module 2 ( L.10-15 )
1/82
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
83 Terms
liquid dosage form ( limitation )
bulkiness
leakage from container
physical & chemical stability < solid dosage form
shelf-life ( require special storage )
difficult to administer
what is solution
chemically & physically homogenous mixture of more substance
solution = solute + solvent
exam pf solution
two or more gases
solid in liquid
liquid in solid
factor to consider when formulation liquid dosage
route of administration
disease state
intended patient pop ( age/type )
physicochemical property of drug
what is solubility
max conc.. or amount of substance ( solute ) that may be completely dissolve in given amount of solvent ( at given temp and pressure )
( solid in liquid )
solubility of liquid in liquid
miscibility = solubility of liquid in another liquid
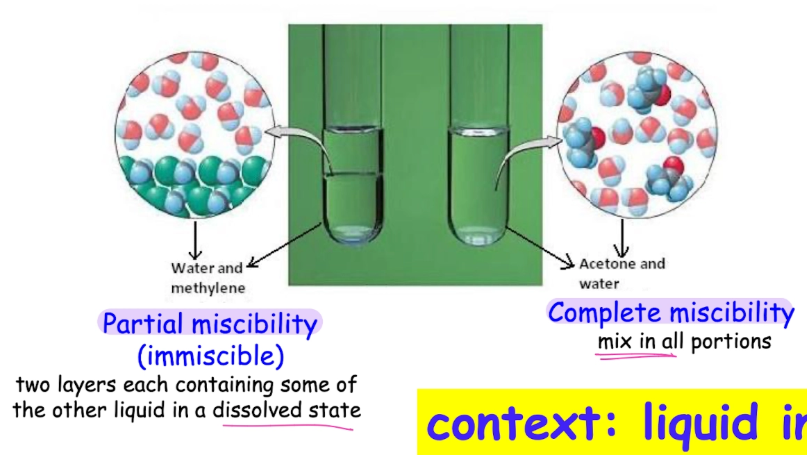
why solubility important ( when formulation drug into liquid form )
formulation
influence design , chose of excipient , dosage type , manufacturing process
stability
influence its chemical stability
poorly soluble drug = precipitate or degrade
why solubility important ( when administration into body )
bioavailability
drug must dissolve for absorption & to exert therapeutic effects
affects rate of dissolution & extent of absorption into blood stream
poor solubility = slow or incomplete drug release . DE absorption , DE bioavailability
dosing
affect dose require to achieve desired therapeutic effect
poor solubility = inaccurate dosing
type of solubility
intrinsic ( So )
solubility of PURE , in-ionise drug in give temp
not incl salt , ionisation and co-solvent
serve as baseline
apparent ( S )
solubility of drug consider all form in solution
reflect real-world solubility
factor that affect solubility
temp.
pressure
→ significant effect on solubility of gas
→ IN solubility of gas in liquid
nature of solute
nature of solvent
how do we express solubility
solubility is quantitative ( associate w/n. and unit )
ex. percentage , molarity , parts
BCS classification
USO definition = n. of mL of solvent in which 1g of solute will dissolve at specified temp
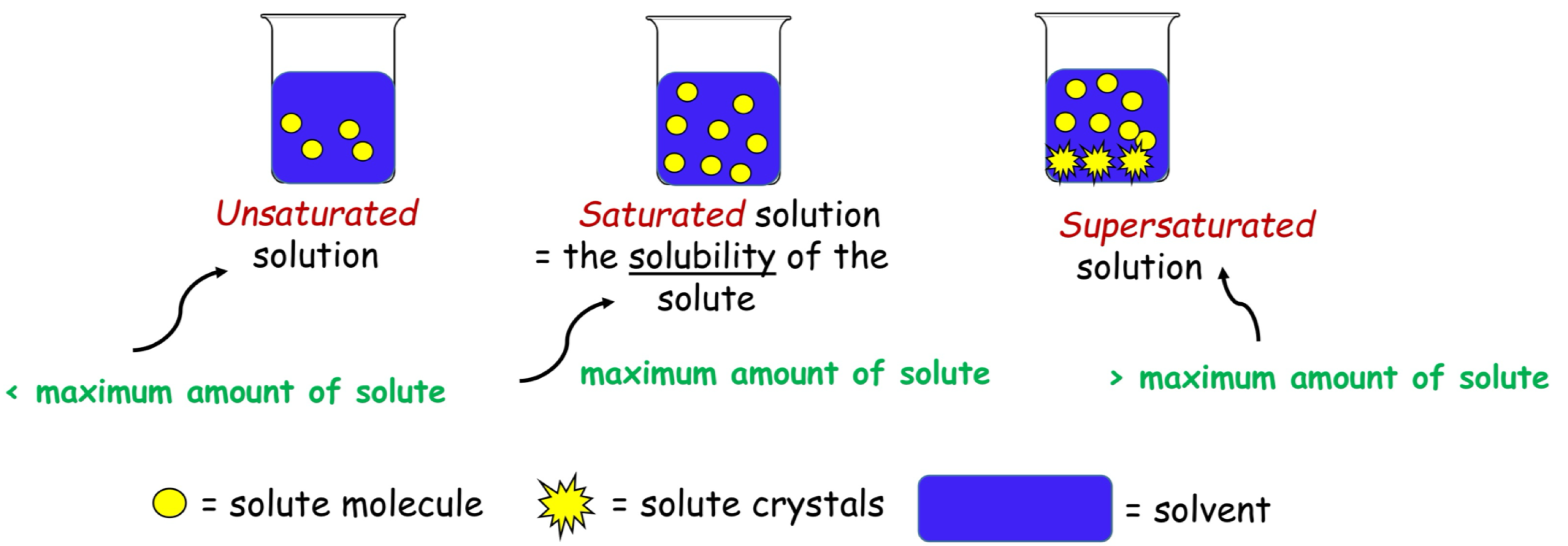
degree of saturation
amount ofd solute dissolves in solvent relative to max amount that can be dissolve ( specified temp and pressure )
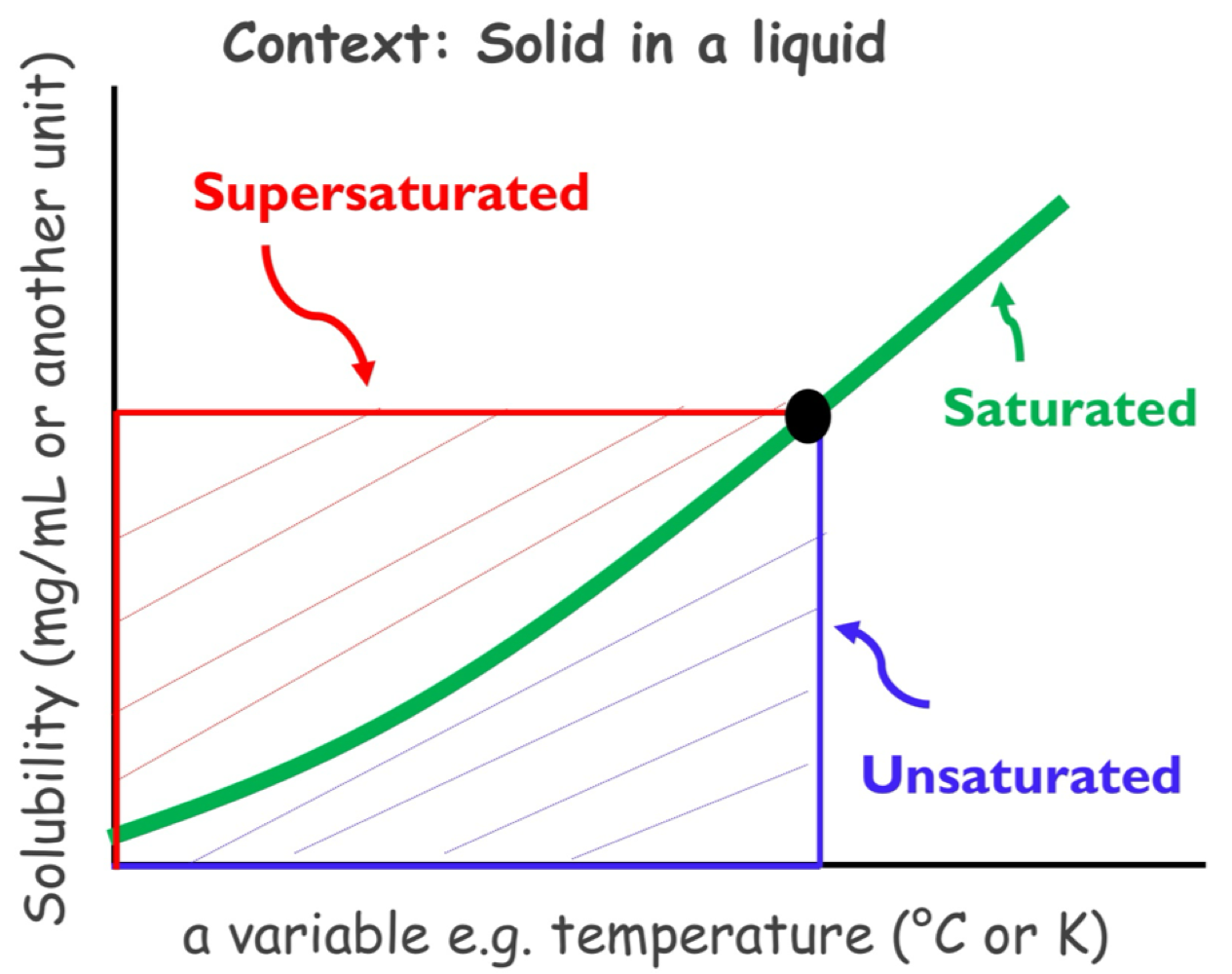
solubility curve
graphical representation = how solubility change of substance against any relevant variable ( ex. w/ temp , pH , solvent )
for solid , solubility IN w/IN temp. ( curve moves upward w/IN temp )
"study saturated or unsaturated, useful predicting dug behaviour”
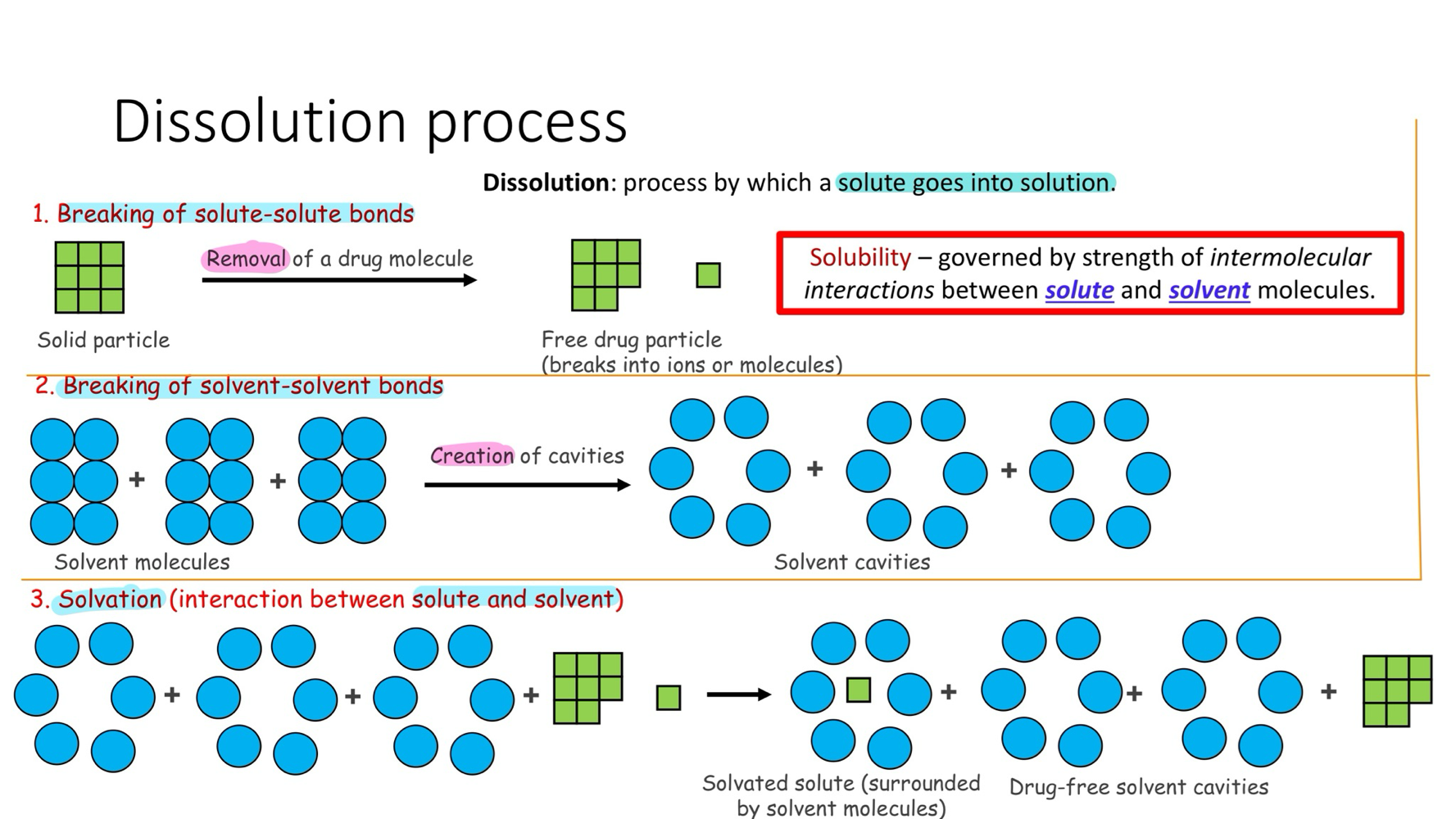
dissolution & salvation & hydration ( definition )
dissolution = process by which substance goes into solution
salvation = attraction and association of molecule of solvent w/molecule or ions of solute
hydration = when water is used as solvent
solubility VS dissolution rate
solubility = max amount of solute that will dissolve in give solvent
thermodynamic property → tell how much drug can dissolve , nut nit how fast it dissolve
dissolution rate = speed at which dissolution occur
kinetic property → tell how fast drug goes into solution
type of dissolution process
break of solute-solute bonds
breaking of solvent-solvent bond
salvation ( interaction b/e solute & solvent )
factor affect dissolution rate
temp
→ IN , IN dissolution rate of most solid ( solution absorbs E )
agitation/stirring /mixing
→ move dissolve solute away from surface of solid , IN dissolute rate
particle size reduction ( IN SA to volume )→ IN dissolution rate ( more solute is exposed to solvent )
predicting solubility during pre-formulation
goal → optimal formulation design
challenge → poor solubility is major cuz of drug development failure
pole of pre-formation studies
→ access drug solubility early
→ identify potential formulation challenges
→ Guide section of formulation strategies
outcome
→ DE development time & cost
→ improve & regulatory sucess
solubility of solute in given solvent
for solute to dissolve in solvent , bond b/w solute must be broken and new bond must formed b/w solute & solvent
compound dissolves due to intermolecular force b.w solvent & solute
like dissolve like
used to predict solubility of solute in solvent
predicting solubility
solubility→ governed by strength of intermolecular interaction b/w solute & solvent
solute must interact with solvent
nature of solvent ( factor affect )
polarity
H-bond
pH ( if solute is weak electrolytes
solvent polarity
dielectric constant
→ measure of how mush substance become polar , when place in external electric field
tell you how good solvent is @separating & stabilising charge
help predict ow well solute will dissolve in it
predictor ( rough measure ) of polarity
polar ( ex. water )
seme-polar ( ex. glycerol )
non-polar ( ex.octane )
polarity of water ( refresher )
polar covalent bonds b/e O & H
O is more electronegative→ pull electron closer → permanent dipole
polar solvent & solubility of ionic solute
polar solvent = high dielectric constant & (+ ) /(-) dipole
ionic solute = strong electrolyte
mechanism = ion-dipole interaction
→ solute attraction to =/- dipole on solvent , inter ionic attraction weaken , ionic lactic breaks , ions dissociate, solvation → ions stabilise in solution
ionic molecular and charge strength
ionic compound → generally water soluble , bit ionic bond side in strength
( water CANT break all ionic bond )
attraction b/w ions in solid < F. of attraction b/w ions and water = compound does not dissolve
charge strength → magnitude of charge on an ion ( solute )
→ how strongly it interact w/water → show solubility
w/small charge = higher solubility
w/large charge = lower solubility
solubility of ionic compound ( general rule )
cation & anion in ionic compound can be used to predict it’s AQ solubility
( as charge IN , solubility will change ( IN/DE ) )
polar solvent & solubility od neutral compound
mechanism = dissolve by formation H-bond
polar group ( solute ) are attracted to polar solvent ( strong dipole ) → solute-solute bond break → solute stabilised in solution
non-polar solvent & solubility of non-polar solute
not DE interionic attraction in ionic crystals or H-bond → not dissolved polar solute
mechanism = via van der Waals F
solvent surround solute → temporary dipole → weak wan der Waals attraction → solute disperses throughout solvent → solubility
semi-polar solvent and solubility
- & + dipole charge has polar and non-polar → intermediate polarity
polar = interact via dipole=dipole or H-bond
non-polar = via van der Waals
nature od solute
chemical nature
charge strength
ability to form H-bond w/solvent
hydrophobic ( hydrophilic )nature of molecule , ration of polar to non-polar group )
degree of ionisation of functional group
physical nature
solid state ( crystalline ) form
ability to form H-bond w/solvent ( nature of solute )
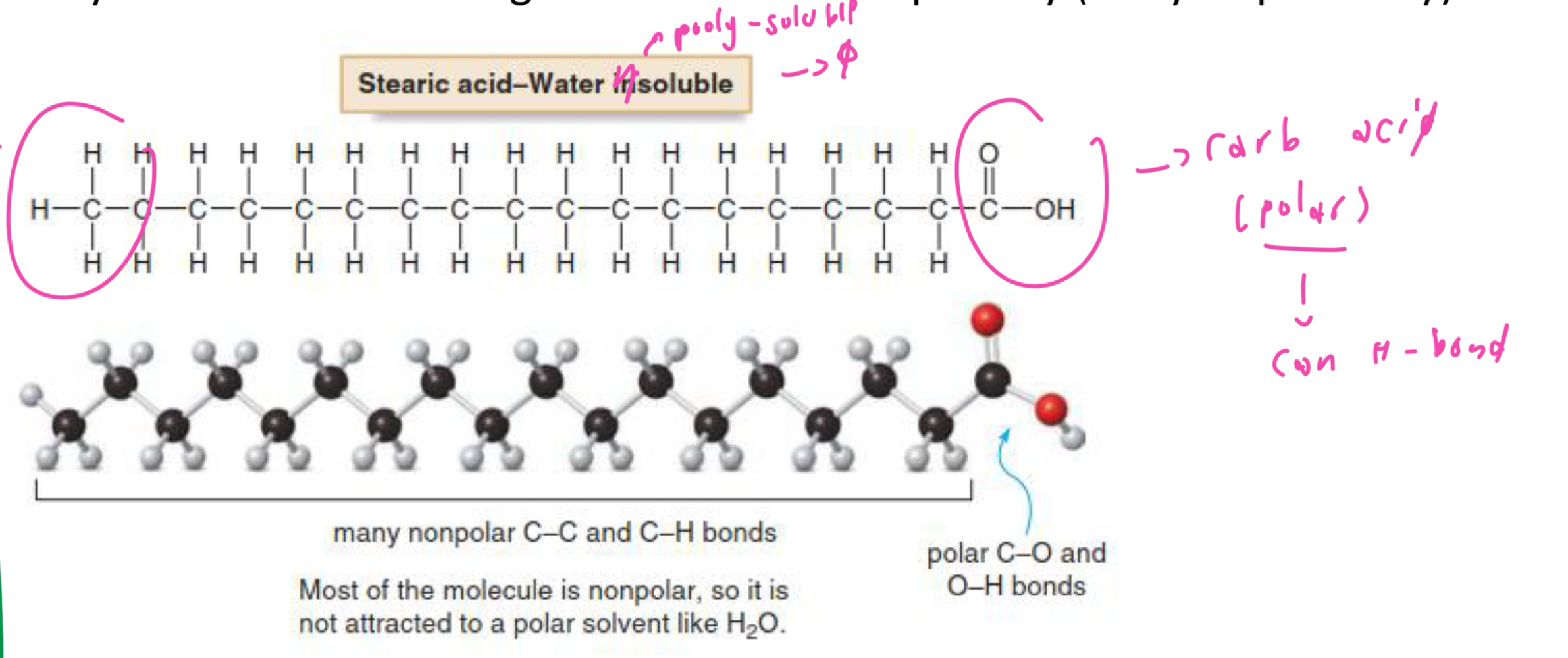
H-bond form when H-atom attach to electronegative atom (F , O , N )
determined by examining structure feature of both solute and solvent
look for polar functional group
→ like dissolve like rule , polar solute can interact strongly with/polar solvent via H-bond
hydrophobic nature of solute
ration od polar to non-polar
→ solubility of substance change w/its size and polarity
IN H-bond , IN hydrophilicity
degree of ionisation & solubility
fraction of solute that dissociate into ion in solution
solute
strong electrolyte( ionic compound )
weak electrolytes
( IN ionisation , IN ions in solution → solubility)
degree of ionisation ( strong electrolytes)
complete ionised → HIGH solubility
ionice compound
strong acid & base
degree of ionisation ( weak electrolytes)
weak electrolytes = weak acid & base
weak electrolytes are poorly water soluble
highly influenced by pH
→ weak acid are more soluble in base
→ weak acid are more soluble in acid
solubility challenges of weak electrolytes
( most of drug are weak electrolytes)
poor solubility
→ difficult of formulation as liquid dosage form
→ drug must solubilised adequately for efficient absorption
→ precipitate out of solution at certain pH oe ion conc. change
strategies to improve AQ solubility
modify solid state ( co-crystals , amorphous )
salt-formulation
co-solvency
pH adjustment
complexation
surfactants/micelles
colloidal delivery system or nanoformulation
solubility and solid state of solute
solid
→ Amorphous ( lack order )
→ Crystal ( highly ordered )
crystal
→ polymorphs = diff crystalline form of same molecule
→ Pseudo-polymorph ( solvate/hydrate ) = contain water or other solvent as guest molecule
crystalline
crystalline form
→ lower E state = most stable
→ rigid structure & stronger = low solubility ( DE H-bond w/water )
strategies to improve solubility
→ chess most suitable polymorph to balance b/w process ability and stability
amorphous
amorphous form
→ high E state = unstable
→ solubility > crystalline form ( IN H-bond due ri semi-random state )
strategy to IN solubility
→ convert drug from its crystalline from into its amorphous
improve solubility via salt formation
60% of drug are formulates as self ( converting drug into salt form by adding ionise group )
dissociate on AQ environment to ON AQ solubility
most preferred approach to IN AQ drug solubility of weak electrolytes
presence of acidic or basic functional group is essential
selecting salt type
dictate largely by physiochemical properties of API but also
→ formulation/route of administration
→ drug release profile & desire bioavailability
→ regulatory consideration
salt of basic
salt of acidic
improving solubility by co-solvency
mixture of solvent used to enhance drug solubility
work together to IN solubility beyond each individual solvent could achieve on its own
important in formulation od injectable solution
adjust solvent polarity
→ DE water -water H-bond
→ alter ionisation balance ( pH adjustment maybe require )
→ IN drug 0solvent interaction = IN solubility of unionised drug
limitation and selection consideration ( co-solvency )
limitation
risk of precipitation on dilution ( after administration)
selection consideration
biocompatibility ( route-administration)
dose limits
toxicity at high conc.
effect of dilution on drug solubility by co-solvent
dilution DE solubility
→ DE linear w/dilution
→ solubilising capacity of co-solvent formulation DE exponentially
consequence of precipitation
→ pain
approaches to minimise precipitation
→ DE drug load formulation
→ adjust rate of administration
pH to achieve solubility requirement
2 parameter when formulating weak electrolytes drug
solubility of drug at desired pH
pH that must be maintain to prevent precipitation of drug from solution of known or desired conc.
→ predict by pHP equation - calc solubility of weak electrolyte ( drug ( at given pH of solution
buffer are used to adjust / maintain pH
definition of complexation
complexation = interaction between/w one or more molecule of 2 compound ( ex. ligands and substance )
mechanism of interaction in complexation
covalent ( coordinate )
non-covalent
cause of complexation
drug or solute can interact with/other component in formulation
( ex. other drug , excipient )
bor biological compound
deliberate or unintentional complexation as a result of
change in pH , solvent
chelation
adsorption
interaction w/storage container
in-vivo
drug incompatibility
consequence of complexation
( physical and chemical properties of complexing SPP are alter )
change in solubility, efficacy , stability . taste , absorption or elimination
type of complexes
coordination complexes-
→ metal complexes
molecular
→ small-small molecule
→ small -large molecule
→ large -large molecule
coordination complexes-
complexes form via coordinates bond
electron rich ( ligand ) bonds w/electropositive atom ( substate ) by donating its pair of electron
coordination covalent bond VS covalent bond
coordination covalent bond = covalent bond in which BOTH electron come from same atom
covalent bond = form by 2 atom charge pair of electron
molecular
complexes form by multiple attractive non-covalent interaction ( H-bond , electrostatic attraction, van der Waal F or hydrophobic )
coordination complexes ( metal complexes )
most commode type of coordination complex
central metal atom or ion ( cation ) surround by (-) charge or neutral molecule possessing lone pair electron
ligand senate pair of electron
metal ion accept pair of electron ( ex. substrate )
metal complexes ex. anti-cancer drug
platinum , Palladium , gold
metal ion bond to essental biomolecule
→ DNA , protein , enzyme
→ inhibits their function , cuz cell death
metal complexes ex. clinical implication
x. tetracycline ( antibiotic )
small - small molecule complexes
molecule bearing functional group w/opposite polarity can interact w/each other in solution
small - small molecule complexes ( ex. drug-caffeine )
ex. drug-caffeine complexes ( IN drug solubility)
→ solid state = brevet crystallisation or create amorphous forms
→ co-solevnt = interact w/both drug molecule and water )
small-small molecule ( ex. Procaine Pencilline G )
combination of penicillin (antibiotic ) and procaine ( anaesthetic )
→ salt complex , DE penicillin, cuz slo absorption, longer half-life , prolonged therapeutic effect , DE frequency of administration
small-small molecule ( ex. aminophylline )
aminophylline > water-soluble than theophylline = more suitable for IV administration
small-small molecule ( ex. cation/antion )
precipitate ( insoluble complex )
electrostatic attraction b/w oppositely charge ions
small-large molecule
enzyme-substance ( drug ) interaction
drug binding to plasma protein ( can’t interact w/its receptor )
→ DE drug solubility
→ DE volume of distribution
→ DE elimination
association complexes ( ex. micelles )
inclusion/occlusion complexes
ex.cyclodextrin-dug complexes
large-large molecule
complexes b/w nucleotides base in DNA molecule via H-bond
inclusion complexes
complex in which molecule ( host ) w/ cavity can accommodate substance ( guest )
ex.cyclodextrin( CD ) complexes
pharmaceutical application of cyclodextrin( CD ) complexes
IN solubility
IN stability
IN bioavailability
DE tease masking
→ poorly soluble drug not automatically able to for, inclusion complex w/ CD BE size , shape & polarity and potency at low dose
effect of cyclodextrin( CD ) complexes on drug solubility
appropriate choice of host and guest = high selectivity → IN solubility
release of guest from cyclodextrin( CD ) complexes
non-covalent , so guest molecule continually associated from host
large guest = slower formation & dissolution
major mechanism of release
→ dilution
→ competitive displacement
→ change in ionic strength & temp
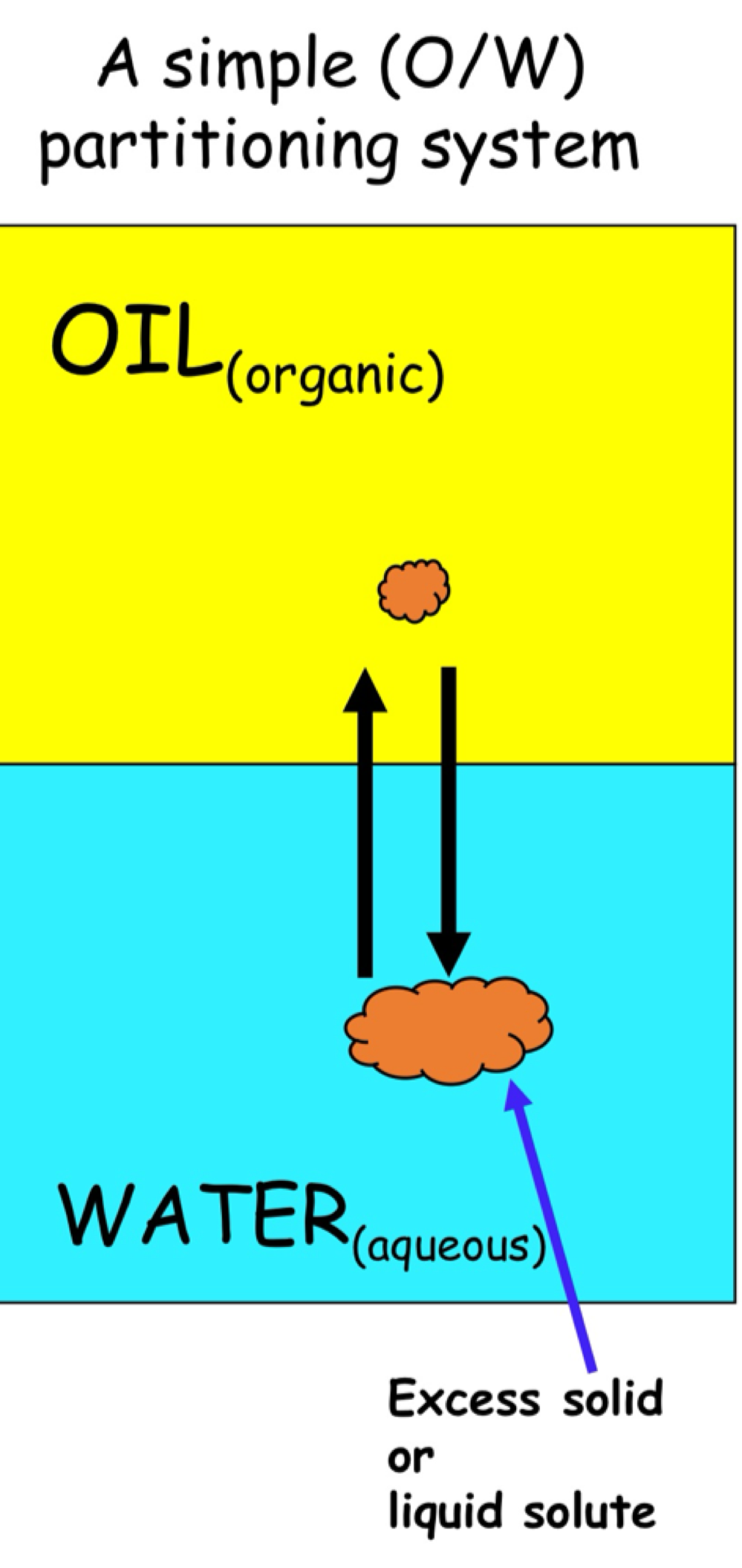
partitioning ( definition )
process of drug’s movement over time across membrane
= distribution of molecule ( solutes ) b/w immiscible solvent
Swater ⇌ Soil
solute distributes or partition in defines conc. ration = partition constant or partition ratio
affinity for polar & non-polar
affinity for water ( polar ) = never enter bloodstream
affinity for lipid ( non-polar ). = never dissolve in water
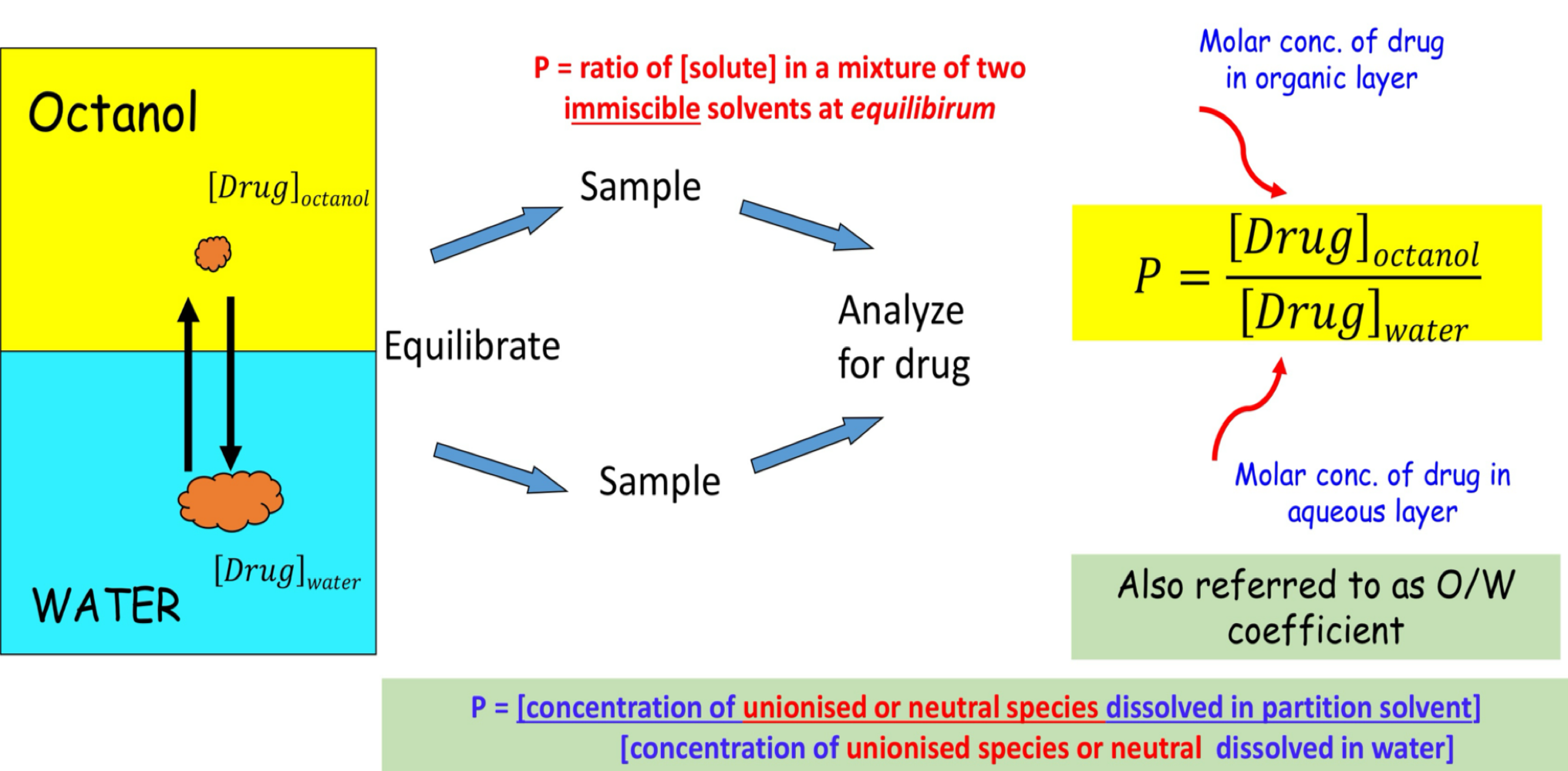
partition coefficient ( P )
to predict likelihood that drug will dissolve in water and cross cell membrane ( quantify relative affinity for each other )
how is partition coefficient ( P ) determined
logP
partition coefficient ( P ) value vary ( small - large )
relative lipophilic / hydrophilic or lipophilicity of compound ( unionised or neutral SPP )
help estimate both drug solubility & permeability
log P value
logP ( - ) → hydrophilic
logP = 0 → balance
logP ( + ) → lipophilic
lipophilicity change as function of pH for ionisable compound
lipophilicity of ionisable compound
logP = measure lipophilicity Drugunionied
many drug are ionisable → exist as mixture of ionised & unionised
proportion of each form depend on pH ( vary across body )
ionise form is more water-soluble and less membrane-permeable = inaccurate prediction of drug behaviour
lipophilicity isn’t fixed for ionises → change w/pH
logD = measure lipophilicity of ionisable compound ( where partitioning is pH dependent )
→ non-ionise = log P = logD at all pH
→ ionise = logD change w/pH
P vs D ( lipophilicity )
P = true partition coefficient , when all drug is in unionised form
D = apparent partition coefficient , when AQ phase is ionise due to pH
pharmaceutical impotence of partitioning
logP help in formulation dosage from
→ balance of lipophilicity & hydrophilicity for drug candidate
→ predict of AQ solubility
→ adsorption to packing
logP help to predict ADME
→ lipphilicity = predictive of frug permeability across biological membrane ( → bioavailability)
logP help in formulation dosage from
partitioning affect preservative efficacy
→ microorganism grow in AQ phase
→ optimum in formulation , essential to resist microbial growth
( unionise form = bacterial penetration )
partitioning & preserving efficacy
conc. in AQ phase ( region of bacterial growth )
DE , if preservative partitions into immiscible oily phase
( unionise form = cross bacterial wall )
logP help to predict ADME
lipophilicity & drug behaviour in body ( major determinate of ADME )
bioactivity vs logP → parabolic relationship
fluid/tissue diff in pH , size , nature content
( lipophilicity can change over pH range )