Heme #2 Exam #2
1/184
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
185 Terms
CYTOGENETICS
cytogenetics
-study of the structure of chromosomes an the relationship between chromosome abnormalities and diseases states
-congenital: effects every cell in the body
-mosaic abnormalities are only sen in some cells or tissues, depending on when it happened in embryonic development
-acquired: after birth where a stem cell becomes genetically abnormal
abnormal cells often undergo functional changes that improve their ability to survive and reproduce like:
-loss of contant inhibition
-loss of apoptotic potential
-increased growth factor recognition
-loss of tumor supressor gene expression
-increased cell cycle progression
Chromosome basics
-46 in normal cells (23 homologous pairs)
-22 sets are autosomal
-2 sex chromosomes (XX or XY)
centromere
-heterochromatin, very repetitive sequences
-although repetitive, each centromere has its own unique sequence
-kinetochore forms at the centromere during cell division and is the attachement point for spindle fibers
-sister chromatids join at the centromere
-chromosomal segments without centromeres are lost during cell division
telomere
-tip of the chromosome
-protective cap to prevent degredation
-composed of repetitive TTAGGG sequences
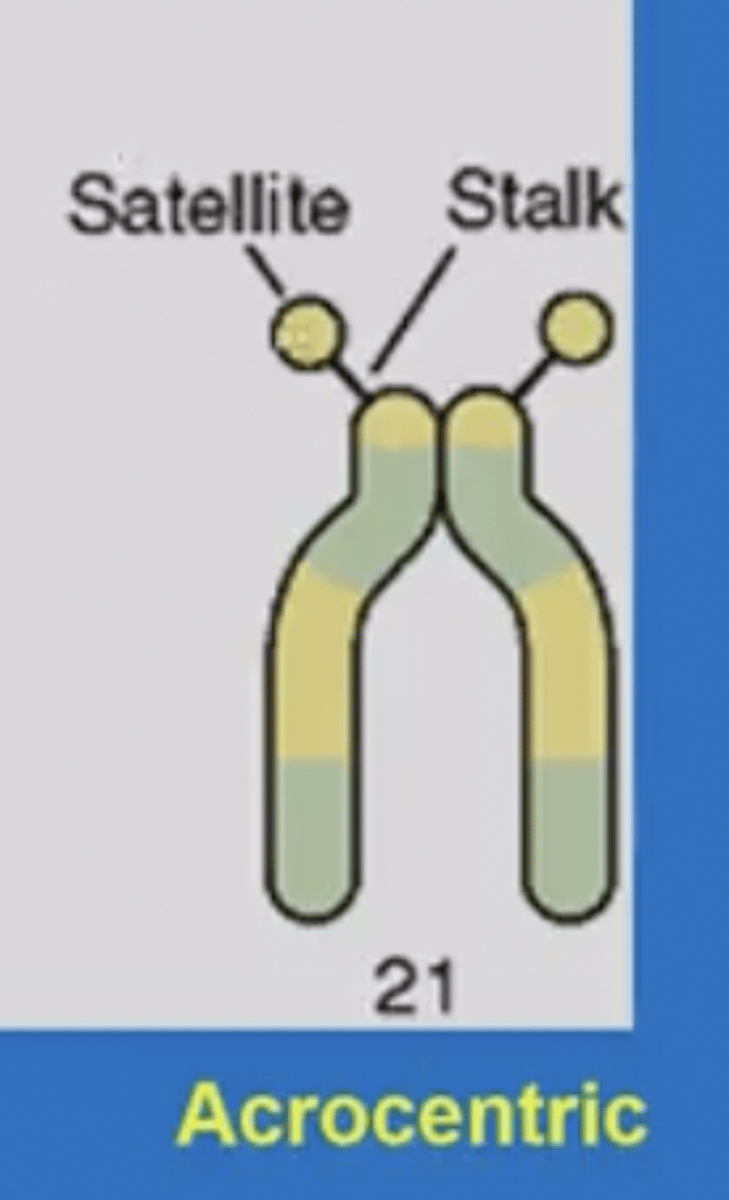
satellite chromosome region
-only in chromosomes 13-15, 21,22
-small dots at the tip of the P arm
-contains coding regions for rRNA
-assists in building nucleioli
-p arms are above centromere (short)
-q arms below centromere (longer)
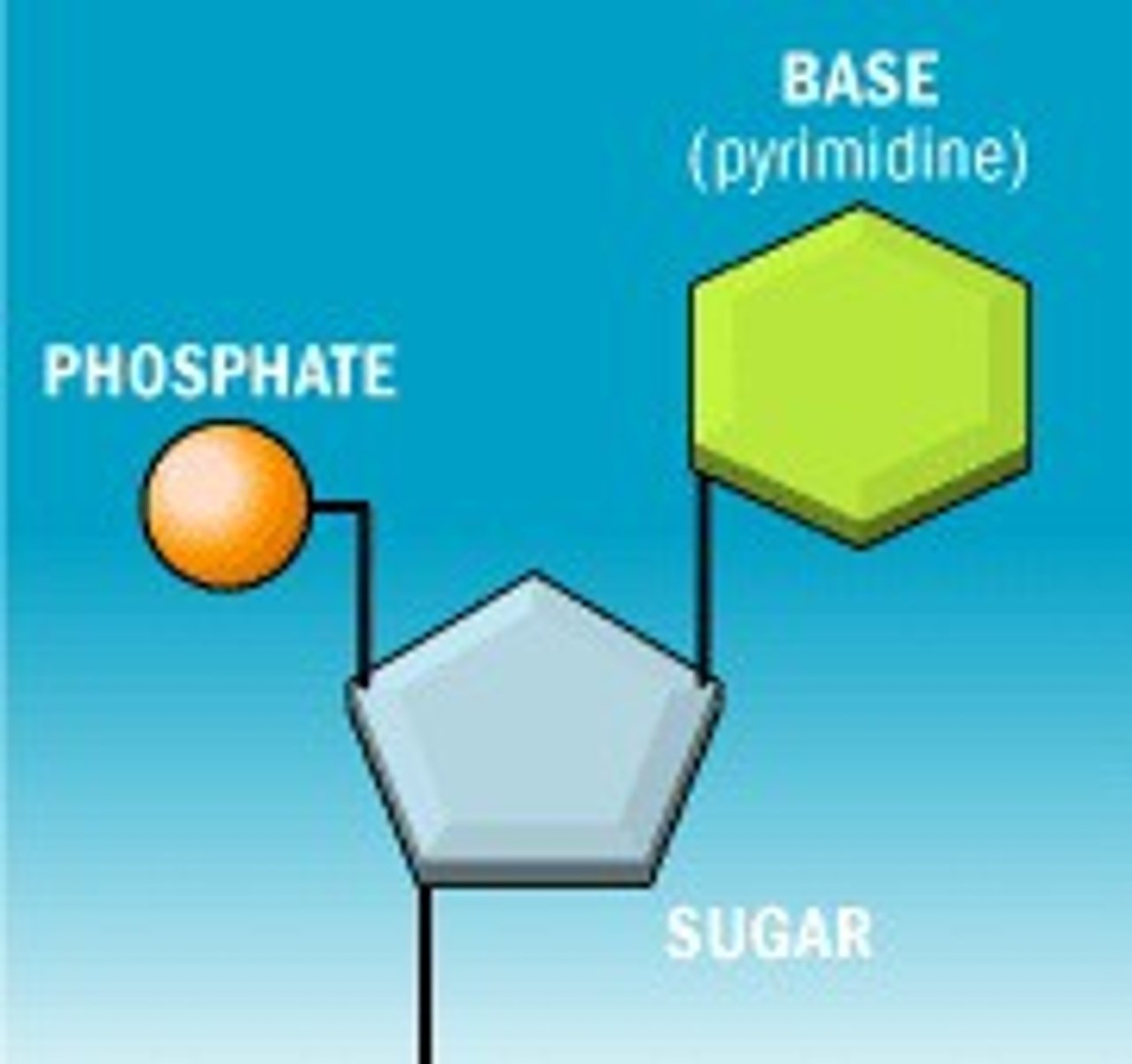
nucleotides
-Basic units of DNA molecule
COMPONENTS:
1) 1/4 nitrogenous bases (ACTG)
2) phosphate group
3) 5 carbon sugar (ribose)
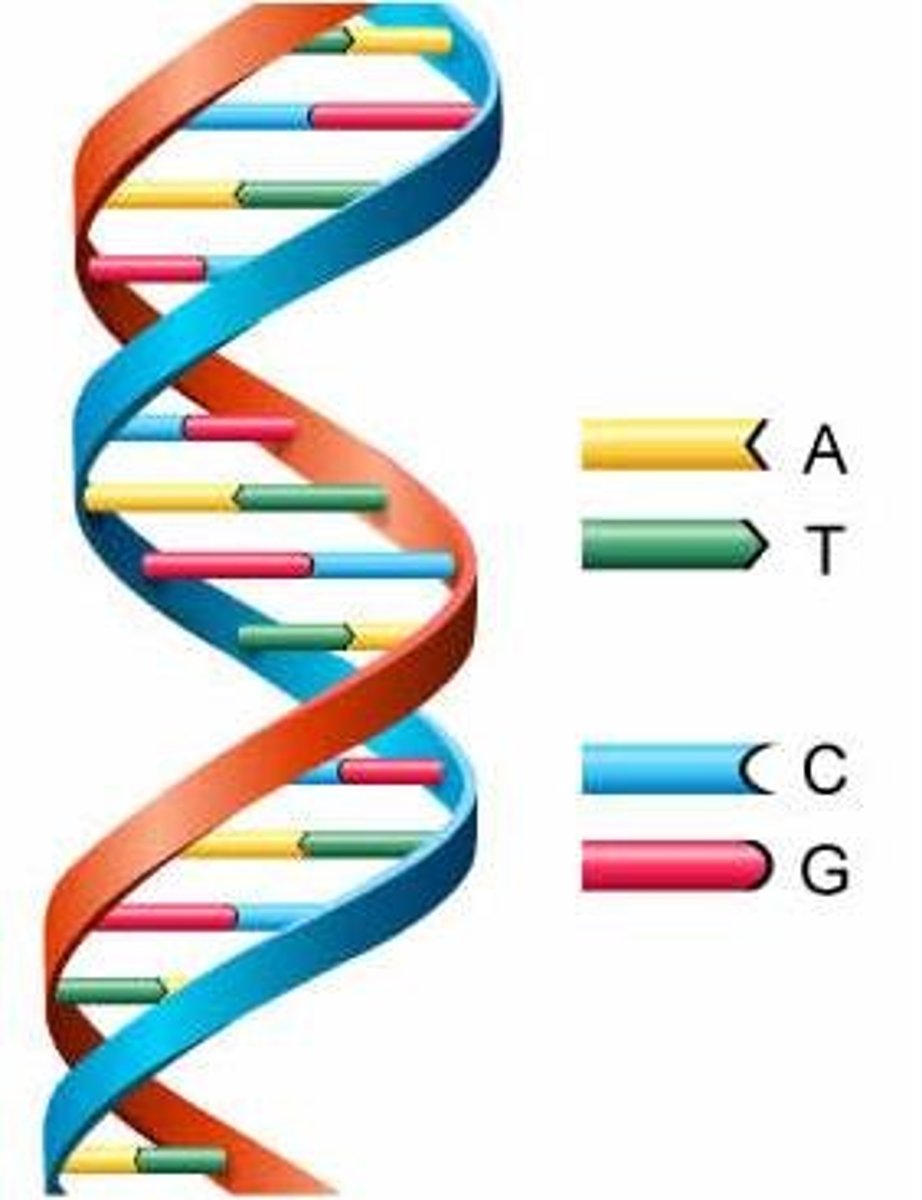
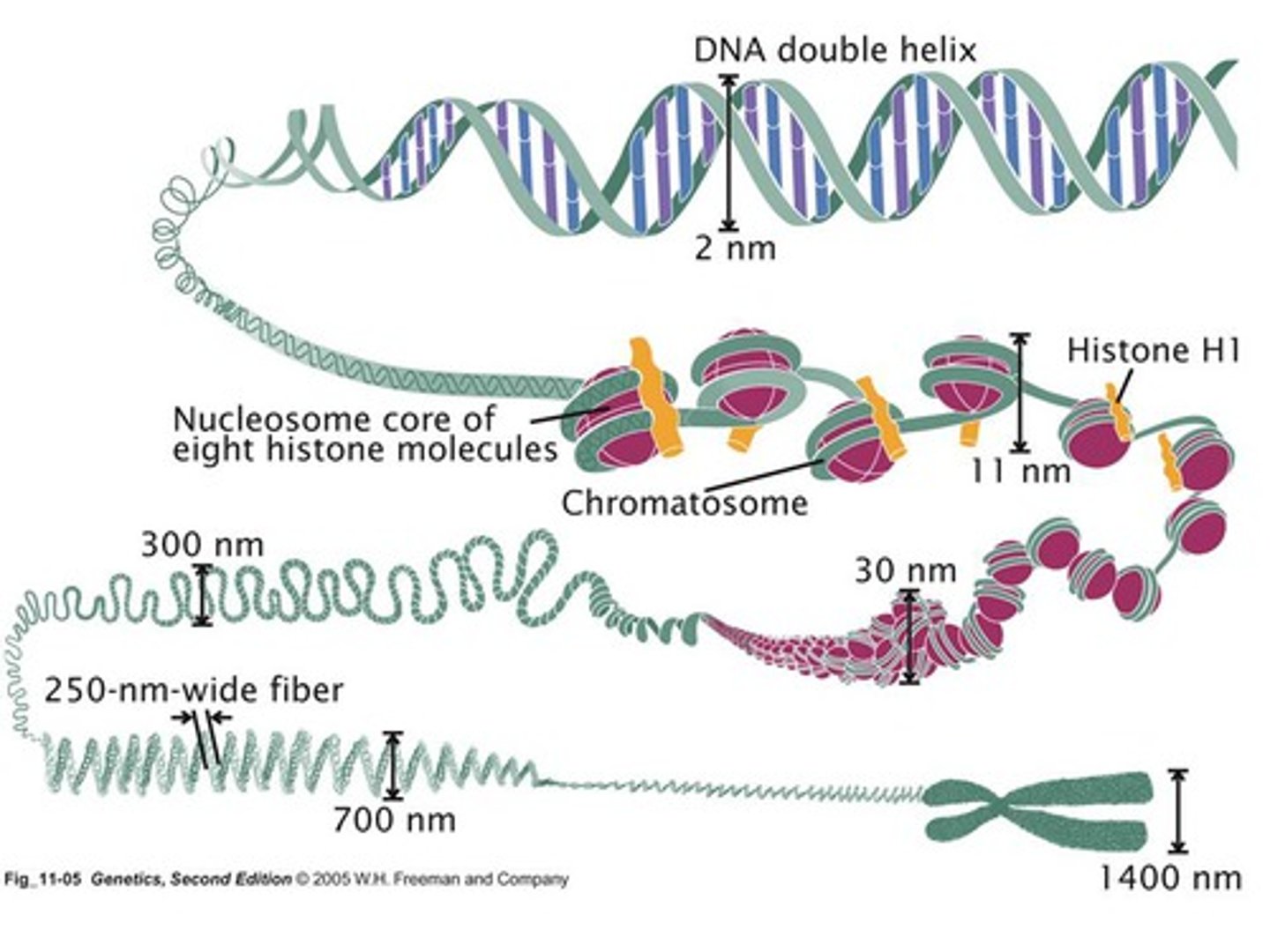
double-helix
Shape of DNA
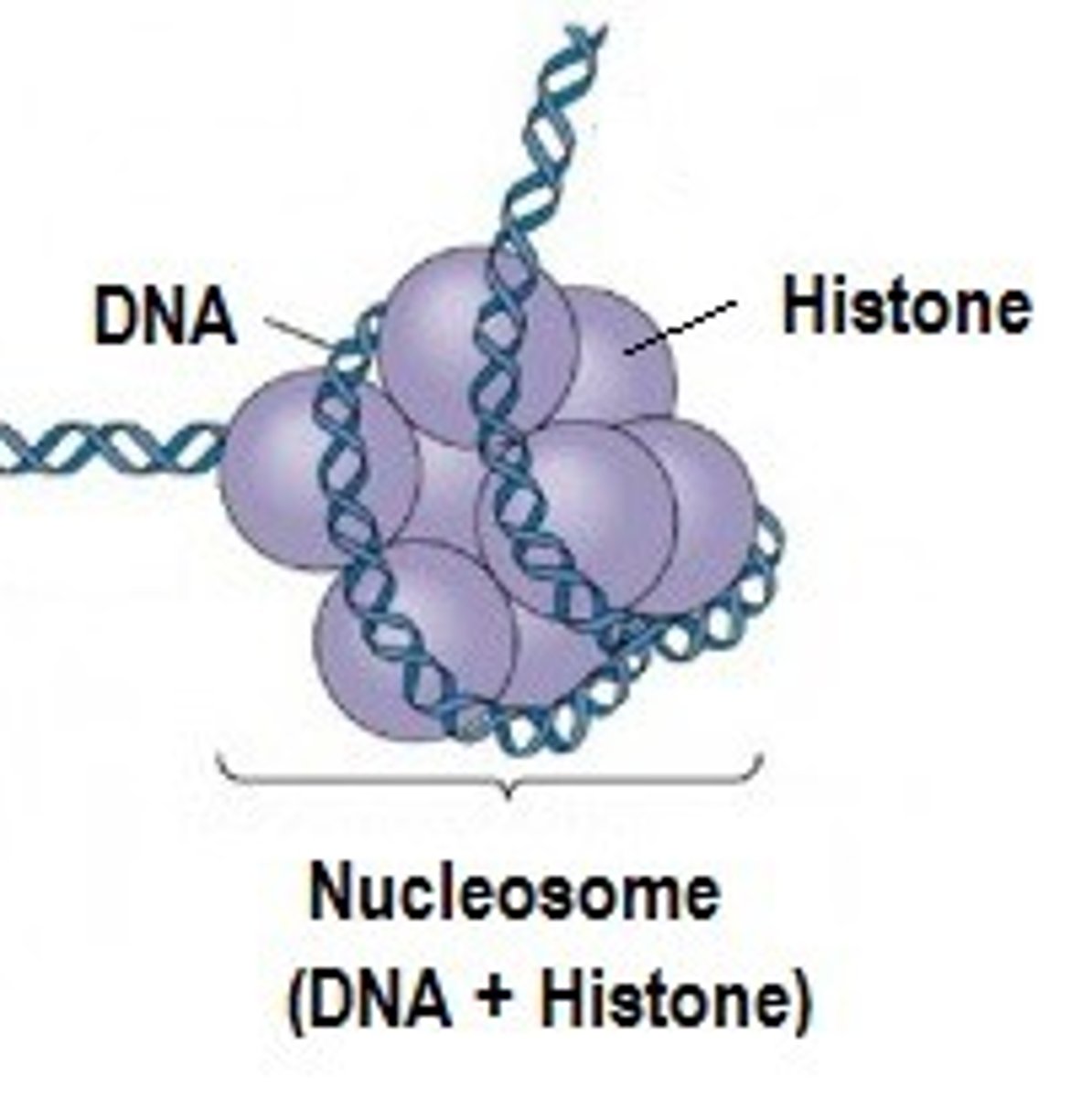
nucleosome
-DNA coiled around histones
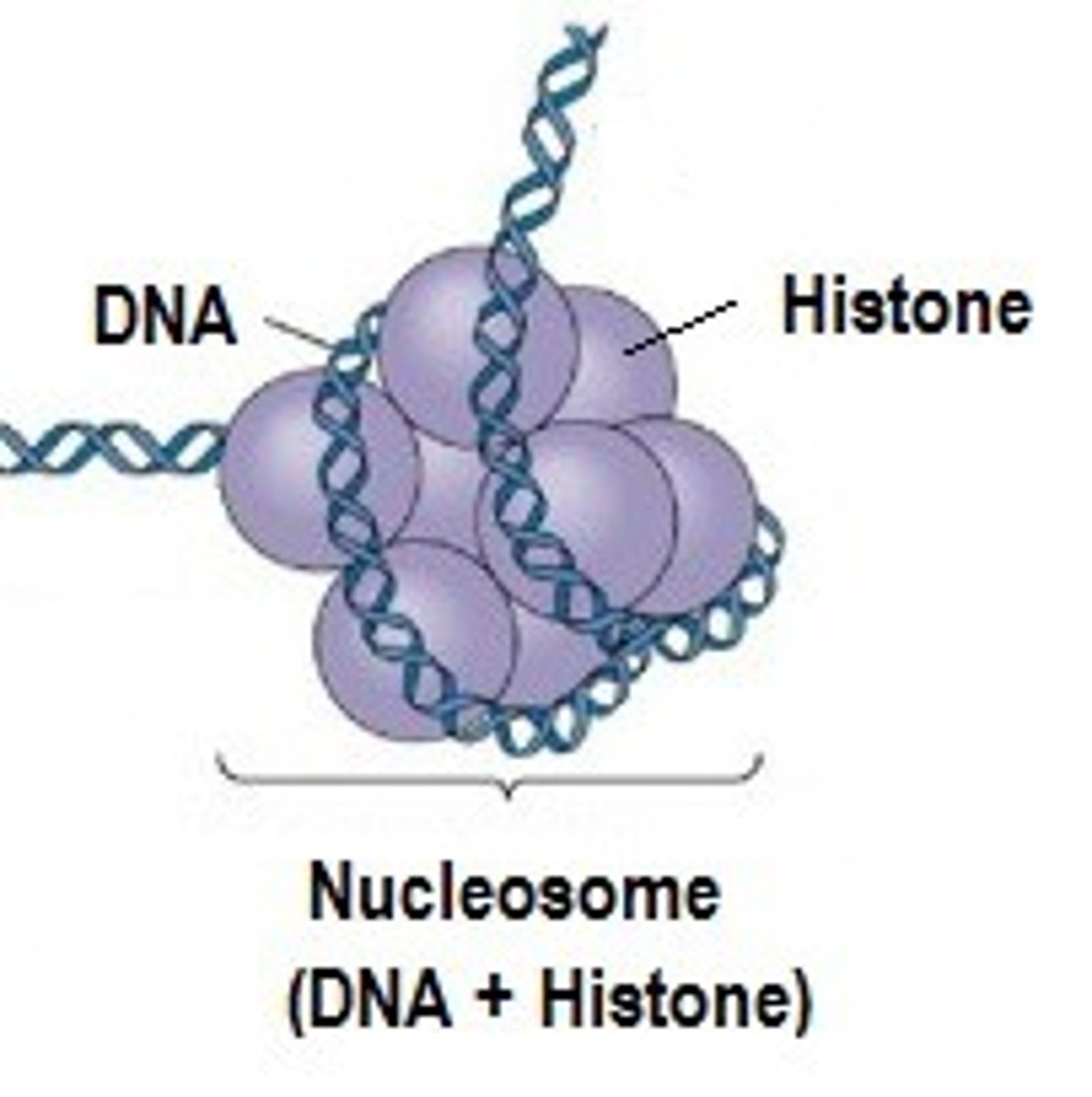
histone
protein molecule around which DNA is tightly coiled in chromatin
gene
chromatin
-nucelosomes twist and wrap to become chromatin
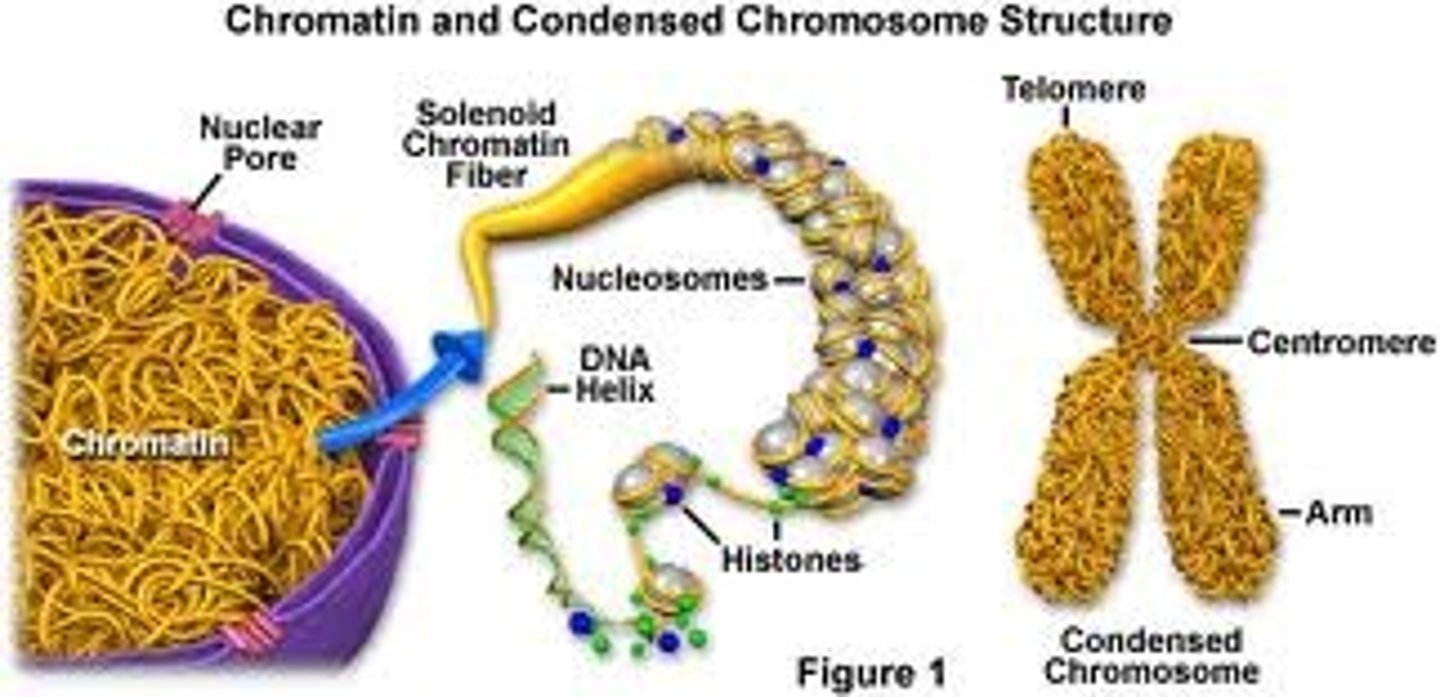
chromosome
-nucleic acids and protein together
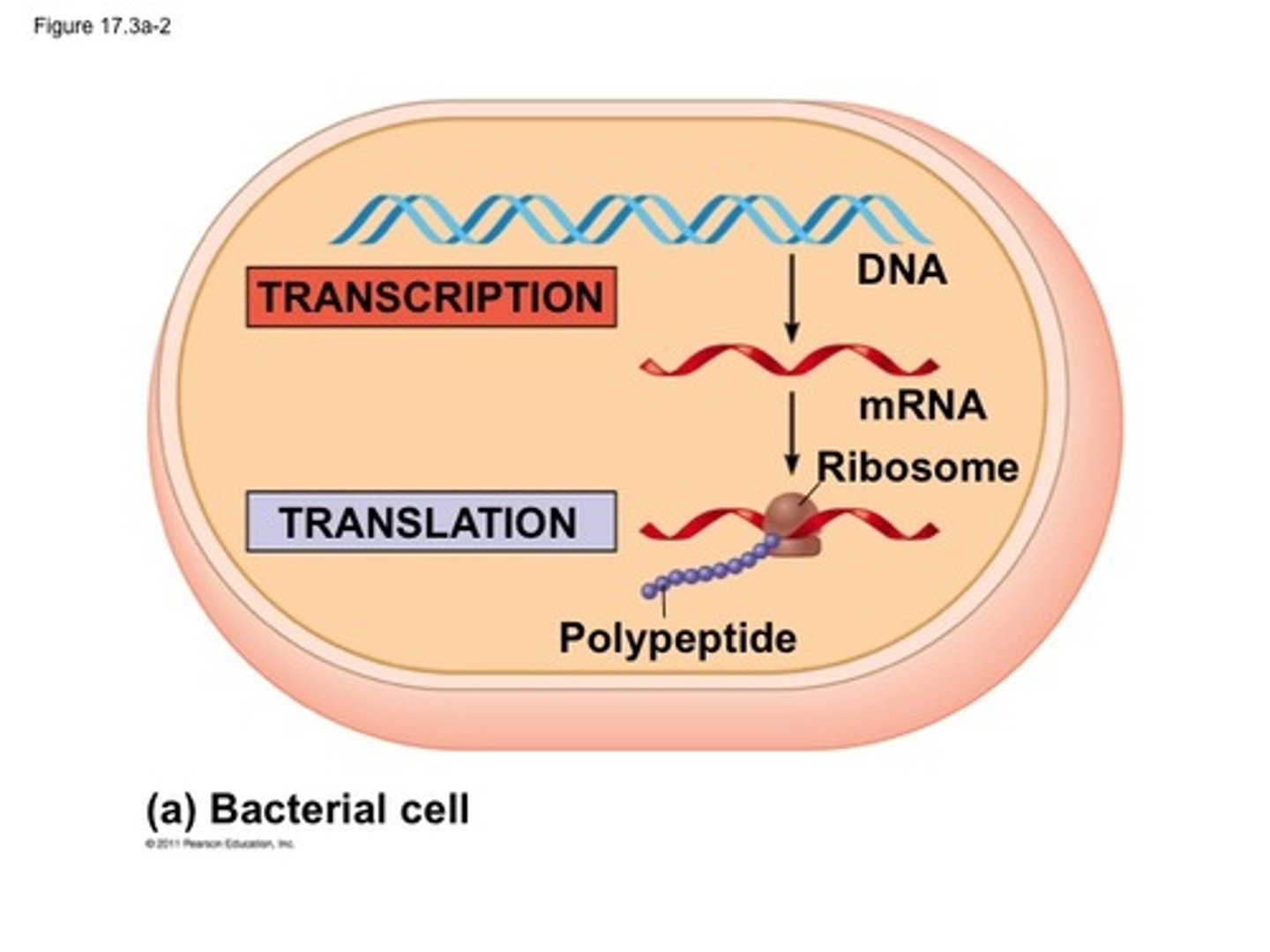
transcription vs translation
DNA --> mRNA in nucleus is transcription
mRNA --> tRNA --> AA --> protein is translation
euchromatic regions vs heterochromatic regions
EUCHROMATIN:
-lightly staining because loosely coiled
-active gene transcription
-rich in GC nucleotides
HETERCHROMATIN:
-darkly staining with G-banding because tightly coiled
-low active gene #
-rich in AT nucleotides
metacentric
-centromere in middle
-equal P and Q

sub-metacentric
-centromere off center
-shorter P than Q

acrocentric
-centromere close to end
-forms satelleties

deletion
-loss of material from chromosome
-interstitial: within an arm, no telomere involvement, requires 2 breaks
-terminal: involves telomere, requires 1 break
duplication
-exact segment is present twice usually right next to each other
translocation
-exchange of material betwen 2 chromosomes
-Balanced/reciprocal: 2 chromsomes exchange a segment
-Unbalanced: segment of one is moved to another without receiving something in return
-Robertsonian- fusion of two acrocentric Q arms
inversion
-segment of chromosome flipped within itself
-pericentric: includes centromere
-paracentric: no centromere included
ring chromosomes
-two breaks occur, one in each arm of the chromosome
-telomeres are lost and the remaining ends are fused
isochromosome
-break occurs just below the centromere so one chromosome is lost
-tips of remaining q arms fuse together and when chromosome lengthens again it is like a mirror image
aneuploidy
Abnormal number of chromosome (any deviation from 2n, not a multiple of n though so like +/- 1
polyploidy
-an extra set of chromosomes (multiples of n)
triploidy is 3n gaining an extra of ALL
hyperploidy
higher than 2n but not a mulitple of n (aneuploidy?)
hypoploidy
-lower than 2n but not a multiple of n
pseudoploid
cell with 46 chromosomes but not normal (46, XX, -7, +21 i think means missing a 7 but an extra 21)
trisomy
gain of an extra chromosome (1,1 --> 1,1,1,)
nomenclature
chromosome # --> arm --> region --> band --> sub-band
monosomy
loss of chromosome (1,1 --> 1)
Nomenclature Rules
1. total # of chromosomes COMMA
2. sex chromosome complement (XX, XY, XXY, etc.) COMMA
3. parenthesis are around chromsome # and breakpoints of abnormal chromsomes
4. if sex chromsome is abnormal, put the normal one first, follow that with a comma, and description of the abnormal sex chromsome
EX: 46, X, del (X) (p22)
5. it autosome is abnormal, follow sex chromosome naming with a common and then follow that with description of the autosomal abnormality
EX: 46, XX, del(5)(q13q33)
EX: 48, XY, +8, +21
EX: 46,XY,t(9;22)(q34;q11.2)
EX: 46,XX,inv(16)(p13.1q22)
6. when describing single abnormal chromosomes, no puncutation should separate the breakpoints or any other part of description
7. semicolons are used to seprate the chromosome and the breakpoints of a translocation since you are describing 2+ chromosomes
ABBREVIATIONS:
-del = deletion
-one breakpoint is used if terminal
-two breakpoint is used if interstitial
-inv = inversion, requires 2 breakpoints
-t = translocation
-dup = duplication
-i = isochromosome (breakpoint is at center of the mirror image)
-r = ring chromosome
-mar = marker chromosome
cell cycle
1. Interphase
-G1 (growth and metabolism)
-S (DNA synthesis)
-G2 (prepare for cell division)
2. Mitosis
P - chromatin condenses, nuclear membrane degrades
M - chrosomes move to equator
A - sister chromatids pulled apart by spindle fibers
T - nuclear membrane reforms
3. Cytokinesis
-cell membrane reforms around organelles and new DNA
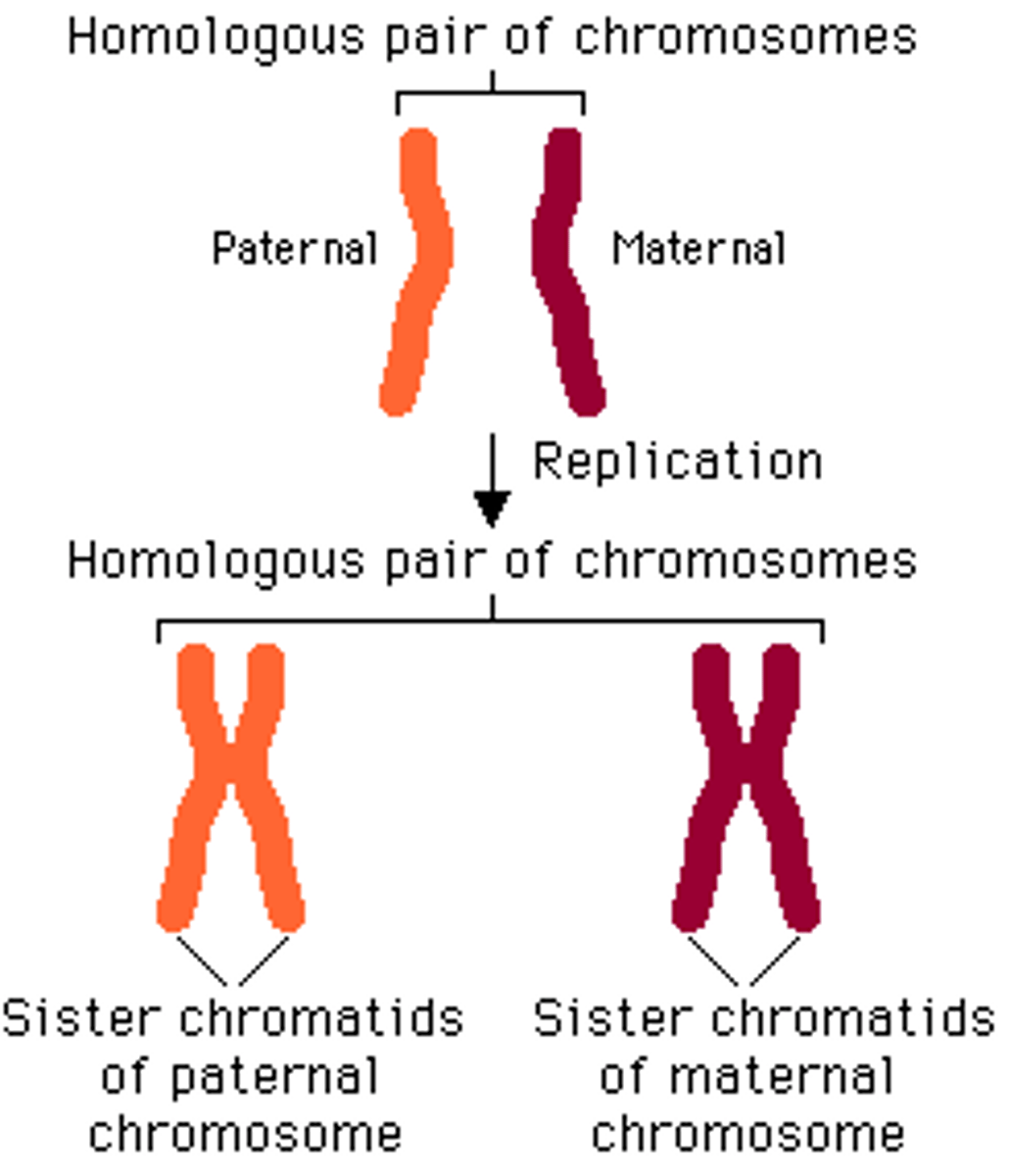
homologous chromosomes
Pair of chromosomes that are the same size, same appearance and same genes.
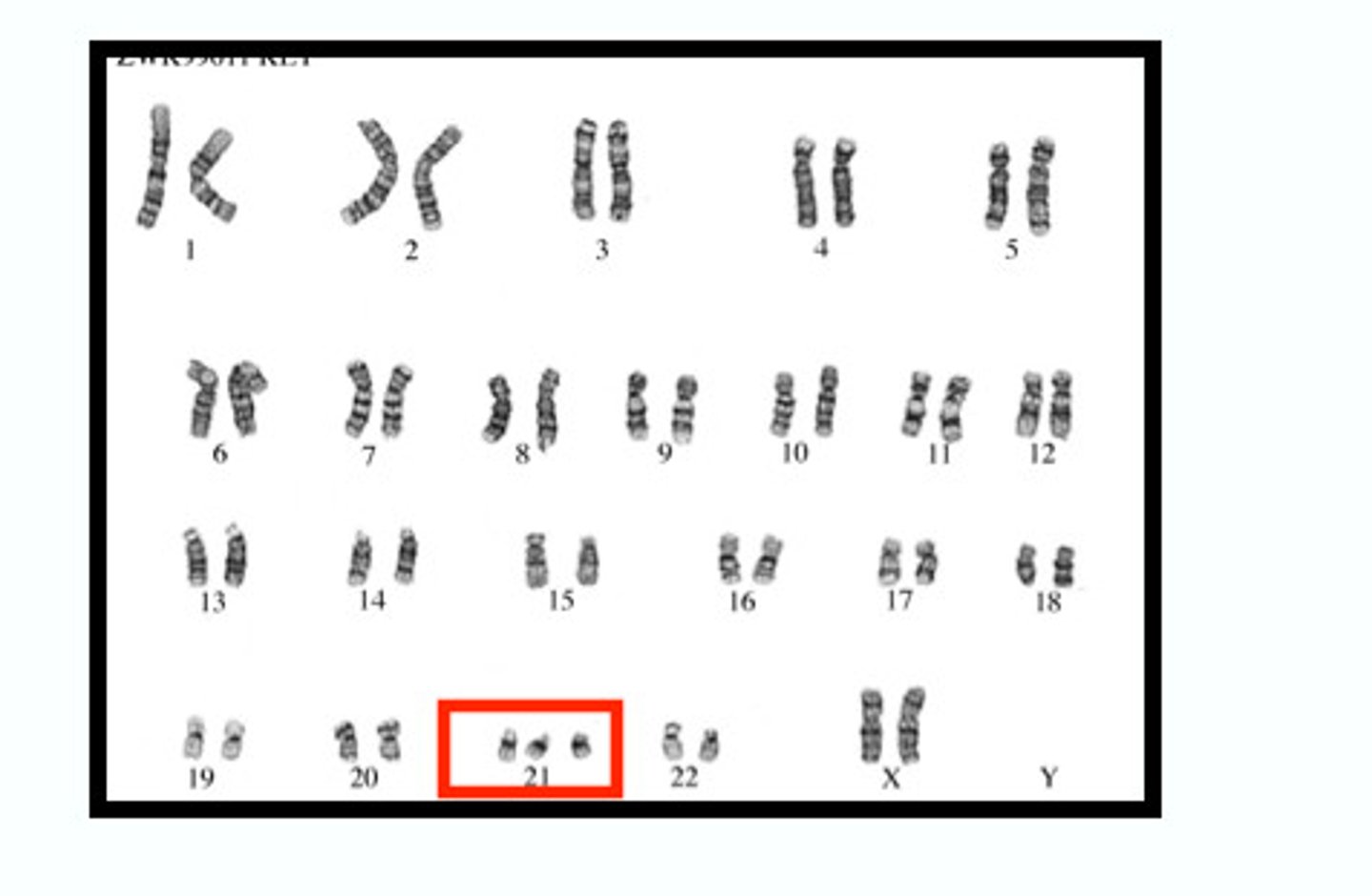
specimens used in cytogenetics
-blood for constitutional and acquired abnormalities
-bone marrow and lymph nodes - acquired only
-amniocytes and chorionic villi
-productions of conception (miscarriage)
-tumors -acquired
-CSF - acquired
-sodium heparin is best for anticoagulant!!
-PHA added to promote division of T-lymphs
-colcemoid added to arrest cells in metaphase
-hypotonic solution added to increased cell volume
-centrifuged and supernatant is drawn out
-fixitive added
-fixative repeated after centrifuging and supernanat removed again
-pellet is then resuspended and a drop of specimen is put on the slide and allowed to dry (too fast, overlap, too flow =overspread)
-use phase micrscopy to see
FISH
-fluorescent in situ hybridization
-molecular cytogenetics
-uses fluorescent dye molecules bound to DNA and then hybridized to patient DNA
ADVANTAGES:
-metaphase or interphase cells
-identifies very small anomalies
-does not require living cells, just intact DNA
-larger sample size since analysis is less time consuming
DISADVANTAGES:
-longer analysis and turn around time?
-limited to regions where the probes hybridize
-results can be misleading if done wrong
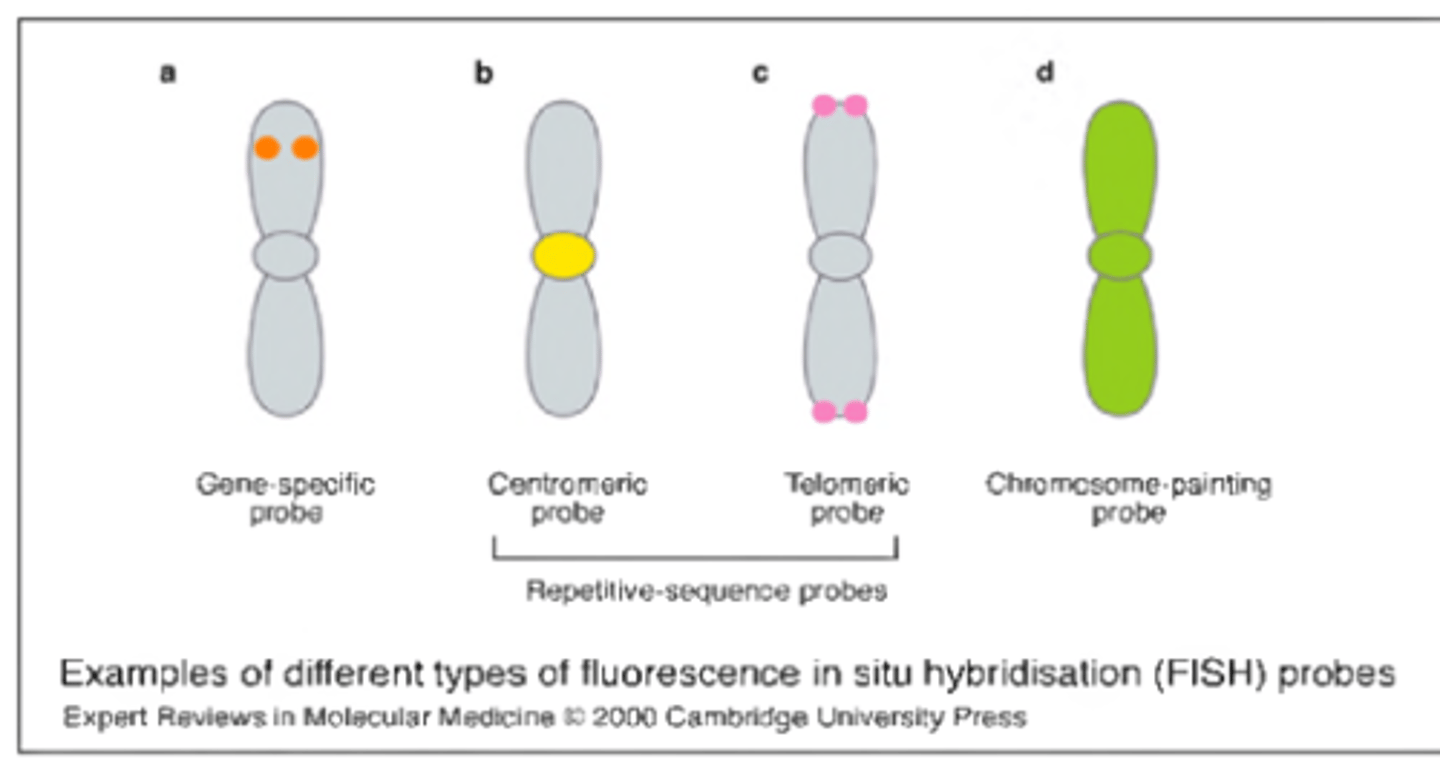
types of FISH probes
-centromere: hybridize to the alpha satellite region of chromsome
-locus-specific: hybridize to specific euchromatic sequence (identifies deletions, translocations, inversions)
-whole-chromosome paints: for a specific chromosome to identify chromsome segments of unknown origin
-sub-telomere: hybridize sequences just proximal to the actual telomeres
-multi-color paints (M-FISH): allow for a karyotype that depicts each autosomal pair and sex chromosomes in own color!
BCR-ABL protein in CML
-production of a hybrid/chimeric protein
-chronic myeloid leukemia
-BCR-ABL protein in CML behaves very different from the normal tyrosine kinase that is encoded by the ABL region
**meaning it never becomes INactive (always active)
**inhibits apoptosis
**becomes less adhesive so released early from BM
-on chromsomes 9 and 22
Chronic Myeloid Leukemia Lesson #11
myeloproliferative neoplasm definition
-acquired hematopoietic neoplasms that have unregulated differentiation and proliferation of stem cells
-in BM and PBS
-affect all 3 cell lines
4 main classifications of MPNs + 3 extra for WHO
GENERAL:
1 - chronic myeloid leukemia (chronic myelogenous leukemia)
2 - primary myelofibrosis
3 - polycythemia vera
4 - essential thrombocythemia
WHO ADDS:
1 - chronic neutrophilic leukemia (CNL)
2 - chronic eosinophilic leukemia (hypereosinophilic syndrome) (CEL/HES)
3 - myeloproliferative neoplasm unclassifiable (MPN-u)
main features of MPNs
-gradual onset
-middle age, older adults (but can happen in younger)
CLINICAL:
-hemorrhage
-thrombosis
-infection
-pallor
-weakness
HEMATOLOGICAL:
-anemia or polycythemia
-LE picture
-leukocytosis
-basophilia
-thrombocytosis (maybe bizarre too)
BONE MARROW:
-hypercellular
-eventually becomes fibrotic with increased reticulin fibers
-when this happens, hematopoiesis moves extramedullary to spleen or liver
Chronic Myeloid Leukemia (CML, one of the 4 main MPNs)
-uncontrolled proliferation of cells mainly granulocytes
-20% of all leukemias
-middle aged usually, peaking 40-60
3 Phases of CML
chronic
accelerated/intermediate transitional/aggressive
blast
chronic phase of CML
-usually presents in this phase
-splenomegaly and leukocytosis
-abdominal pain due to enlarged spleen is important
-left shift with MYELOCYTE BULDGE and basophilia
-few symptoms, usually seen during general exam
-arthritis bc of gout can occur
-controlled with medication regulating fatigue and complications like leukocytosis, splenomegaly, anemia
-lasts 2-3 years but can last long with new treatments
Accelerated phase of CML
-aka intermediate transitional or aggressive phase
-AGGRESSIVE
-10-19% myeloblasts in PBS or BM
->20% basophils in PBS
-lasts 1-1.5 years
-harder to control
-symptoms add bone pain and fever
-additional chromosome abnormalities appear
-medication response is poor resulting in more blasts, promyelocytes, and basophils, low platelets, uncontrollable splenomegaly
Blast Crisis phase of CML (acute leukemia phase)
-BM has >30 or >20% blasts depending on FAB or WHO
-skin or tissue infiltration (extramedullary)
-most blasts are myeloid but 1/3 cases can have lymphoid phenotype
-extreme basophilia and thrombocytosis
-additional chromosome abnormalities are found
-hard to treat, lasts 3-6 months
PBS in CML
-leukocytosis with LEFT SHIFT
-increased in all granulocyte precursors including blasts
-promyelocytes and blasts are usually less than 10% though
-absolute eosinophilia or basophilia
-N/N mild anemia
-platelet counts can be L or N or H
BM in CML
-90-100% hypercellular because of proliferating myeloid precursors
-ME ratio can be 10-100:1
-less than 20% blasts
-Auer rods are unusual to see unless in blast crisis phase
-reticulin stain can show mild fibrosis (where BM elements are replaced with connective tissue)
-megakaryocytes prominant
Chromosome Abnormality in CML
-PHILADELPHIA CHROMSOME (gold standard)
-reciprocal translation between chromosome 9 and 22
-t(9;22)(q34;q11)
-this translocation relocates the oncogene ABL from 9 to 22 in the bcr region
-results in 9 being longer than usualy
-results in 22 being shorter than normal (PHILADELPHIA)
-acquired
-BCR/ABL fused gene can be see with FISH or molecular testing
-you need to see this chromosome abnormality to call it!
-Philadelphia then codes for an abnormal protein called p210 that enhances tyrosine kinase activity which very much supresses apoptosis and increases cell production!
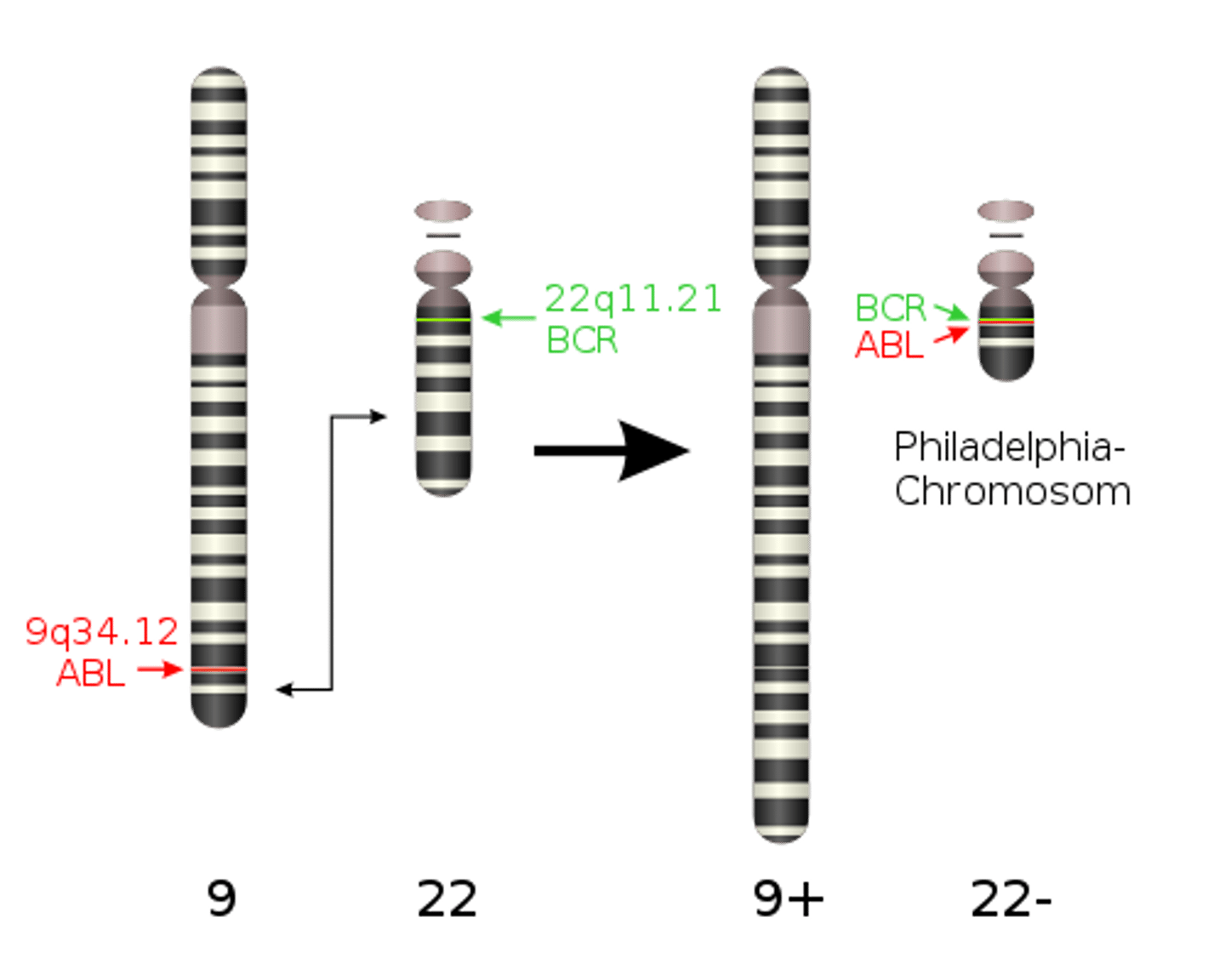
Fish Testing in CML
-test for Philadelphia chromosome
-fluorochrome labeled DNA probes for metaphase cells
-red probe for ABL on chromosome 9
-green probe for BCR on chromosome 22
-IF +: red and green probe are right next to each other (called yellowish fusion signal)
In 95% of CML patients is the philadelphia chromosome, the other 5% have the bcr/abl translocation.
IF NO PHILADELPHIA: they will have poor response to therapy, no basophilia, thrombocytopenia, short survival
LAP score for CML
-Leukocyte Alkaline Phosphatase found in granules of neutrophils segmented and band
-LOW LAP SCORE bc many immature neutrophils which have lower scores
-CML < 40!! (help rule of PV and other MPNs)
treatment for CML
-goal to eliminate all cells containing philadelphia chromosome
-treatments vary by age, BM donor availability, etc.
-Gleevec-imatinib mesylate is treatment of choice now
-allogenic BM transplant after
GIM: improves duration of chronic phase bc it's a tyrosine kinase inhibitor to slow growth, inhibit proliferation, and induce cell death
ALLOGENIC BM TRANSPLANT: transplant is the only known cure for CML usually done in <55 year olds. high mortality rate with this though
leukocyte apheresis can lower WBC if >300 where whole blood takes out the WBCs to temporarily reduce WBC
prognosis of CML
-highly responsive to treatment in the chronic phase
-survival in accelerator phase <1 year
-survival in blast phase ~few months
DETERMINED BY:
-age, symptoms, splenomegaly, anemia, negative philadelphia chromosome, high or low platelets, low megakaryocytes, basophilia, myelofibrosis
-short duration of remission, longer time to reach remission, poor suppression of philadelphia-positive cells by chemo
Chronic Neutrophilic Leukemia
-rare MPN
-leukocytosis WITHOUT immature granulocytes
-elevated neutrophil count
-hypercellular BM, granulocytic hyperplasia
-philadelphia chromosome negative
-LAP increased
-need to rule out other causes of neutrophilia and CML
Chronic Eosinophilia Leukemia (hypereosinophilic syndrome)
-high eosinophils (>1.5), in BM, blood, tissues
-organ and tissue damage because of charcot-leyden crystals that form
-LAP normal
-philadelphia negative
-poor prognosis
-rule out other causes of high eosinophils
-look for signs of organ and tissue damage
LESSON 12: Polycythemia Vera
I love you! You did that other lesson in less than an hour :) do this one and then make cards for them all so far! then done for the day!!
Polycythemia is
increased RBCs
polycythemia vera is
-a MPN with unregulated increased of RBCs in the PBS and BM
-increase in RCM (Red Cell Mass), RBC count, or HGB
*could also have WBC and PLT increases too
relative vs absolute polycythemia
RELATIVE: decrease in plasma volume so more concentrated
ABSOLUTE: increase in red cell mass
3 groups of polycythemia
1. Polycythemia Vera: increase in RBC mass; unregulated production of RBCs due to a clonal hematopoietic stem cell disorder
2. Secondary Polycythemia: increased in RBC mass; explainable or apparent increase in RBCs (smoking, high altitudes)
3. Relative Polycythemia: decreased plasma volume with normal or low RBC mass (dehydration, burns, etc.)
pathophysiology of PV
-stem cell defect causing unregulated and accelerated erythropoiesis
-stem cells are sensitive and very responsive to erythropoietin
Clinical findings of PV
-40-60 year olds
-more males than females
-gradual onset
-increased RBC mass leads to symptoms like blood thickening, headache, visual disturbances, weight loss, itchy skin, venous or arterial thrombosis
-splenomegaly and hepatomegaly with progression
-thrombosis or hemorrhage
-hypertension due to thickened blood
-platelet abnormalities possible
-plethora (red complexion) due to increased RBCs
-can lead to myelofibrosis or acute leukemia
PBS findings for PV
-RBC 6-10
-HGB >18
-hematocrit (>55%)
-increased RBC mass
-N/N cells
-reticulocytes could be slightly elevated
-leukocytosis (12-20) without fever or infection
-neutrophilia with mild left shift
-LAP >100
-thrombocytosis >400 (abnormal morphology and function too)
*if advanced, could also have
Myelofibrosis (LE anemia, dacrocytes)
Acute Leukemia (anemia, low plts, blasts)
BM findings for PV
-not necessary to diagnose PV but is common practice to diagnose
-hypercellular with lots of erythroid and myeloid precursors
-normoblasts may collect in large clusters
-megakaryocytes are increased and enlarged with lobulated nuclei
-fibrosis may be present so reticulin stain is helpful
-iron stores are reduced or absent because it's all being used to make all the RBCs
LOOK AT TABLE/WKSHT TO SEPARATE THE 3!!!
other lab findings in PV
-oxygen sat normal
-erythropoietin levels low bc the body sees all the cells being made, but this doesn't stop PV from making lots of cells still
-URIC ACID INCREASED (due to turnover of NAs from RBCs, could lead to gout)
-Vitamin B12 increased
-25-50% have cytogenetic abnormalities
-JAK2 gene mutation (95% have this!!!!!)
**it is common for PV to have IDA too so high RDW, microcytosis, elliptocytes even with high RBC/Hgb
***thalassemia also has high RBC but PLTs have normal morphology (here they are high and abnormal morph). Thal also doesn't have basophilia
PV JAK2 Gene Mutation
95% of patients have this so helps diagnose
Therapy for PV
-no cure but tx prolongs survival
-therapeutic phlebotomy is the 1st choice of tx though it reduces iron supply and blood volume
-myelosuppressive therapy can be used along chemo or radiation to extend quality and length of life
-untreated have survival 6-18 months are dx
-treatment can be >10 years
-thrombosis is a common complication and major caused of death in PV
-15-20% go to myelofibrosis
-15-20% go to acute leukemia
Secondary Polycythemia
-increased in RBC mass with no changes in other lines
CAUSES:
-increased erythropoietin bc of hypoxia, high altitudes, COPD
-tumors excreting erythropoietin
-familial polycythemia (high oxygen affinity hemoglobins so less o2 to tissues)
-neonatal polycythemia bc of intrauterine hypoxia
-defective o2 transport bc of smoking or pollution in environment
relative polycythemia
-dehydration, hemoconcentration or GAISBOCK'S syndrome
-high Hgb and Hct bc of low plasma volume
-STRESS erythropoiesis = Gaisbock's syndrome found in hypertensive, overweight nervous males that smoke and drink alcohol
-WBC, platelets, iron stores, O2 sat, and BM cellularity are all normal. cytogenetics normal too
PRIMARY MYELOFIBROSIS & ESSENTIAL THROMBOCYTHEMIA
Primary Myelofibrosis
-MPN with unregulated proliferation of hematopoietic cells AS WELL AS extramedullary hematopoiesis and systemic bone marrow fibrosis
*aka chronic idiopathic myelofibrosis, etc.
-though to happen when a defect or mutation occurs in the hematopoietic stem cells.
-all 3 cell lines CAN be affected by typically it is only two
-as BM becomes fibrotic, the liver and spleen take over hematopoiesis and become enlarged
clinical findings in myelofibrosis
-50-70 year olds
-gradual onset and can be asymptomatic at first
-then fatigue, weakness, bleeding/bruising, night sweats, extremity pain, bone pain, pain in ULQ because of enlarged spleen, gout, renal stones
-
lab findings in primary myelofibrosis
-LE picture
-N/N anemia with DACROCYTES, nRBCs, anisocytosis, polychromasia, basophilic stippling
-WBC can be variable with some blasts, immature granulocytes, and maybe high baso and eos
-LAP score normal (maybe slightly elevated)
-NO PHILADELPHIA
-platelets variable, could be atypical, bizzare, large, hypogranular
-low PLTs come with disease progression, could see circulating micromegakaryocytes (those cells with platelets blebbing off looks just like a nucleus!!!)
-could become panctyopenic with progression
BM findings in primary myelofibrosis
-fibrosis in the BM makes aspirations hard and result in a dry tap (aspirate with no units)
-hypercellularity and fibrosis
-clusters of large, atypical megakaryocytes may be seen
Reticulin Stain
-demonstrates reticulin fibrosis in MPNs and HairyCellLeuk
-reticulin fibers stain black and it is graded
0 = no fibers
1 = occasional
2 = fibers throughout most, no coarse
3 = diffuse fiber netweork with ropy coarse fibers, no mature collagen
4 = diffuse, coarse fiber with some collagenization
other lab findings in PMF
-alkaline phosphatase increased
-lactate dehydrogenase increased
-uric acid elevated (like in PV)
-B12 elevated (like in PV)
Prognosis/Therpay for PMF
-no cure, survival 4-5 years
-worse prognosis of all MPNs
-infection, thrombosis, hemorrhage are common causes of death
-10-15% go to an acute leukemia
-Tx reduces symptoms so like blood transfusions for anemia, androgens/corticosteroids, chemo to reduce spleen size and fibrosis, radiation to reduce spleen size, thalidomide to reduce spleen size, splenectomy, stem cell transplant (allogenic)
-MYELPHTHISIC anemia is similar and should be ruled out (breast cancer specifically i remember about this)
DIAGNOSE:
-fibrosis >1/3 of BM
-splenomegaly
-LE picture
-no increased RBC mass
-no phil + chromosome
Essential Thrombocythemia (ET)
-affects megakaryocytes
-PBS shows platelets >1000
-thrombosis and hemorrhage are common
-can affect all 3 cell lines though, platelets are just more affected
clinical findings in ET
-50-60 year olds
-bleeding or bruising/nose bleeds
-thrombosis
-slight splenomegaly
-headache, dizziness
-weight loss
lab findings in ET
-PLT >600, usually >1000
-PLTs are mostly normal looking but some giant or hypogranular are possible
-might see megakaryocyte fragments
-ABNORMALLY FUNCTIONING PLTs
-anemia proportionate to bleeding, so N/N anemia with some anisopoikilocytosis possible
-WBC usually normal, maybe elevated (dif normal, maybe some high eos and baso)
-LAP normal to increased
-no philadelphia chromosome
BM in ET
-megakaryocytes are increased and clustered in BM
-enlarged megs with more lobulation
-hyperplasia in megs and some granulocytes too
-if hemorrhaging, PT could have increased RBCs too
-"marked hyperplasia in all 3 cell lines" according to the lab
-iron stains have normal to increased iron
other testing in ET
-B12 increased
-uric acid increased
-LDH increased
-cytogenetic abnormalities are rare but could
-molecular gene:
1) 65% have JAK2
2) 20 have CALR
3) 5 have MPL
4) 10 are triple negative
Prognonsis and therapy for ET
-life is normal but bleeding and hemorrhage could cause death
-ET can go to PV or acute leukemia overtime
-therapy to reduce thrombosis complications and lower PLTs
-palteletpheresis works well and anticoagulants like aspirin can inhibit platelet function
-anagrelide is an antiplatelet drug that works well
Et differential diagnosis
-need to differentiate from reactive or secondary thrombocytosis
-reactive thrombosis is usually caused by an infection, inflammation or carcinoma
Reactive
-PLT can reach 1000 but only temporarily
-WBCs are RBCs are normal in reactive thrombocytosis but not in ET always
-splenomegaly is not in reactive
-PLTs function normally in reactive
ET diagnostic criterai
-thrombocytosis >600
-megkaryocytic hyperplasia
-no causes for reactive thrombocytosis
-no philadelphia chromosome
-hemoglobin <13 or normal RBC mass
-stainable iron in BM
-no fibrosis in BM
-no LE picture
MPNs differentiating them!
-high RBCs can help show CML vs PV
-ET has high PLTs but so can PV but RBCs won't be high
-PMF has really high RECTIC fibers in BM
-basophilia is mostly in CML
Starting Myelodysplastic Syndromes (MDS)
myelodysplastic syndromes (MDS)
-stem cell disorders distinguished by PB cytopenias and dysplastic changes in the BM
-BM hyperplasia with INEFFECTIVE hematopoiesis are why you get cytopenias (BM is producing cells but they are being destroyed before reaching the PB)
aka MDS =
-smoldering leukemia
-preleukemia
-dysmyelpoietic syndrome
MDS with FAB are characterized by
-% of blasts in BM
-% of blasts in PB
-if ringed sideroblasts are there or not
-% of mononcytes present
-extent of cytopenias
-degree of dyspoiesis
5 types of MDS according to fab
- Refractory Anemia (RA)
- Refractory Anemia with Ring Sideroblasts (RARS)
- Refractory Anemia with Excess Blasts (RAEB)
- Refractory Anemia with Excess Blasts in Transformation (RAEB-t)
-Chronic Myelomonocytic Leukemia (CMML)
**FAB doesn't include newer diagnostic methods like WHO does, so WHO is used more
WHO uses more
morphology, clinical, molecular, cytogenetic, and immunophenotypic features
WHO Groups of MDS
-MDS with single lineage dysplasia (MDS-SLD)
-MDS with ring sideroblasts (MDS-RS)
-MDS with multilineage dysplasia (MDS-MLD)
-MDS with excess blasts (MDS-EB-1 or MDS-EB-2)
-MDS with isolated del (5q)
-MDS, unclassifiable
the revision in 2016 with WHO Classifications was
name change from refractory anemia to MDS
Etiology/Causes of MDS
-stem cell defect that causes abnormal maturation and function of hematopoietic cells
-premature destruction (ineffective hematopoiesis) causing cytopenias in the blood
-often idiopathic but can be caused by chemo/radiation, benzene, smoking, viruses
**very common acquired BM failure syndromes in adults!
PBS in MDS
WHITE BLOOD CELLS
-neutropenia
-monocytosis possible
-early blasts, promyelocytes, myelocytes, and metas
-dysplastisc features, hypogranular neutrophils, pseudo-pelger huet nuclei, enzyme defect in neutrophils, ring-shaped nucleus
**pseudo means not every nuetrophil will be round or dumbell shaped, usually more round when acquired)
**hypogranular neutrophils can look like monocytes but less blue cytoplasm, more pink
**ring nucleus in neutrophils
**LOW LYMPHS
*monocytosis can happen in many MDS i think??
RBCs
-macrocytic anemia
-oval macrocytes
-dimorphic RBCs
-low reticulocytes
-anisocytosis,poikilocytosis (dacrocytes possible)
-nRBCs
PLTs
-any count possible
-giant, hypogranular, or fused plts
-abnormal adhesion and aggregation
Clinical Findings in MDS
-weakness/fatigue bc of anemia
-bleeding/easy bruising bc of low platelets
-infection bc of neutropenia
CMML will have hepatosplenomegaly but the others won't!