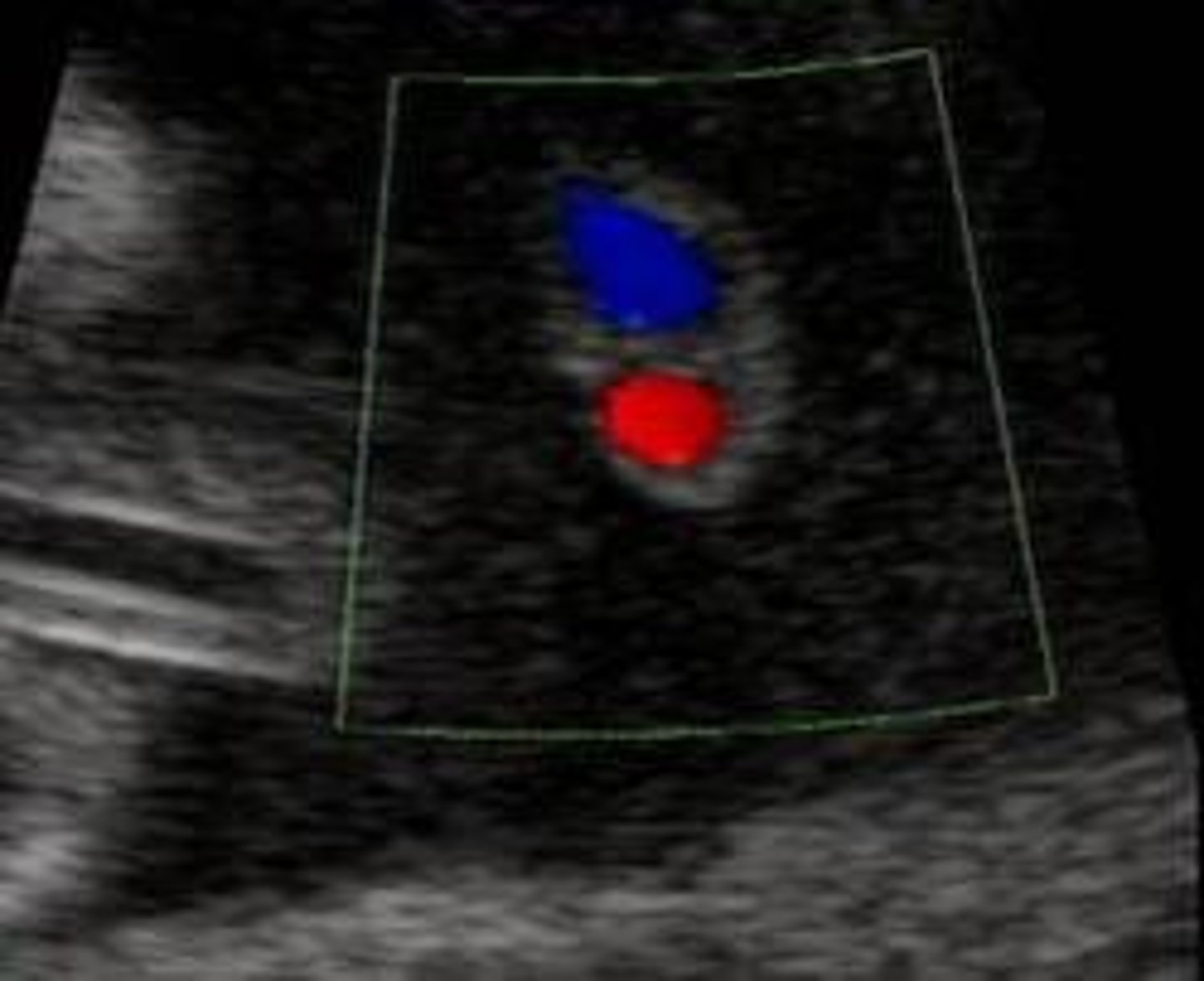
SUA differential
Approximately 65%-80% of all SUAs are isolated and do not impact survival. T18, T13, other aneuploidy. More common in multiple gestations.
hydronephrosis / pylectasis
Hydronephrosis is the backup of urine into the fetal kidneys, suggestive of a urine outflow obstruction. This process can occur for several reasons including T21, ectopic ureter, megaureter, and uroteric-pelvic junction. It is more common in male fetuses than female.
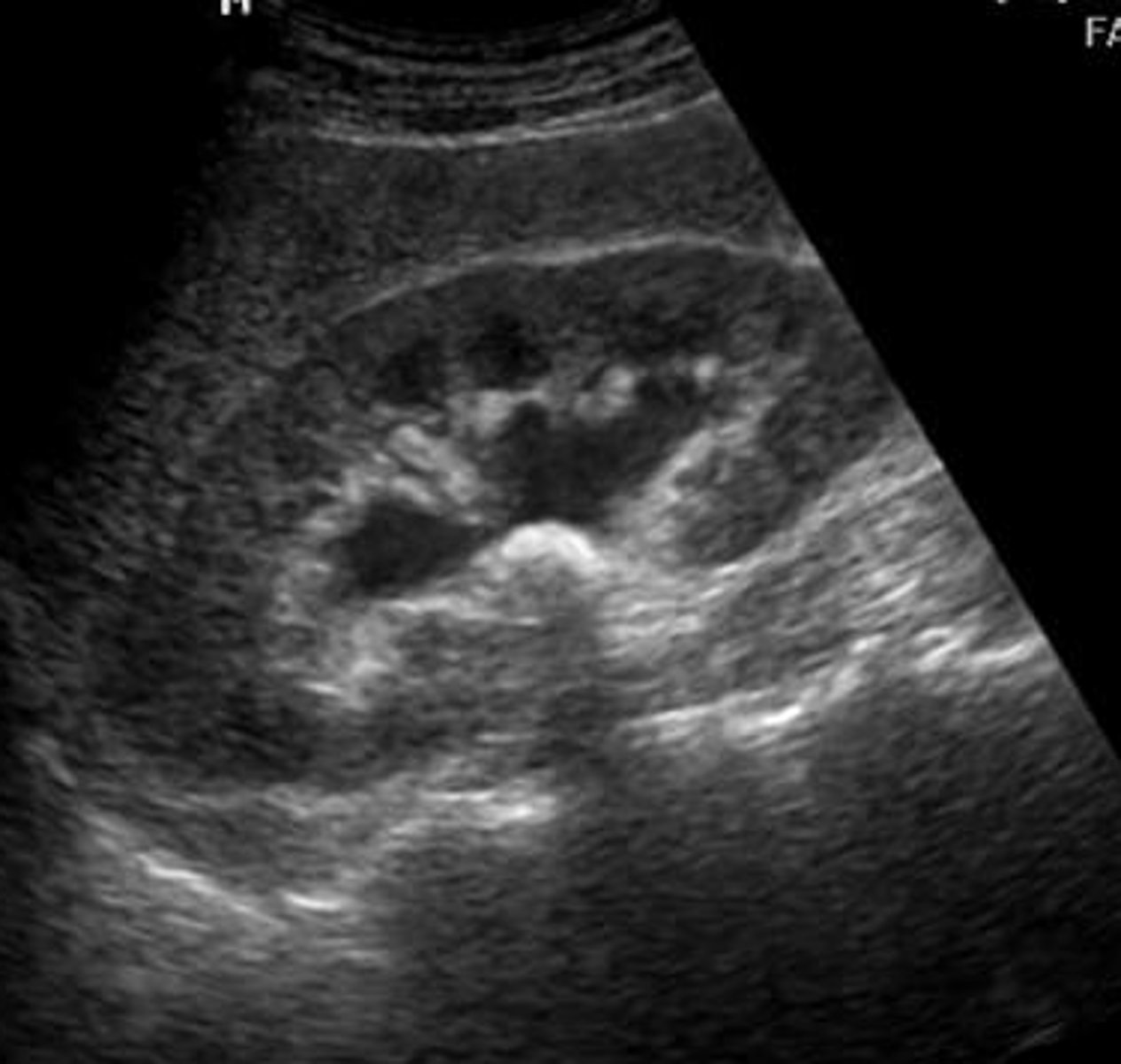
hydronephrosis differential
Most mild cases of hydronephrosis are not related to genetics. There is an AD condition, hereditary hydronephrosis, in which affected individuals typically develop uretero-pelvic junction related kidney obstruction en utero. This condition may be considered if there is a family history of hydronephrosis.
renal agenesis
Renal agenesis (RA) is when one or both kidneys do not form and are missing. This results from a developmental failure of the ureteric bud and the metanephric mesenchyme.
Renal Agenesis differential
Sporadic, T18, single gene disorders. Unilateral RA tends to be sporadic or associated with AD disorders. bilateral RA tends to be sporadic or associated with AR disorders. T18 can result in renal agenesis, though horseshoe kidney is a more common T18 finding
Polyhydramnios
The excessive accumulation of amniotic fluid during pregnancy. Fluid levels are influenced by fetal urination, lung liquid production, fetal swallowing, and intramembranous and intravascular absorption.
Polyhydraminos Differential
Polyhydramnios is a common complication of multiple gestation. Other differentials include: aneuploidy, gestational or pre-existing diabetes, esophageal atresia, duodenal atresia, myotonic dystrophy, fetal anemia, twin-to-twin transfusion, immune hydrops, maternal infection, aneuploidy, maternal hypercalcemia, Bartter syndrome
oligohydramnios
Low levels of amniotic fluid, which can be related to problems with the fetal urine outflow tract, or related to trauma, infection, or leakage.
oligohydramnios differential
kidney / bladder malformations, poor fetal growth, going >2 weeks past due date, twin-to-twin transfusion syndrome
megacystis
Megacystis is an enlarged bladder. It has a similar etiology to hydronephrosis, but can be diagnosed earlier and is more likely to result in spontaneous abortion. The main cause is bladder outlet obstruction. This process can occur for several reasons including ectopic ureter, megaureter, and uroteric-pelvic junction.
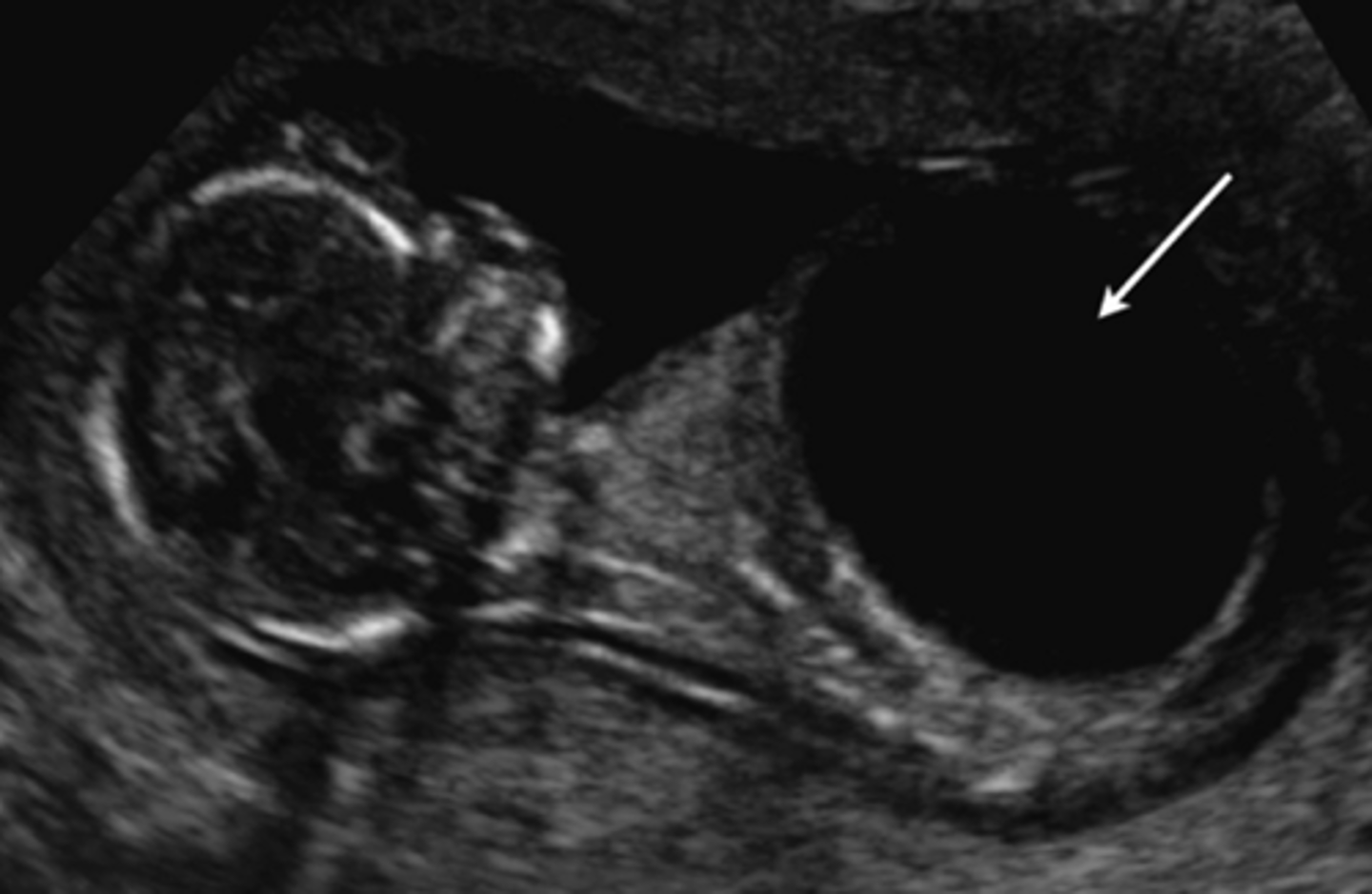
Anal atresia
Imperforate anus is a rare inborn abnormality characterized by the absence or abnormal localization of the anus. The rectum or the colon may be connected to the vagina or the bladder by a tunnel (fistula).
anal atresia differential
Most often sporadic. Genetic causes include VACTERL (which does not have genetic testing), Cat Eye Syndrome
shortened long bones
3 definitions of fetal shortened long bones: biparietal diameter (BPD) to femur or humerus length (FL, HL) ratio of greater than 1.5 standard deviations above the mean for gestational age; observed to expected (O/E) femur and humerus length ratio of <0.92 and <0.90, respectively; femur and/or humerus length less than the fifth percentile for gestational age.
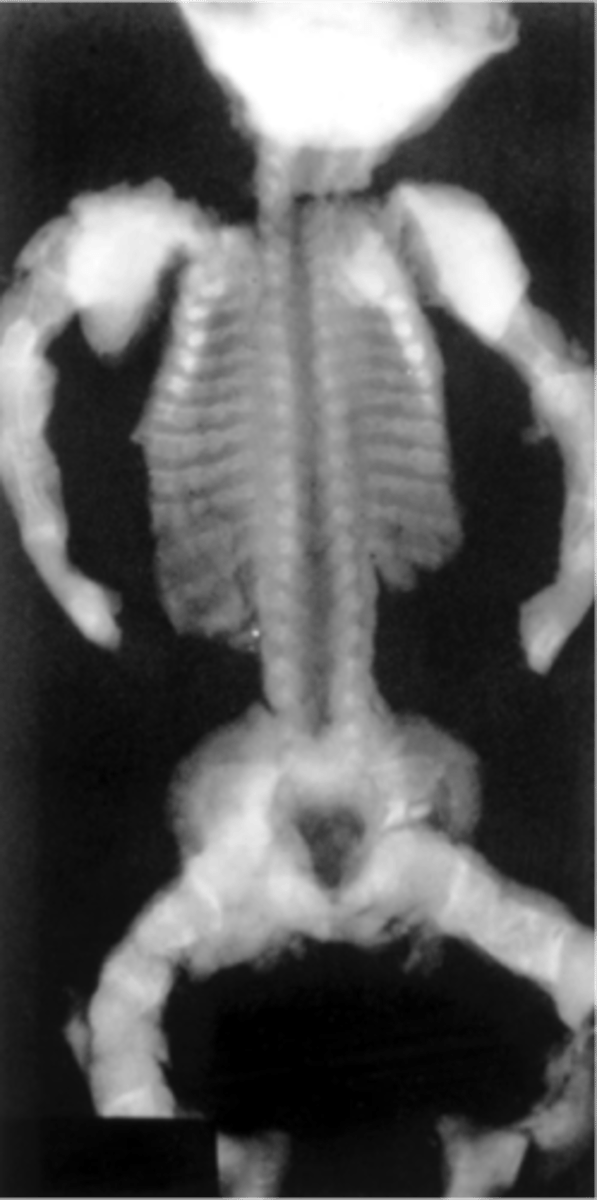
shortened long bones differential
Systemic bone disease or chromosomal disorder. Slightly increased risk of T21. Wide variety of skeletal dysplasias associated with shortened long bones, but diagnosis is difficult prior to birth
Polydactyly
Polydactyly is an abnormality characterized by extra fingers or toes, it can be syndromic or non-syndromic. When it occurs by itself with no other abnormalities it is inherited in an autosomal dominant pattern. There are many different genes associated with nonsyndromic polydactyly including HOX genes, hedgehog pathways, FGFs, and more.
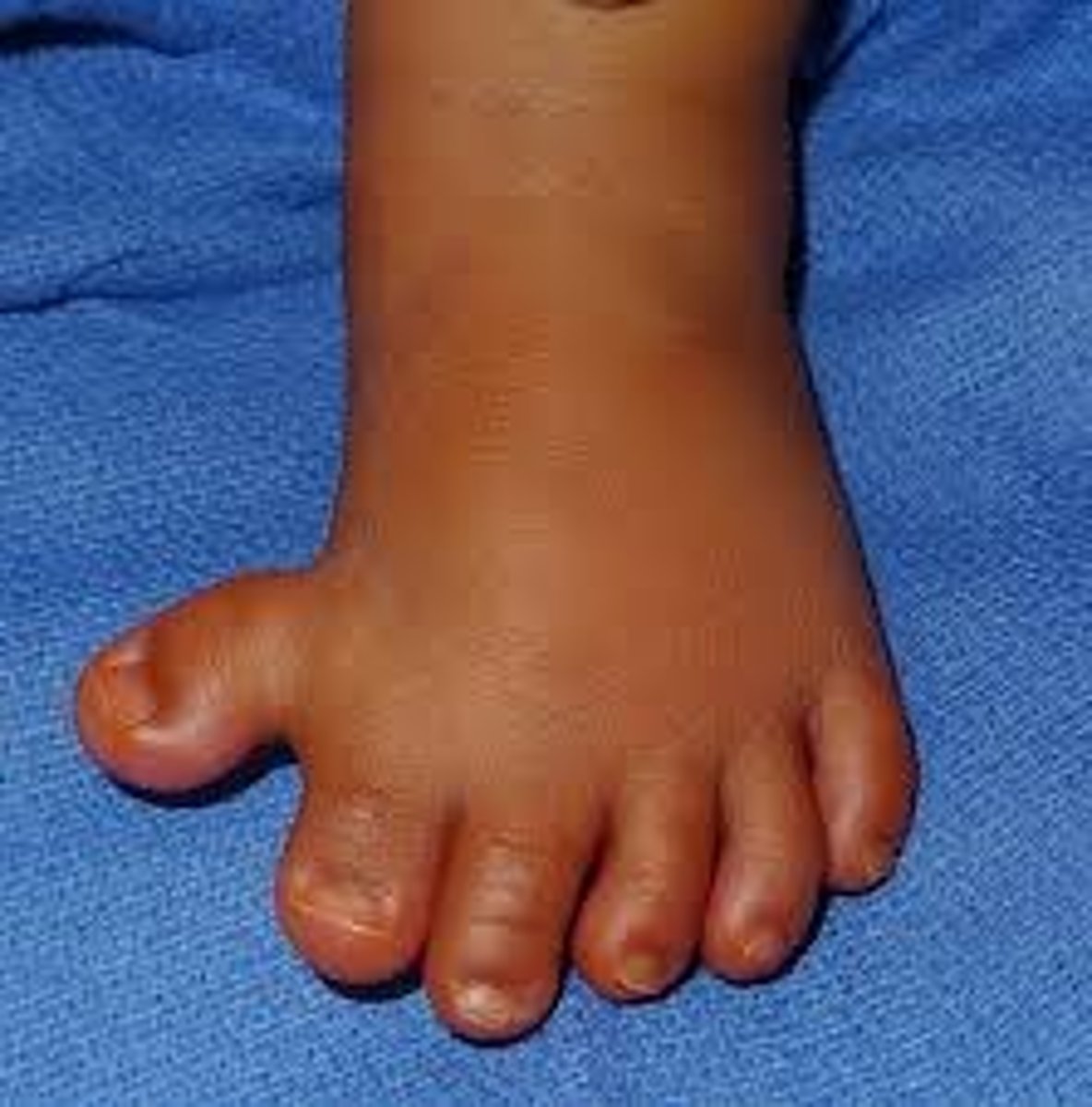
Polydactyly differential
Isolated polydactyly, Trisomy 13, Trisomy 18, Meckel Gruber, Bardet-Biedl, Smith-Lemli-Opitz
Post-axial polydactyly
extra fingers including vestigial remnants that are on the ulnar side of hand or lateral side of foot. The most common type of polydactyly, and the most frequently associated with genetic syndromes
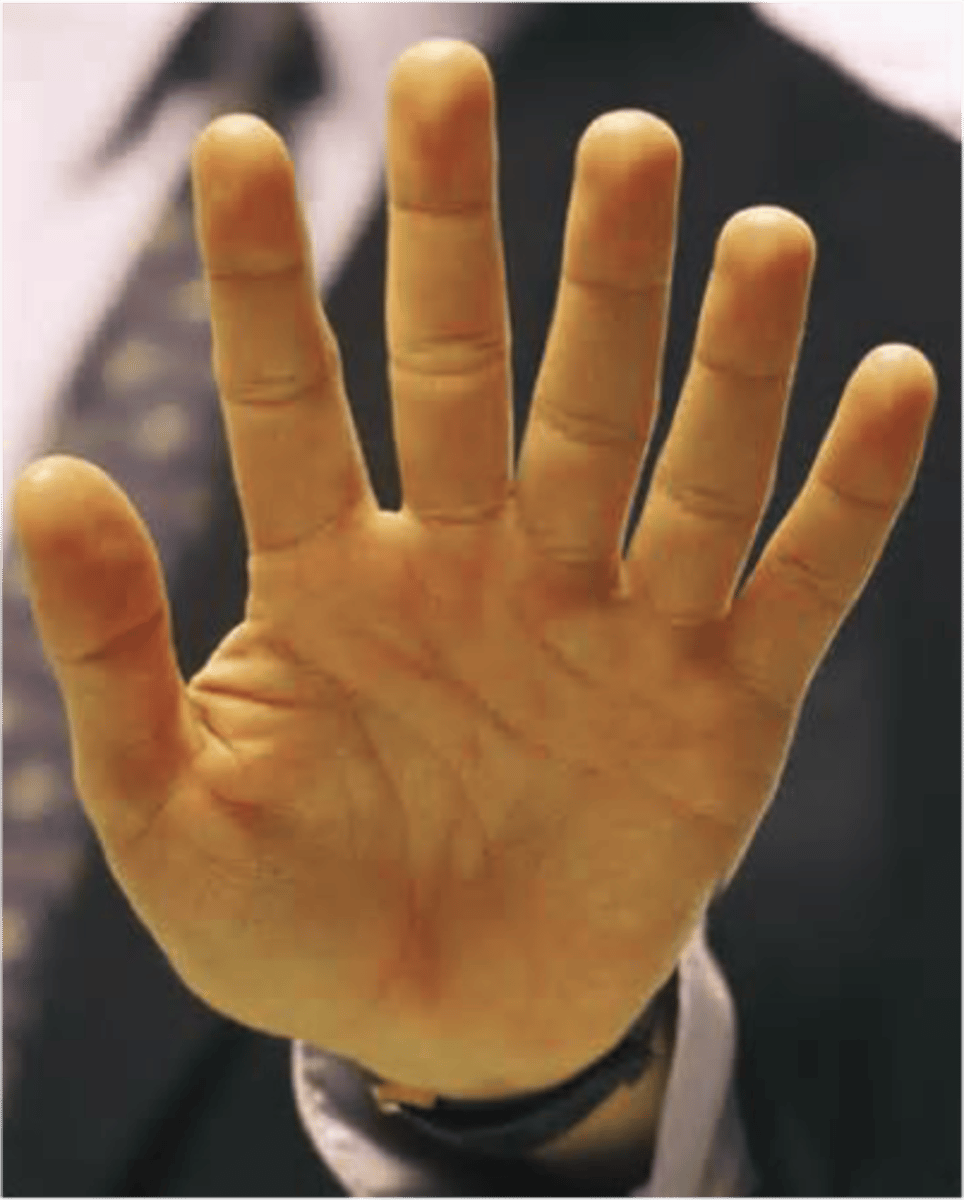
Pre-axial polydactyly
duplication of the thumb(s) or of the halux (great toe)
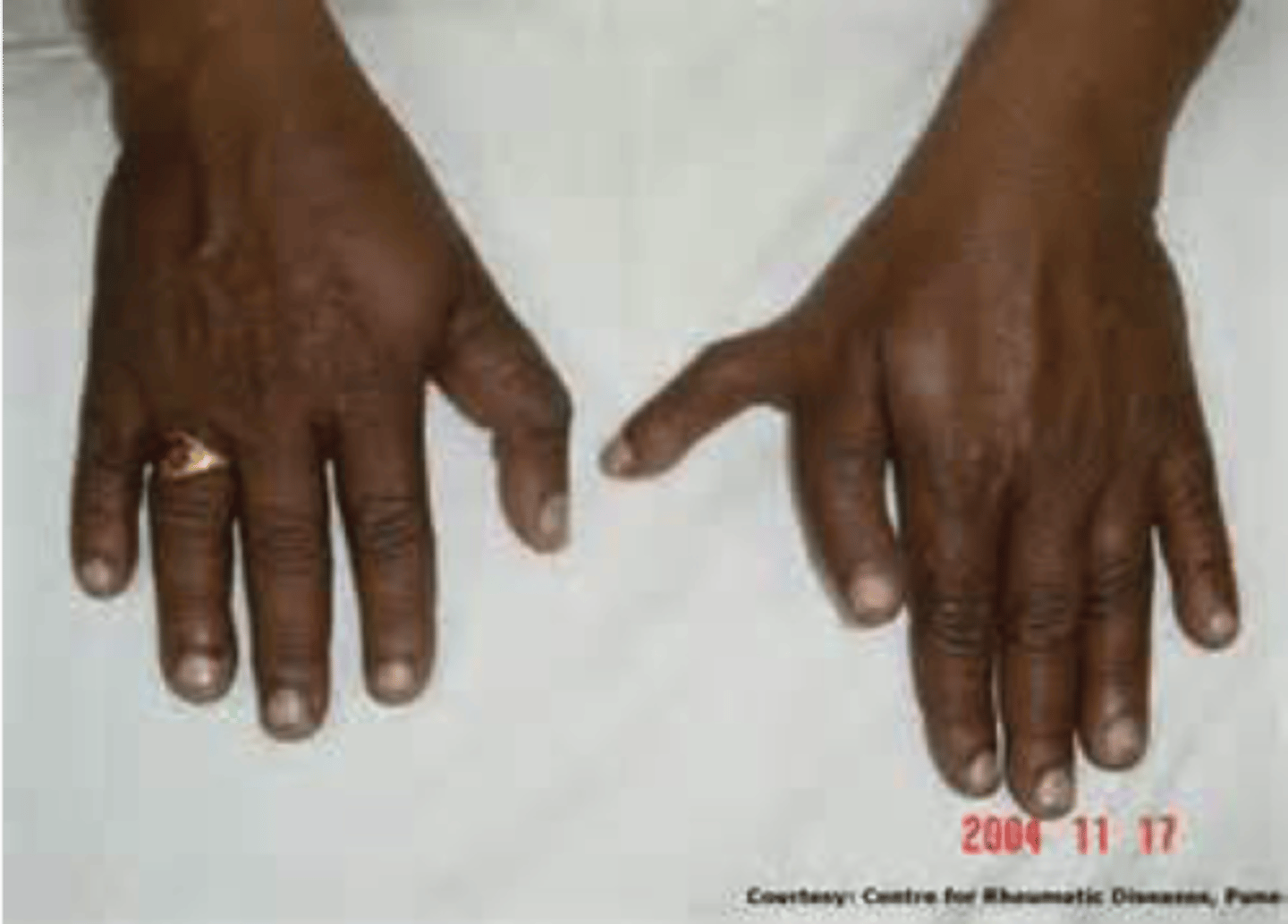
Central Polydactyly
least common hyperdactyly, in which a middle digit is duplicated
Immune Hydrops
Fetal hydrops (heart failure) due to Rh incompatibility. Uncommon due to the implementation of rhogam
hydrops fetalis (definition)
Hydrops fetalis is fetal heart failure evidenced by abnormal amounts of fluid accumulated in two or more body areas of an unborn baby. It often occurs in the abdomen, around the heart or lungs, or under the skin.
nonimmune hydrops
Fetal hydrops caused by congenital fetal anomalies & infections-- anything other than Rh incompatibility
nonimmune hydrops differential
HbBarts Hydrops Fetalis, Turner, Sly Syndrome, skeletal dysplasias, arthrogryposis multiplex syndromes, multiple pterygium syndrome
IUGR numerical definition
Bottom 10th percentile of weight of fetuses of a given gestational age
IUGR differential
Early-onset IUGR is often due to chromosomal abnormalities, maternal disease, or severe problems with the placenta. Late-onset growth restriction (after 32 weeks) is usually related to other problems. The differential is huge-- chromosomal anomalies, other genetic disorders, maternal exposure / illness, etc
prenatal CF findings
Grade 2 "bright as the bone" echogenic bowel (2-3% risk of CF when this finding seen). Ultrasound has very low CF detection. Even the smallest carrier screens look for CF status
newborn / childhood CF findings
frequent / large / greasy stools (a result of malabsorption), failure to thrive, meconium ileus, jaundice due to blockage of the bile ducts, excessive salt in sweat. Chronic respiratory infection with Pseudomonas aeruginosa, fungi, and mycobacteria all increasingly common over time. Inflammation of the upper airway --> frequent runny nose, recurrent siniusitis, nasal obstruction, nasal polyps. Shortness of breath, chronic productive cough that produces sputum, malabsoption, diabetes, pancreatic insufficiency, low weight, wheezing, digital clubbing, cyanosis, coughing up blood, pulmonary heart disease, collapsed lung.
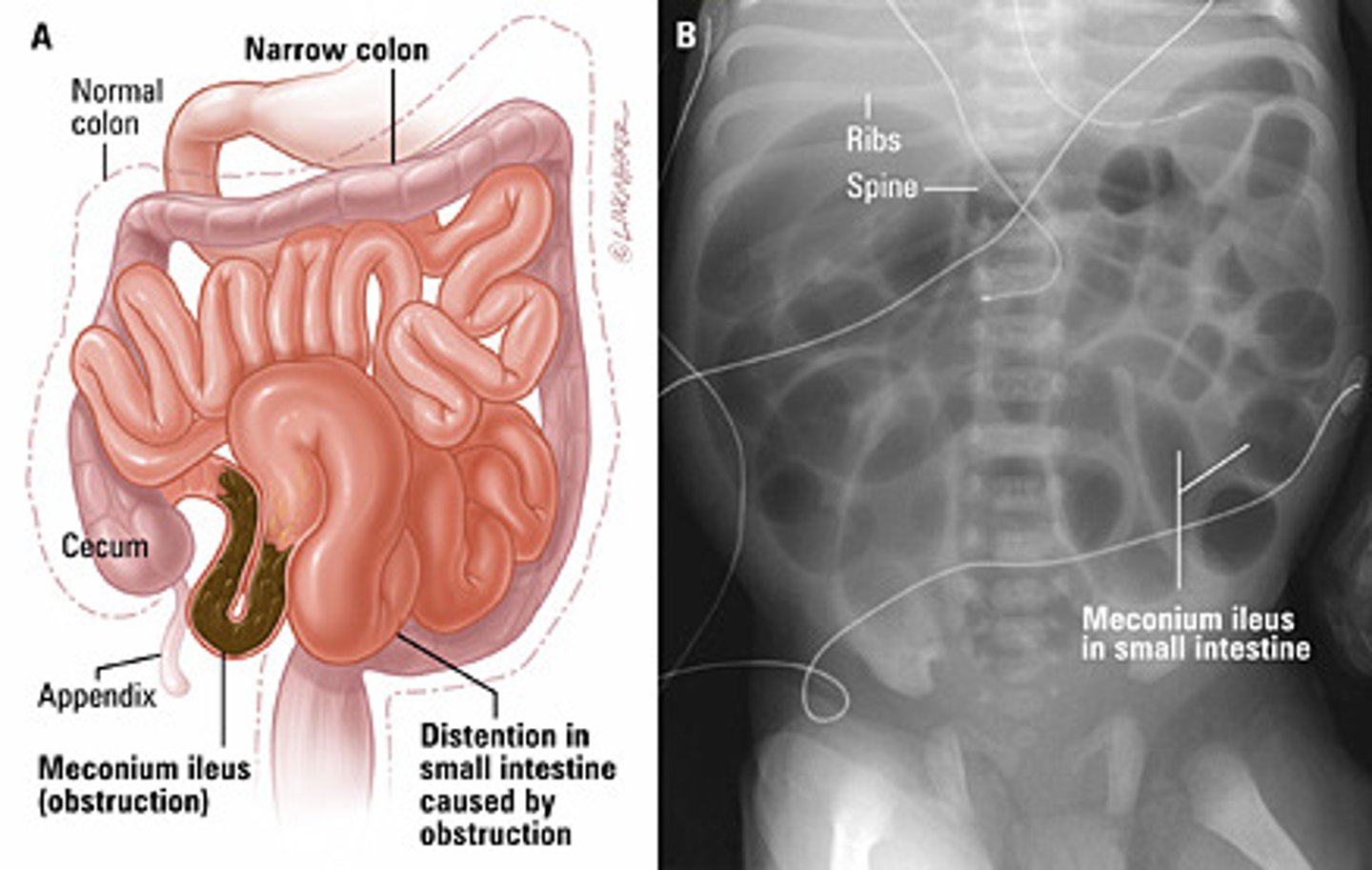
Meconium ileus
small-bowel disorder marked by the presence of thick echogenic meconium in the distal ileum. Essentially diagnostic of CF in the neonatal period
CF carrier rate
1/16 in French Canada / Cajun populations
1/25 in Western European populations
Ivacaftor
CFTR gate-opener for people with GATING mutations
Trikafta
combo of three drugs: CFTR transporter + CFTR corrector + Ivacaftor to open the gates). Only works if affected person produces some CFTR protein.
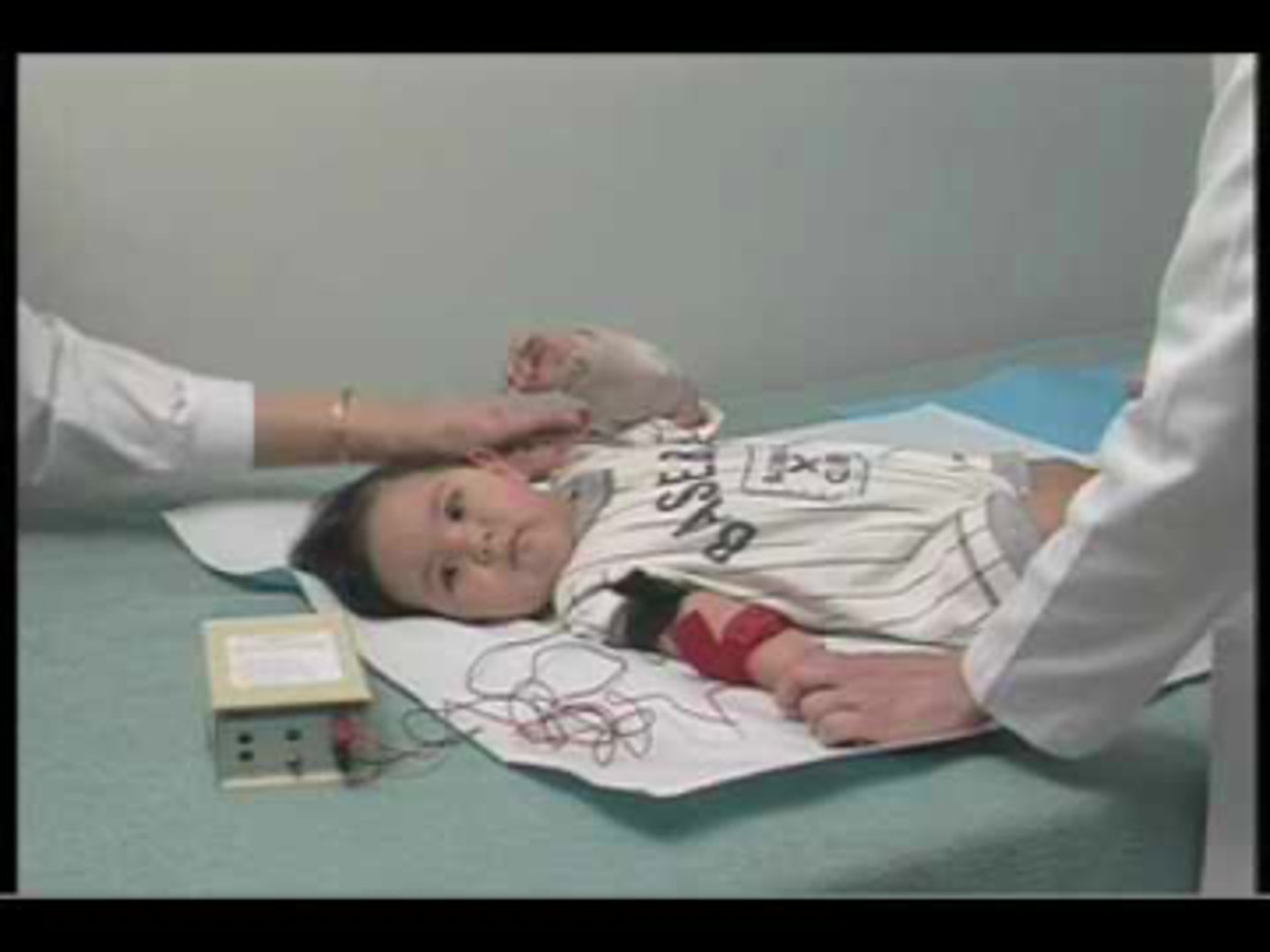
CF sweat test
Gold standard for CF diagnosis. Diagnostic range: >60 mEq/L Cl-. TAT: same day
CFTR class 2 mutation
Most common mutation type, includes deltaF508. Results in a misfolded CFTR protein, which is not normally transported to the cell surface. Responsive to Trikafta.
CFTR class 1 mutation
nonsense / frameshift / deletion mutations of CFTR. Biallelic type 1 mutations are not responsive to Trikafta or Ivakaftor because no functional CFTR protein is produced.
CFTR genotype-phenotype correlation
Geno-pheno strong for pancreatic function but poor for lung function.
Immunoreactive Trypsinogen
Pancreatic enzyme on the NBS. When elevated, the screen is positive for CF.
CFTR diagnostic
elevated trypsinogen on NBS + either positive sweat chloride OR biallelic CFTR mutations