Lysosomal storage diseases (LDLs)
1/10
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
11 Terms
What are the normal functions of lysosomes?
The lysosome is the cell’s primary recycling and degradation centre. Its mechanism is based on maintaining acidic pH via H+ pumps, and has around 60 different acidic hydrolyses that regulate the lysosome’s functions. Aside from this, it also regulates:
Nutrient sensing and energy metabolism
Cholesterol homeostasis and calcium signalling
The organelle also has Cl- ion channels, maintaining electroneutrality despite the acidic pH, and also has glycocalyx on its membrane to prevent it from digesting itself.
What are the different delivery routes to lysosomes?
Endocytosis: extracellular material is packed to become early endosome, maturing into late endosome before fusing with the lysosome
Macroautophagy: autophagosomes carry bulk biomolecules/damaged organelles
Microautophagy: direct imagination of biomolecules into the lysosome (no previous membrane-packing)
Chaperone-mediated autophagy (CMA): specific proteins are brought to the lysosome by chaperones and internalised via specialised transporters
Lysotrackers can be introduced in cells and end up in organelles with low pH, such as lysosomes, and can be visualised through fluorescence.
How are lysosomal enzymes synthesised and transported?
Lysosomal enzymes are synthesised at ribosomes in the ER
Mannose-6-Phosphate (M6P) tags are attached to the enzymes in the Golgi apparatus
M6P is recognised by M6P receptors (M6PRs) in the Trans-Golgi
Vesicles deliver the enzyme to late endosomes, where they fuse with lysosomes and the enzyme dissociates, as a repose to the pH change, while the M6PRs recycle back to the Golgi or plasma membrane
Additionally, small amounts of enzymes are secreted constitutively. M6PRs on the plasma membrane can capture these and bring them back inside for transport to lysosomes
General principles of LSDs.
LSDs are monogenic disorders, where one gene involved in lysosomal homeostasis can cause disease, and mostly autosomal recessive (3 are X-linked).
Some genes also affect several organs, thus making LSDs clinically heterogeneous.
Typical onset is infancy or childhood; adult-onset forms also occur, but often with milder symptoms
The pathology of LSDs are where deficiencies in lysosomal enzymes or transporters result in malfunction of the lysosome, leading to the accumulation of non-degraded substrates.
As a result, there’s an expansion of lysosomal systems due to the accumulation, which is a major hallmark of LSDs.
Substrate accumulation leads to additional organelle dysfunction (mitochondria, ER, Golgi) and triggers inflammation (macrophage/microglial activation).
Can result in complete cellular dysfunction and eventual cell death.
Treatment options vary, but many LSDs still lack effective disease-modifying treatments
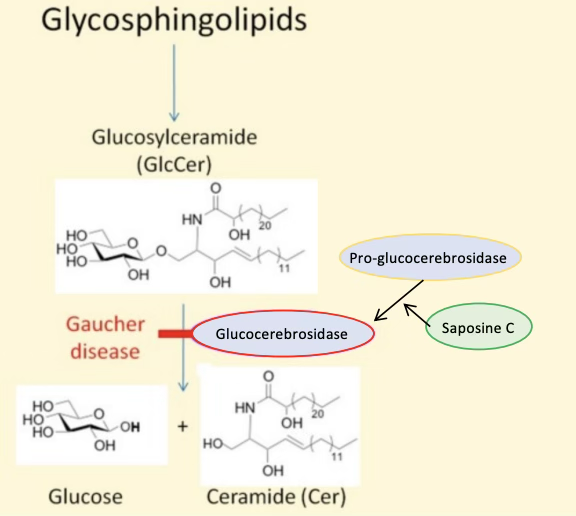
Gaucher?
Caused by mutations glucocerebrosidase or its activator, saposin C, which are enzymes that normally conjugate Glucosylceramide into glucose and ceramide
Results in accumulation of glycosylceramide in macrophages
Macrophages infiltrate bone marrow, spleen, and liver
Symptoms vary, but can include enlargement of spleen and liver, anemia, and bone necrosis due to vascular compression in the bone
Fabry?
X-linked LSD, defecting lipid storage enzyme and resulting in buildup of fatty materials throughout the body
Symptoms include burning paining hands/feet, renal/cardiac issues, and blindness
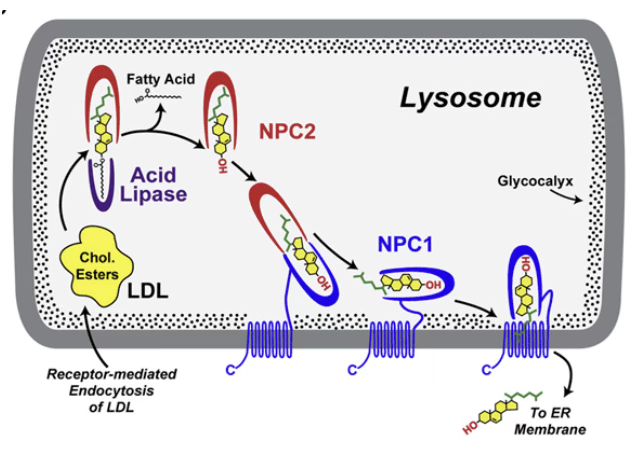
Niemann-Pick C (NPC)?
Caused by mutations in NPC1 or NPC2 coding genes
These are proteins involved in cholesterol distribution in cells, where, in the lysosome, NPC2 mediates the release of cholesterol from LDL, and NPC1 mediates the transport of cholesterol out of the organelle
Defect in these proteins lead to accumulation of cholesterol, which can also cause enlargement of spleen/liver, lung issues, and accumulation in neurones leads to dementia: nicknamed “Children’s Alzheimer’s” as post-mortem brains from children histologically look similar to Alzheimer’s
Neuronal ceroid lipofuscinoses (NCL)?
Generally, lipofuscin is an undegradable auto-fluorescent material that accumulates progressively in post-mitotic cells, and accumulates with age, commonly detected in autopsies of elderly people
NCL is the accumulation in neurones specifically, leading to neurodegeneration, with progressive symptoms resulting in dementia
The symptoms appear at about 1 year of age and death occurs at around 3-8 years
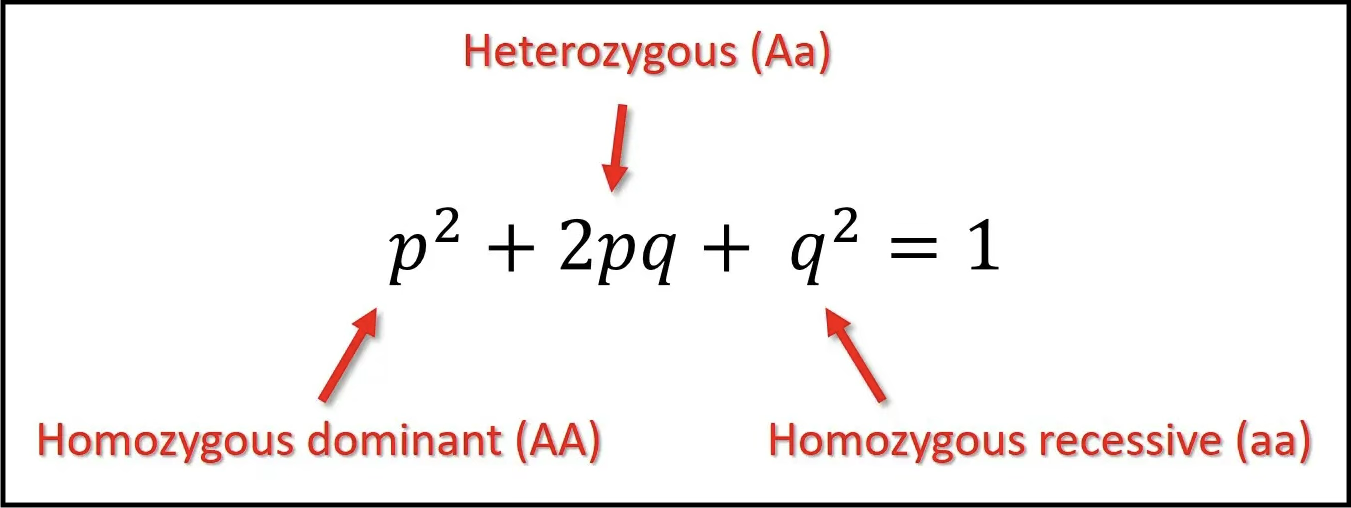
Hardy-Weinberg principle?
An individual has two alleles and can thus be AA, Aa or aa. To calculate the risk of autosomal recessive diseases, the frequency of different genotypes must be known. Hardy-Weinberg's law:
p = frequency of one allele, A
q = frequency of the other allele, a
Used to calculate the frequency of carriers (2pq) in a population.
p + q = 1
p2 + 2pq + q2 = 1
Gaucher example (if prevalence (q2) is 1 in 100,000:
q2 = 1/100,00 —> q = √1/100,000 = 0.003
Thus, p = around 1 (1 - 0.003 = 0.997), and p2 = 0.997
2pq = 2 (1) (0.003) = 0.006
Therefore, the chances of being a carrier of Gaucher is 1 in 6000.
What are the different treatments for LSDs?
Enzyme Replacement therapy
Recombinant M6P-tagged enzymes are infused and taken up by M6P receptors
Effective for Gaucher and Fabry
Disadvantages: cannot cross the BBB, so not accessible to the brain; high cost due to lifetime therapy; potential antibody formation against it
Bone marrow/Stem cell transplant
Healthy donor cells secrete normal enzymes that neighbouring deficient cells pick up
Standard care for Hurler syndrome, but must be done before 9 months of age for neurological benefits
Disadvantages: high morbidity, a problem with immunosuppressive drugs; high mortality; low success for other LSDs
Gene Therapy
Uses viral vectors (like Lentivirus) to deliver a functional gene
One-time treatment, potential cure, would improve quality of life
Risks of insertional mutagenesis and immune response
Other therapies
Substrate reduction therapy (SRT): slows down the production of the substrate that the body cannot break down
Enzyme enhancement therapy: use of pharmacological chaperones to stabilise mutated proteins
Why is it difficult to diagnose LSDs?
Can be an issue for clinicians as it’s difficult to interpret symptoms correctly as there are differences in clinical manifestations from patient to patient. Symptoms are progressive, and children appear normal at birth, making it more difficult. Most LSDs have no cures.