PDA III Parkinson's Disease
1/43
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
44 Terms
PD general features
-Progressive neurodegenerative disease
-3 in 1000 overall, about 1% in people over age 60
-Major symptoms are movement disorders
-Primary neuropathology: death of neurons in the substantia nigra
Major symptoms of PD
-Resting tremor (pill-rolling tremor, looks like they're holding a pill)
-Cog-wheel rigidity
-Bradykinesia: moves around less (aka akinesia, paucity of movement, inertia of movement)
-Postural disturbances, shuffling gait
-Absence of facial expression
-Dementia, esp. in later stages
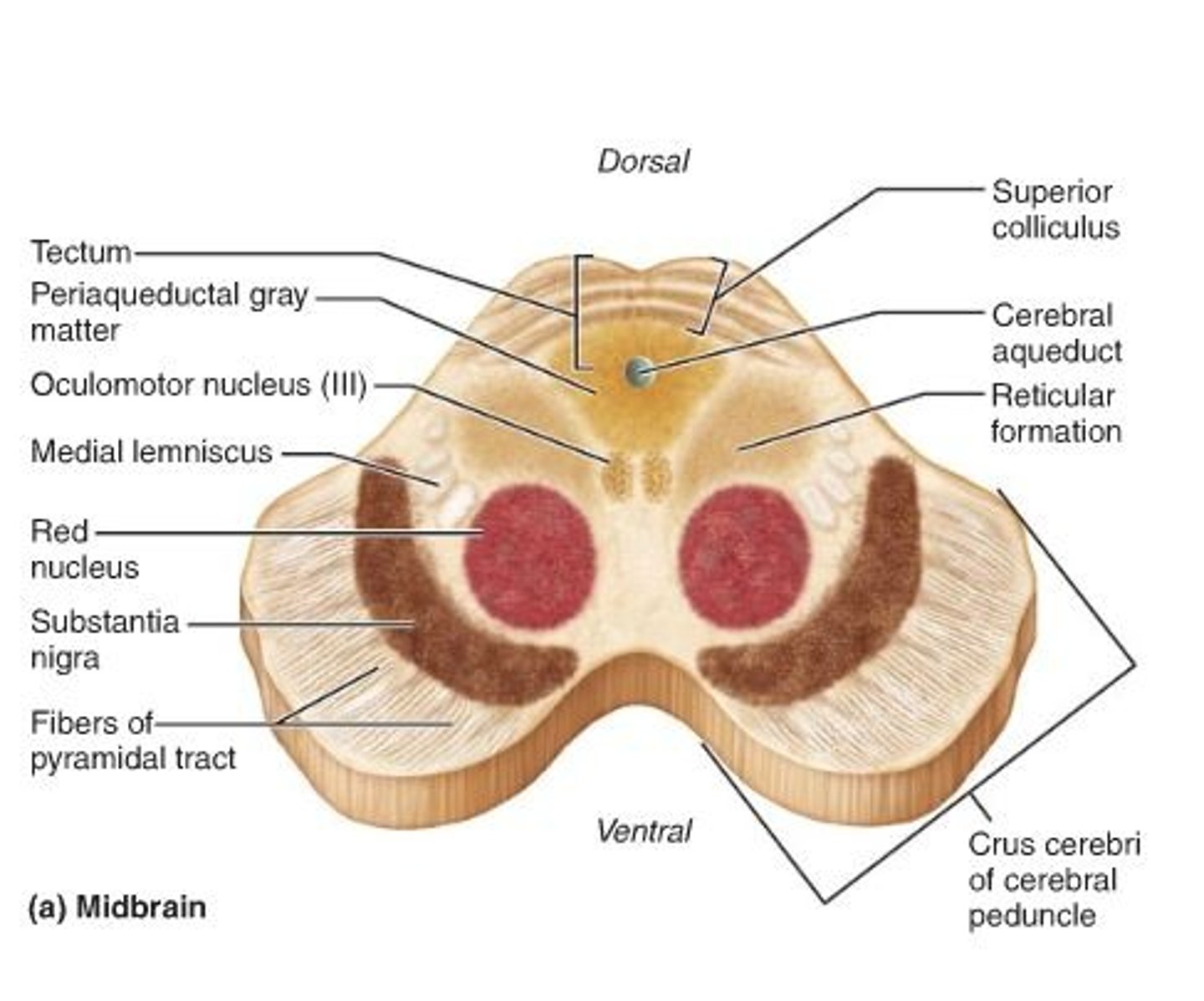
Substantia nigra
-Population of dopamine neurons in the midbrain
-Neurons send their axons to neurons in the forebrain that make up the striatum
-Neurons are black: full of melanin
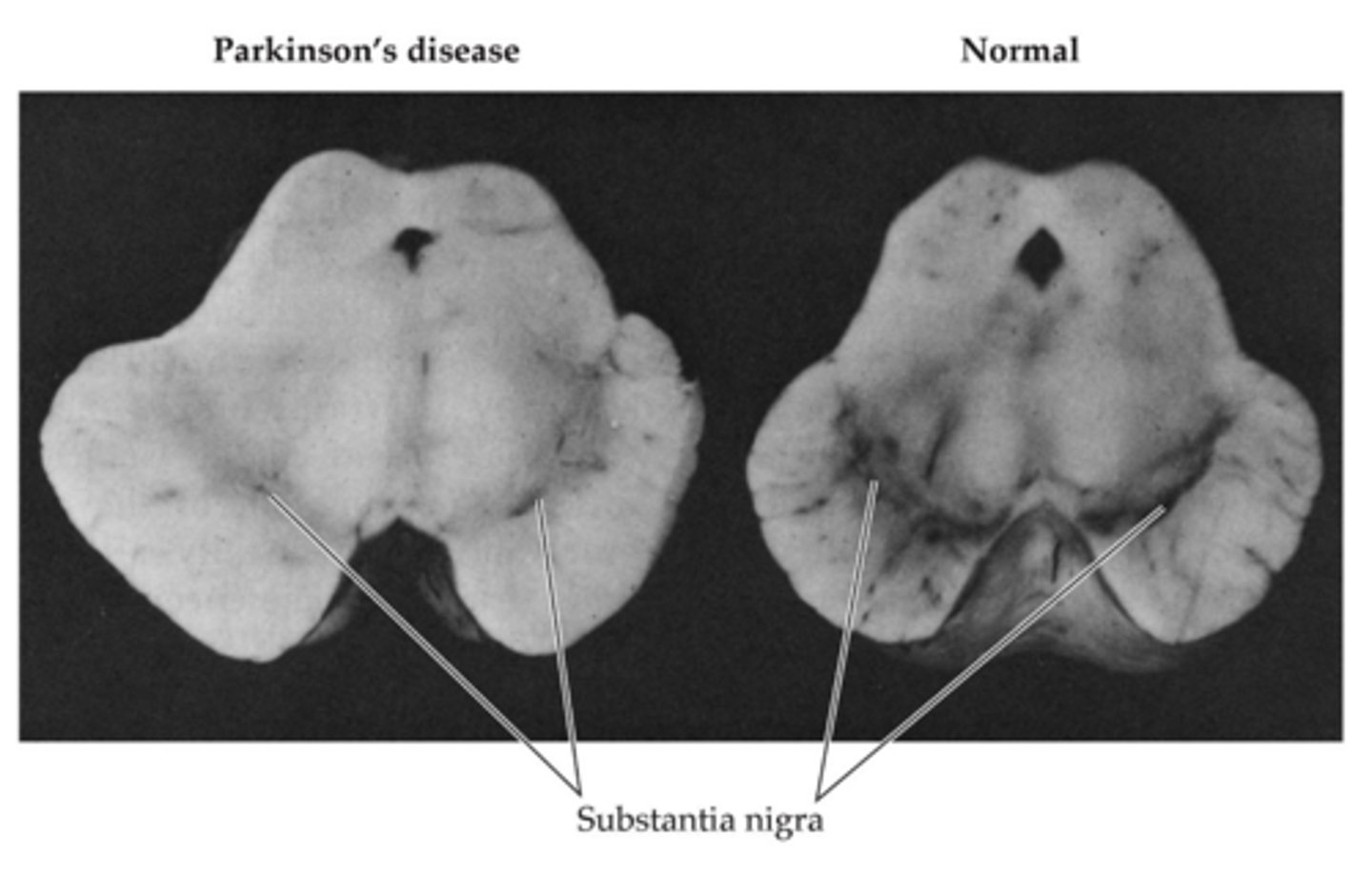
Sub. nigra in PD
-Neurons in the sub. nigra die in PD, neurons can't make it to the striatum
-Death of nigrostriatal pathway
-Significant loss of visible black (loss of neurons)
S. nigra NT
-Dopamine
-Dopamanergic at the striatum
Dopamine in PD
-Loss of dopamine activity in the striatum
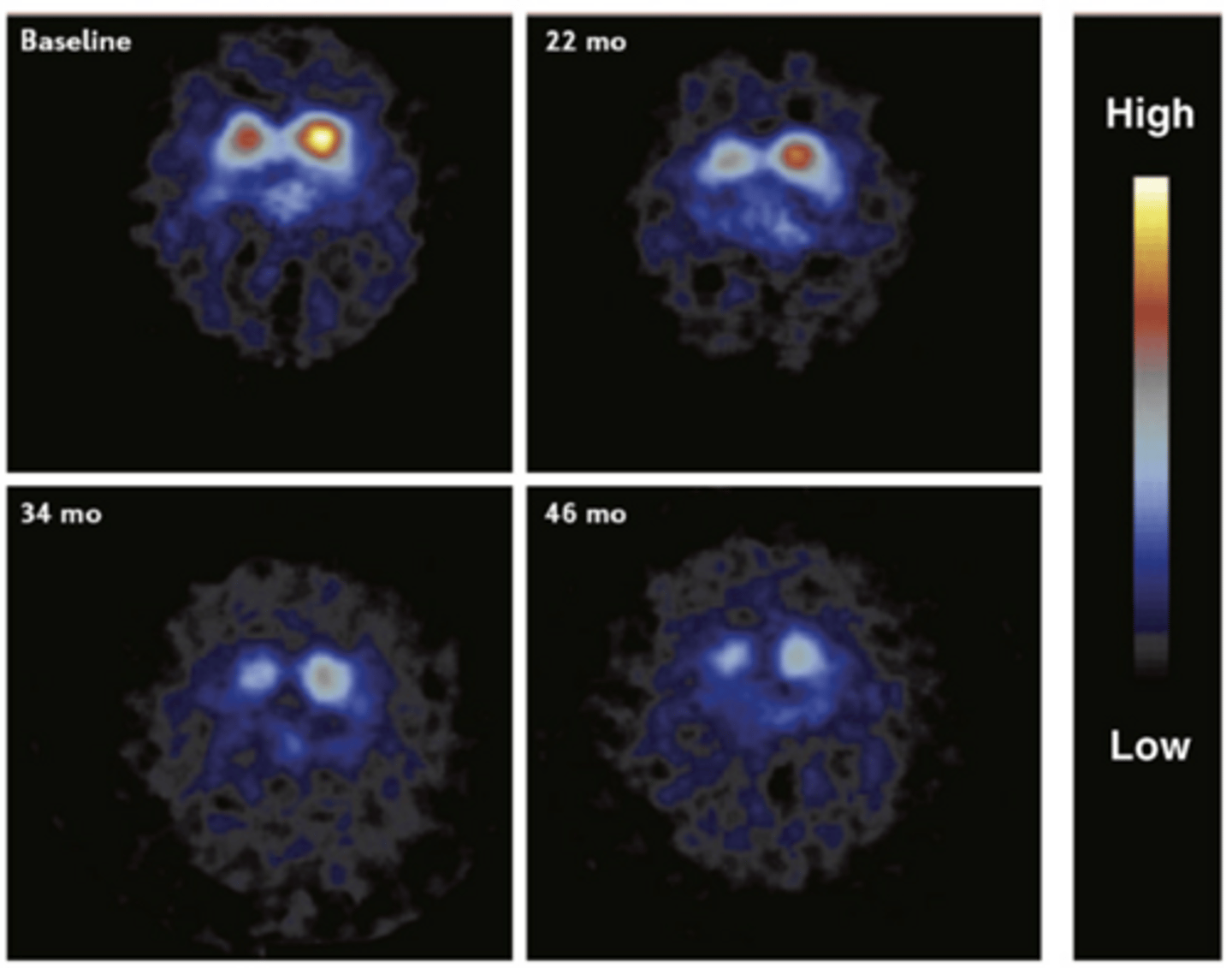
PET scan of the striatum
-Pic is of the two striata
-We have 2 striata on each side of the brain: disease is usually worse on one side than the other
-The PET ligand is binding to the presynaptic dopamine transporter on S. Nigra (we are tracking the disappearance of S. Nigra)
Tracking activity in neural circuits
-Remember glutamate is excitatory and GABA is inhibitory
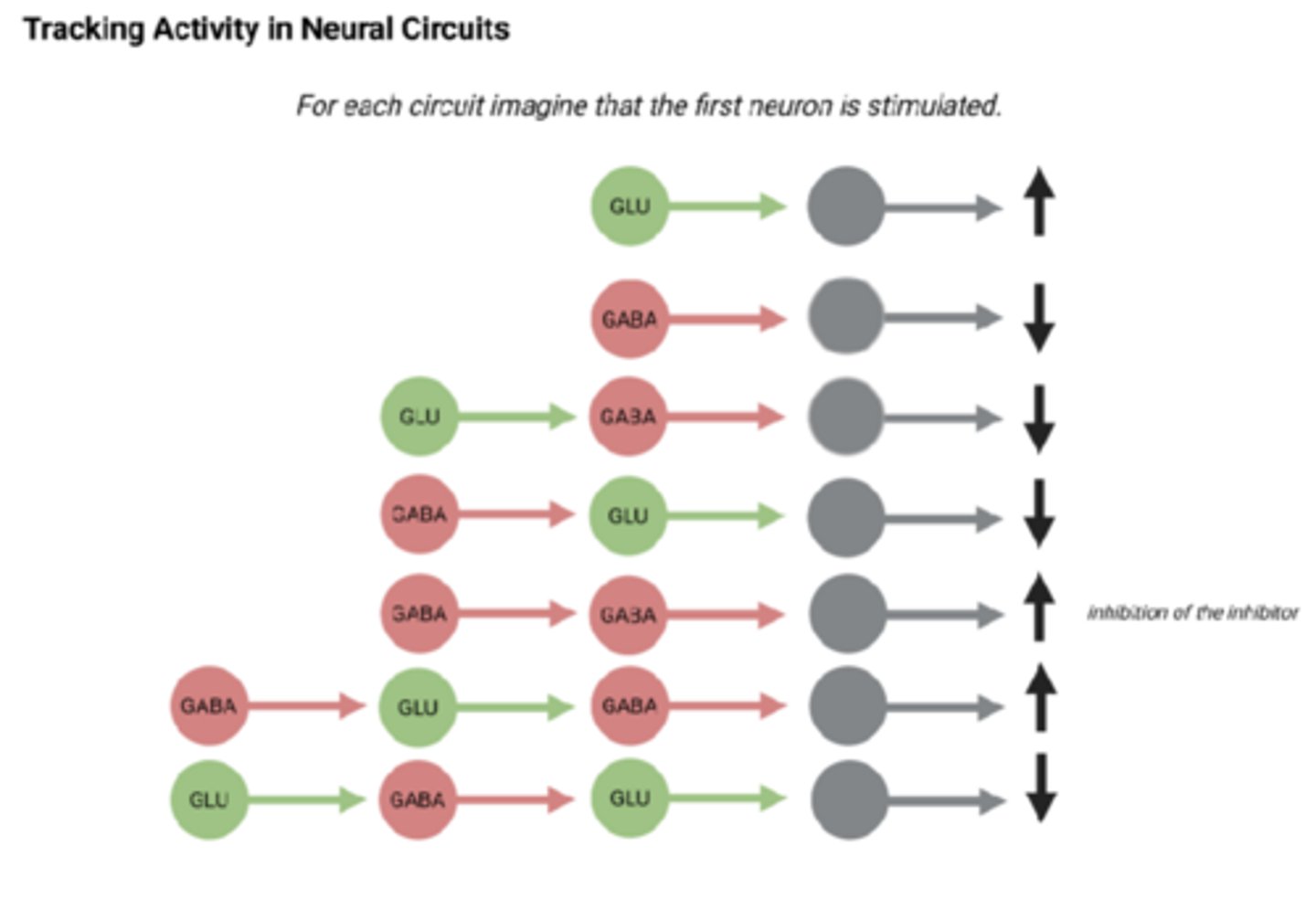
Normal neuroanatomy of the tremor
-Striatal neuron is an interneuron: cell body and axon terminal in one place (striatum)
-Striatal neuron is cholinergic (uses ACh)
-S.N. releases DA, DA releases and is accepted on striatal neurons (Gi coupled D2 receptors on the striatum)
-DA decreases the amount of ACh releasing on GABAergic neurons (Gq coupled muscarinic receptors)
-Decreased ACh means less body movement
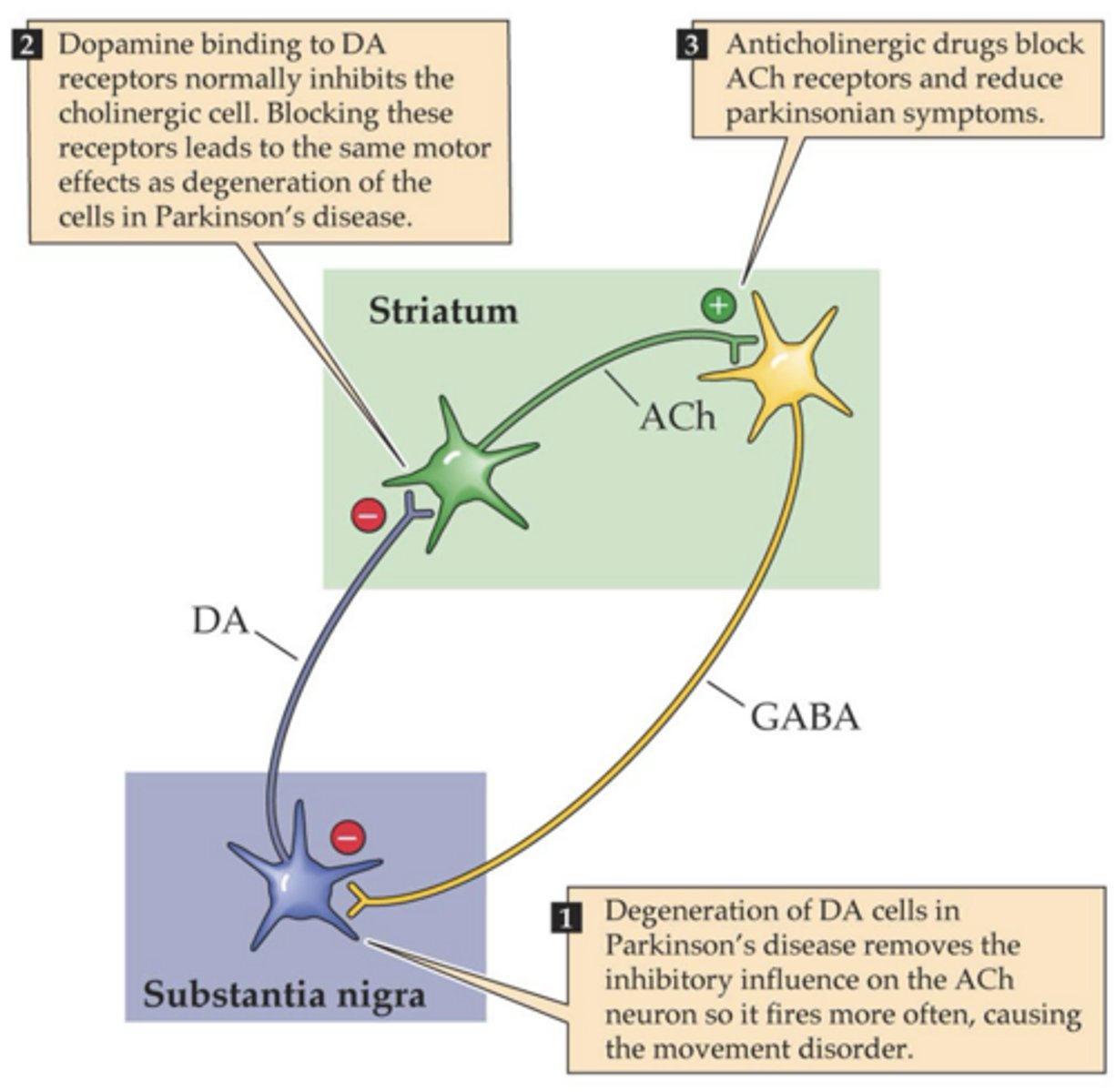
Neuroanatomy of the tremor in PD
-S.N. dies
-S.N. releases less DA
-More ACh is released
-Results in more inhibition SN via GABA
-Look at this more
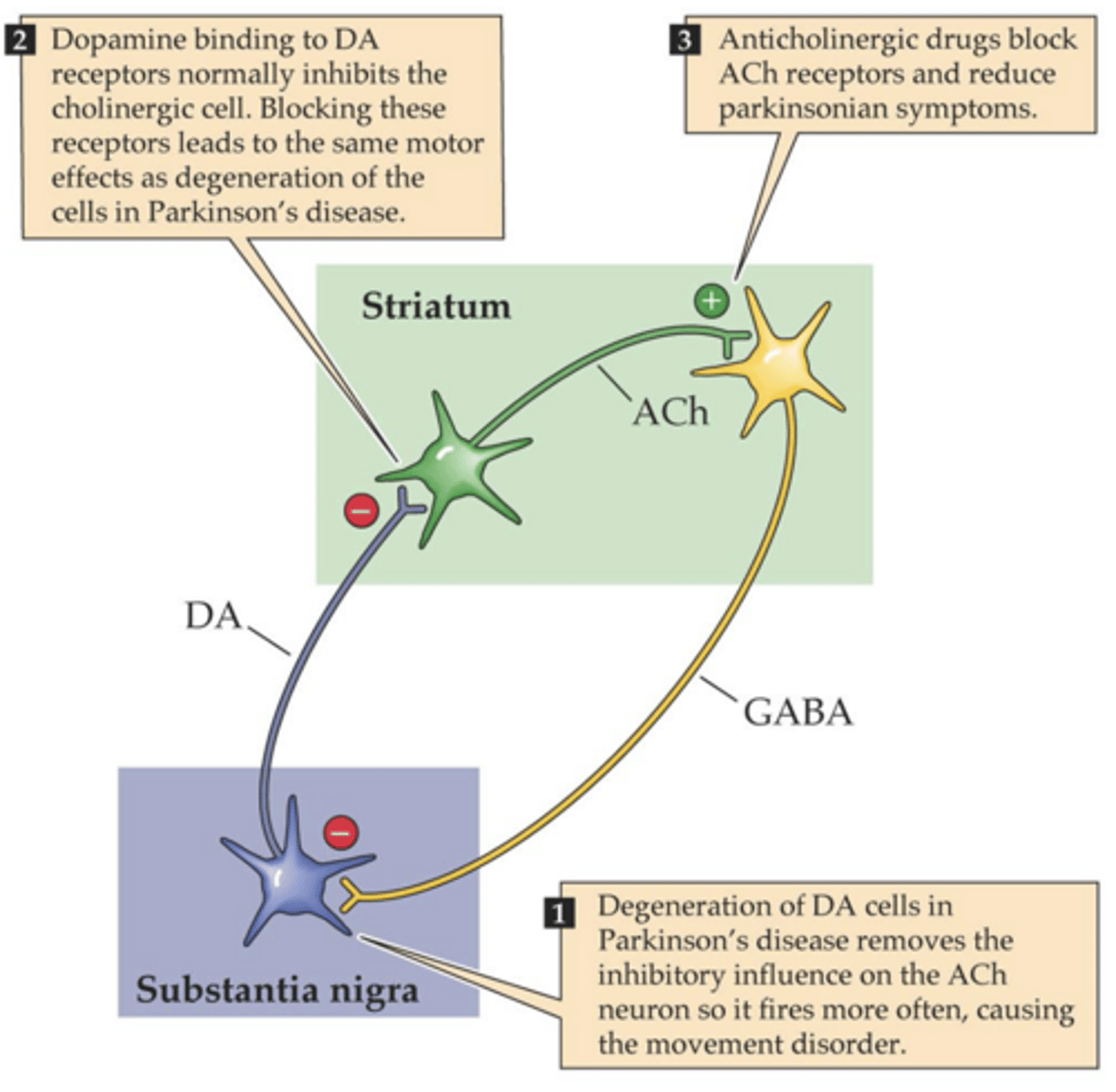
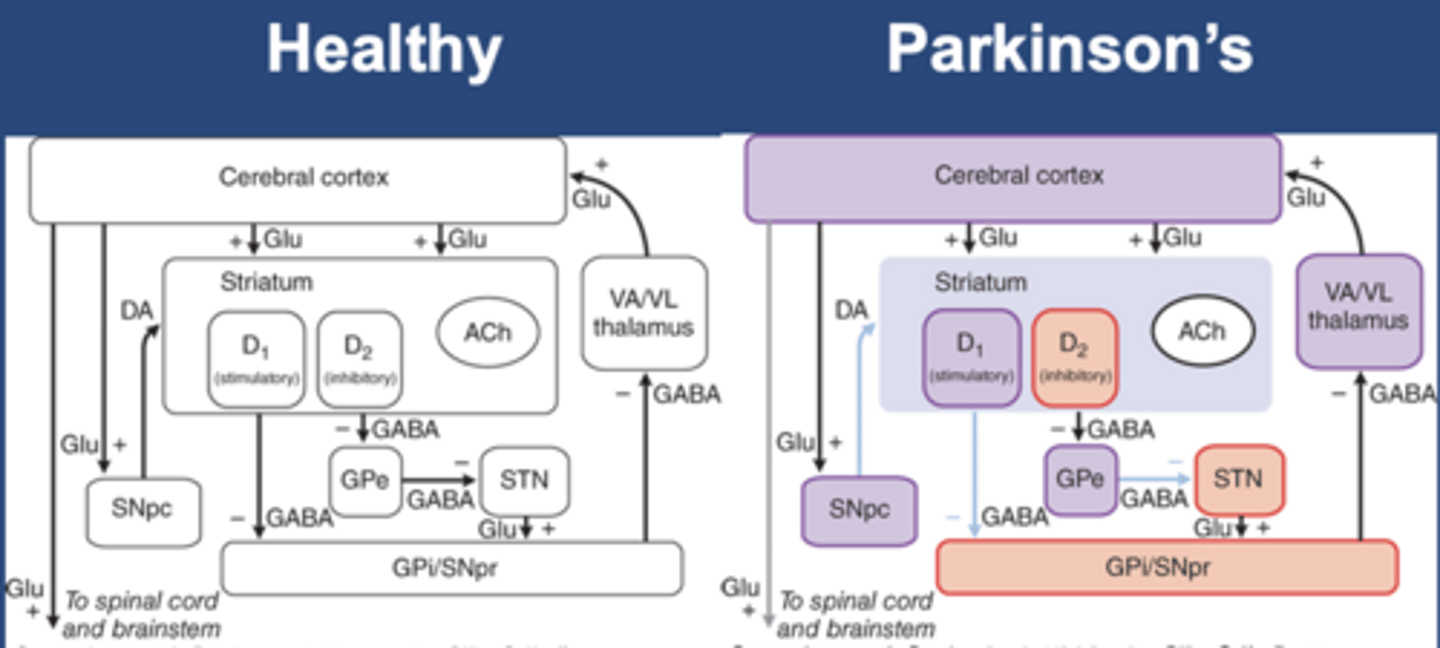
Healthy movement
-Direct pathway and indirect pathway to the globus pallidus
-DA stimulates D1 (stimulatory, direct) and D2 (inhibitory, indirect): both cause excitation in the cerebral/motor cortex
-Excitation in the cerebral cortex causes healthy movement
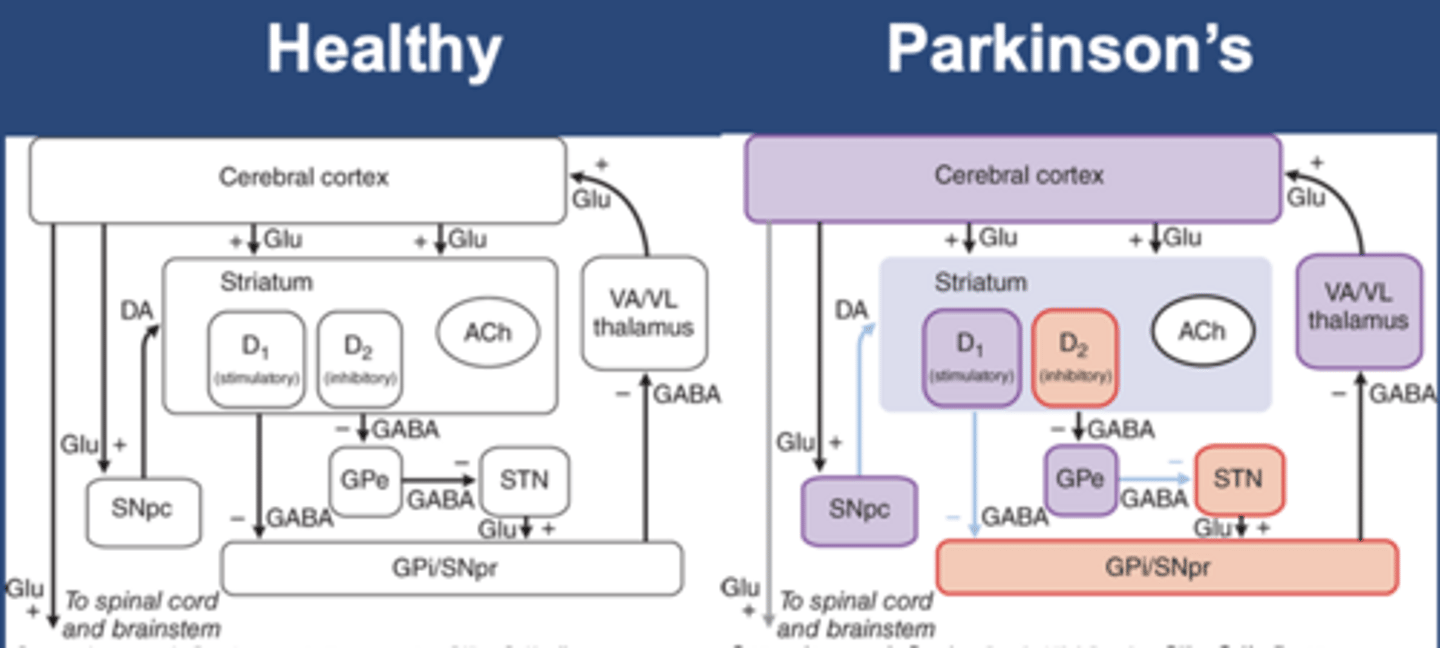
PD bradykinesia
-Slow movement
-Purple: loss of activity
-Red: gain of activity
-Decrease in DA decreases stimulatory D1 and inhibitory D2
-Results in decreased cerebral/motor cortex stimulation
-Less cerebral/motor cortex activity leads to bradykinesia in PD
PD etiology
-Idiopathic: cause is unknown
-Genetic: small minority of cases, tend to have earlier onset
-Drug-induced: drugs block DA, so symptoms look like PD, but there is no S.N. death (parkinsonism, pseudoparkinsonism)
-Toxin-induced (MPTP)
Synthesis of catecholamine NTs
-Tyr comes from our diet
-TH: tyrosine hydroxylase
-TH converts Tyr to Dopa
-Aadc: aromatic amino acid decarboxylase
-Aadc converts dopa to dopamine
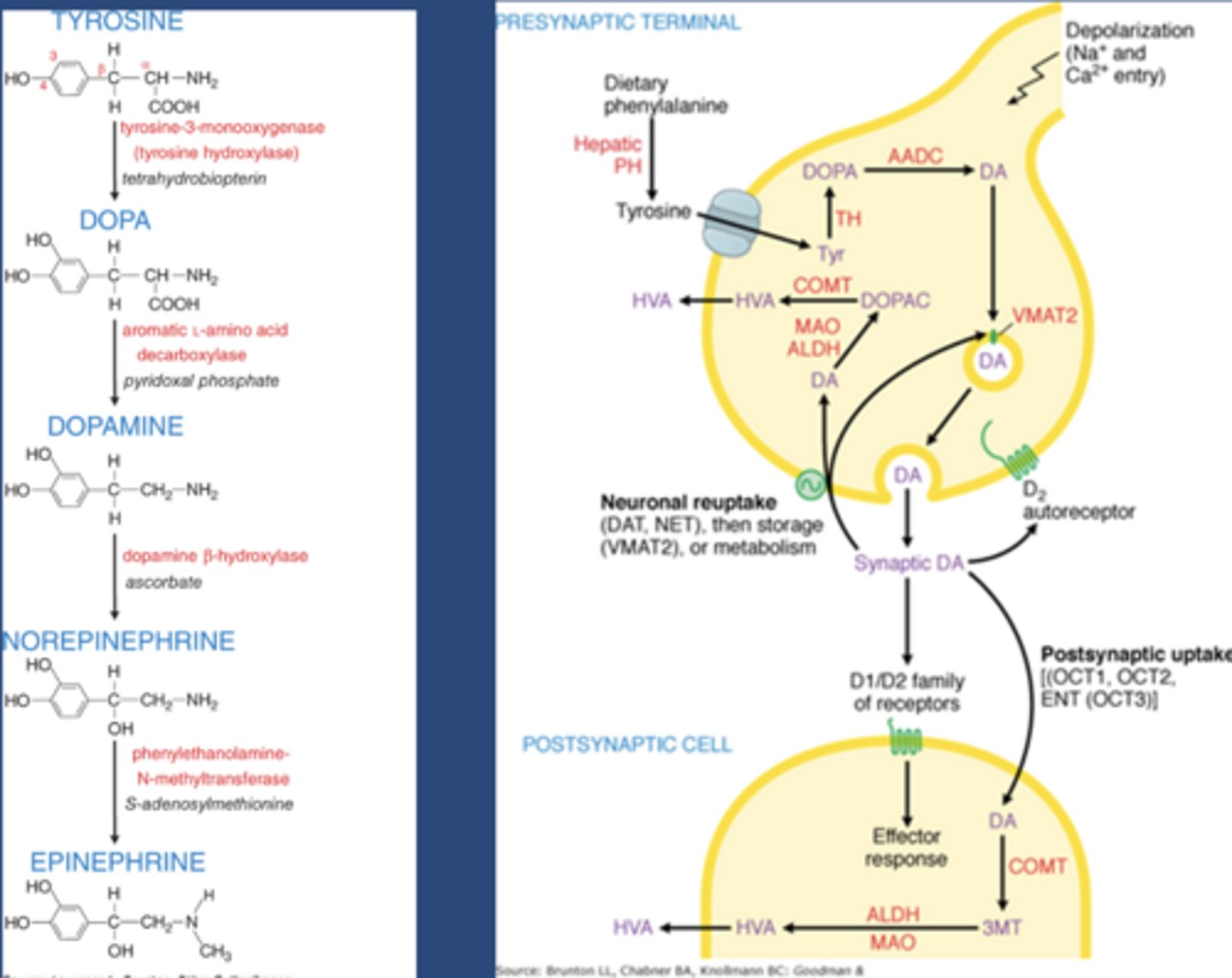
Dopamine storage
-VMAT2: vesicular monoamine transporter
-VMAT2 brings dopamine into vesicles for later release
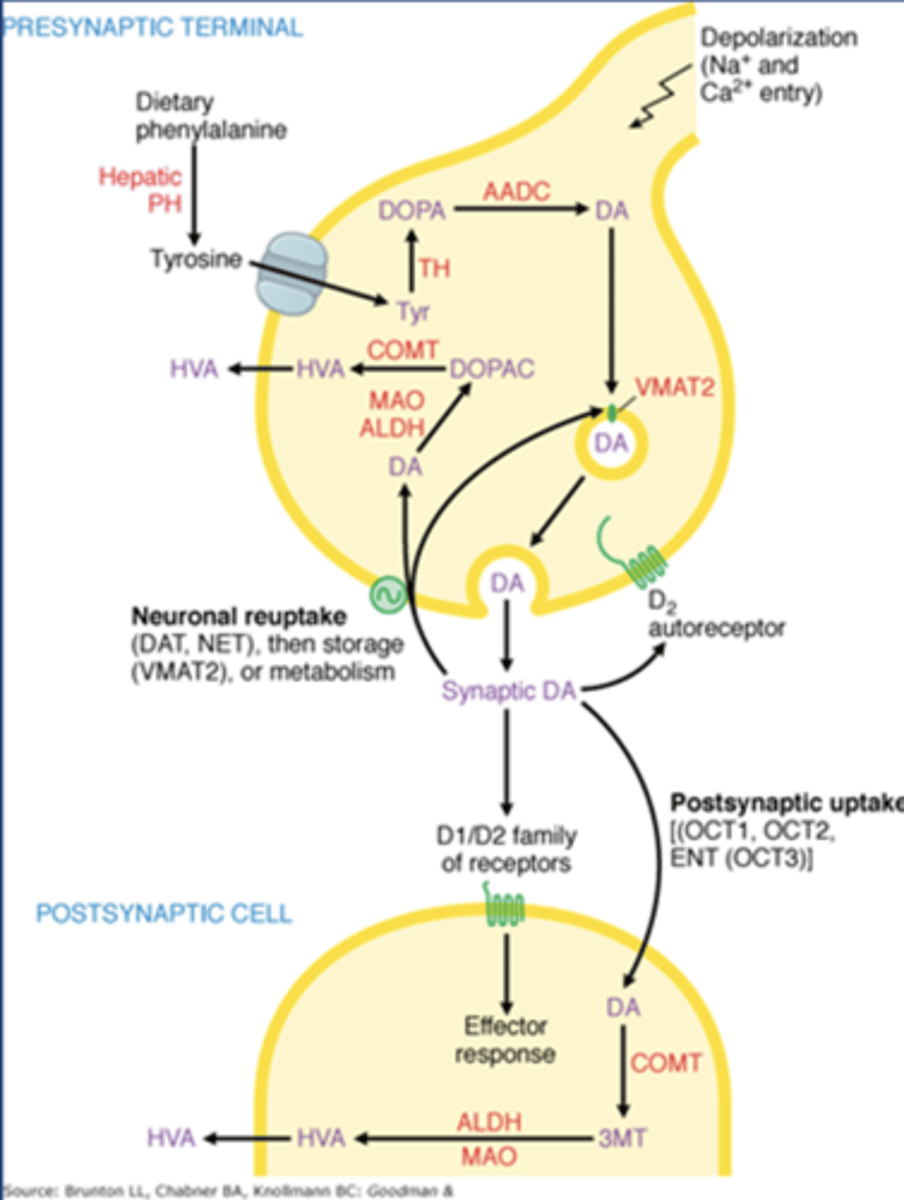
Dopamine release
-Works on D1/D2 receptors on postsynaptic striatal neurons
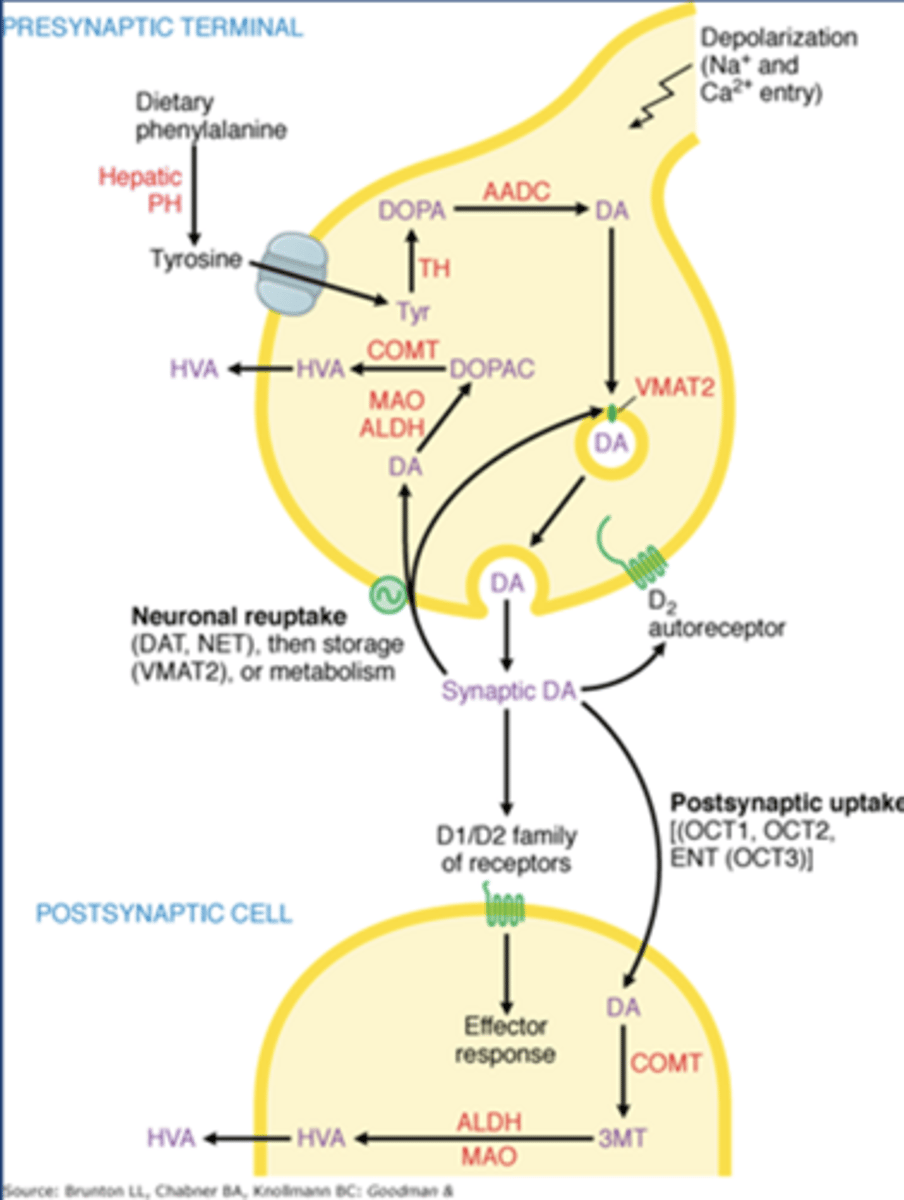
Dopamine autoreceptors
-Gi coupled D2 receptors
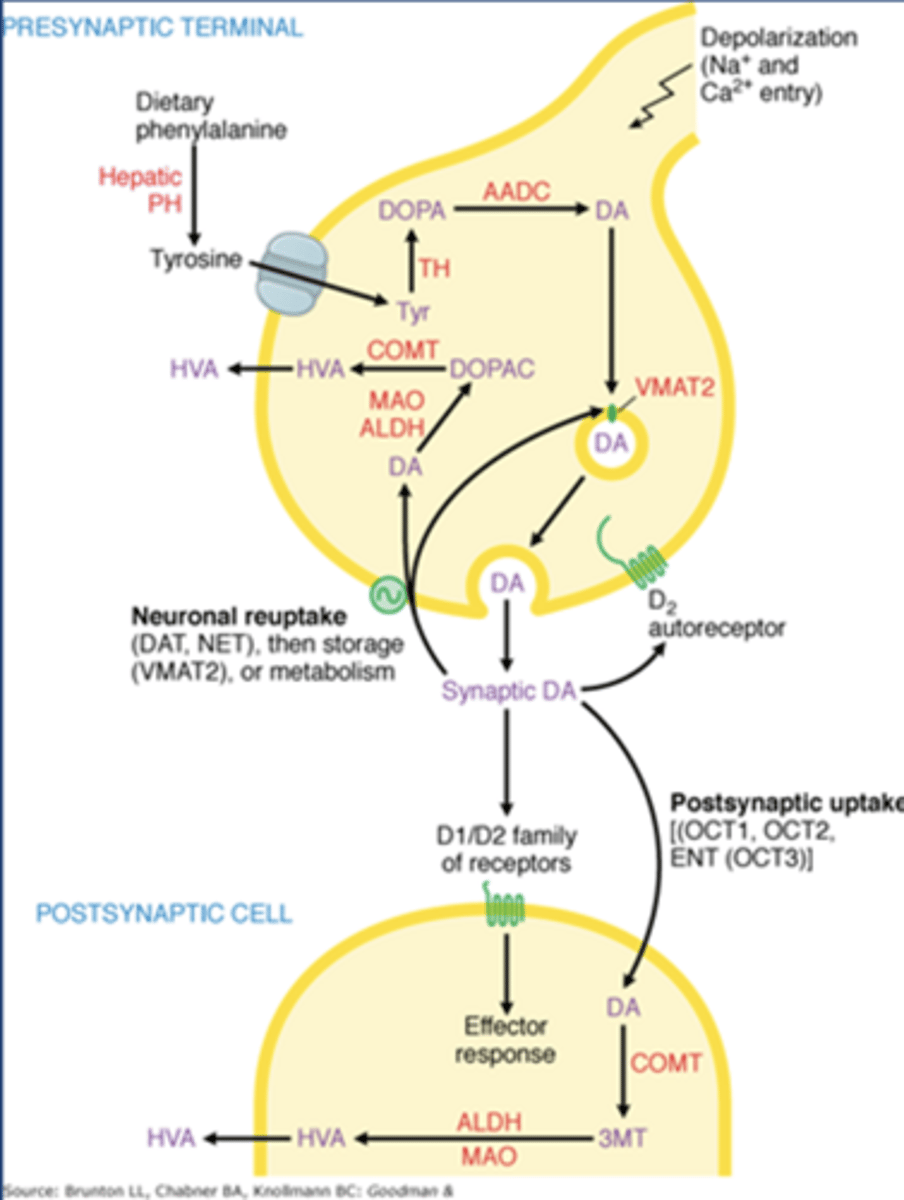
Enzymes that metabolize dopamine
-COMT
-MAO
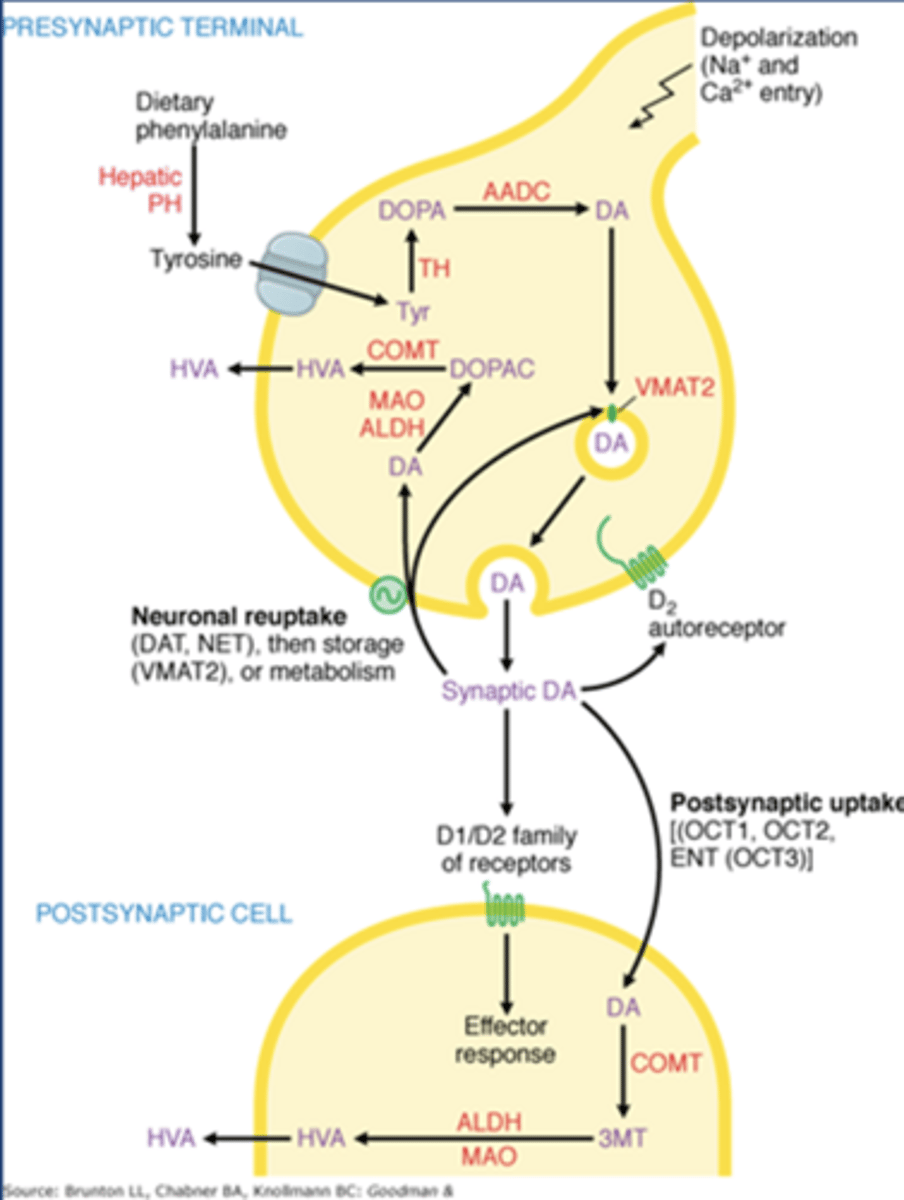
Drugs for PD
-L-DOPA
-Carbidopa
-DA agonists
-Enzyme inhibitors
-Amantadine
-Anticholinergics
L-DOPA
-Amino acid precursor to dopamine
-AADC: Aromatic amino acid decarboxylase
-AADC converts L-DOPA to dopamine (sometimes prematurely in the peripheral tissues)
-L-DOPA is carried across BBB by a transporter, carried into DA neuron by another transporter
-Only 1-5% reaches the brain
L-DOPA acute ADEs
-N/V
-Hypotension
-Arrhythmias
-These are DA-mediated and peripheral
L-DOPA chronic ADEs
-Abnormal, involuntary movement (dyskinesias)
-Nightmares, hallucinations
-These are central in origin: due to L-DOPA being converted to dopamine in the brain
On-off phenomenon
-In regards to L-DOPA treatment
-On: when pt has control of their symptoms
-Off: when pt doesn't have control
-First 1-2 years pt has smooth, day long control of symptoms
-At 2-5 years: peak dose dyskinesia, symptoms return at low plasma conc but before it's time for the next dose
-Reason: therapeutic plasma conc narrows as more and more neurons die
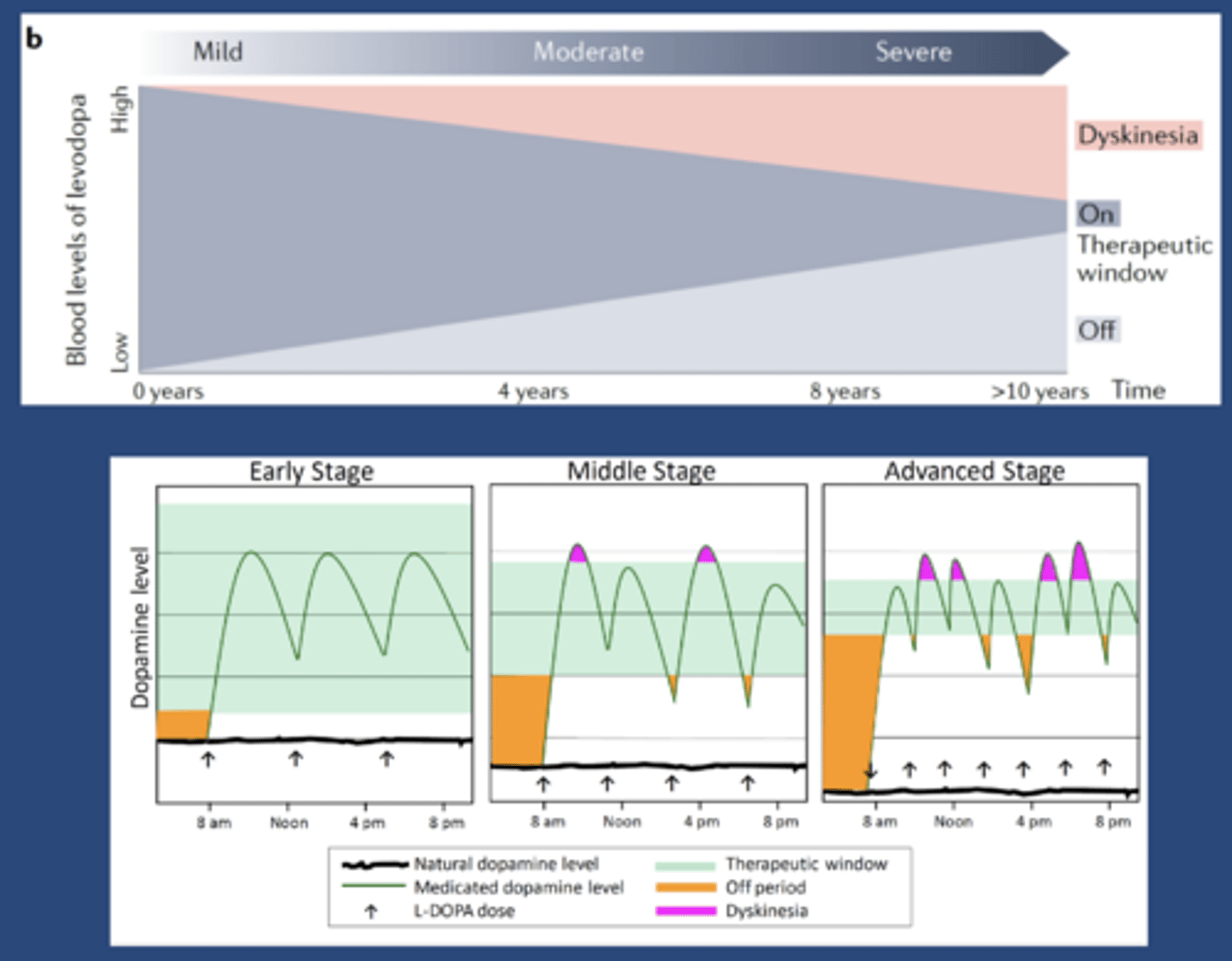
L-DOPA self-limitation
-At some point, there won't be enough dopamine neurons to convert L-DOPA into dopamine
-Destined to fail
Carbidopa
-AADC inhibitor that can't cross the BBB
-Would inhibit L-DOPA changing to dopamine in the peripheral tissues (blocks wasted dopamine)
-3-10 fold more L-DOPA makes it into the brain
-SINEMET (L-DOPA/carbidopa combo product)
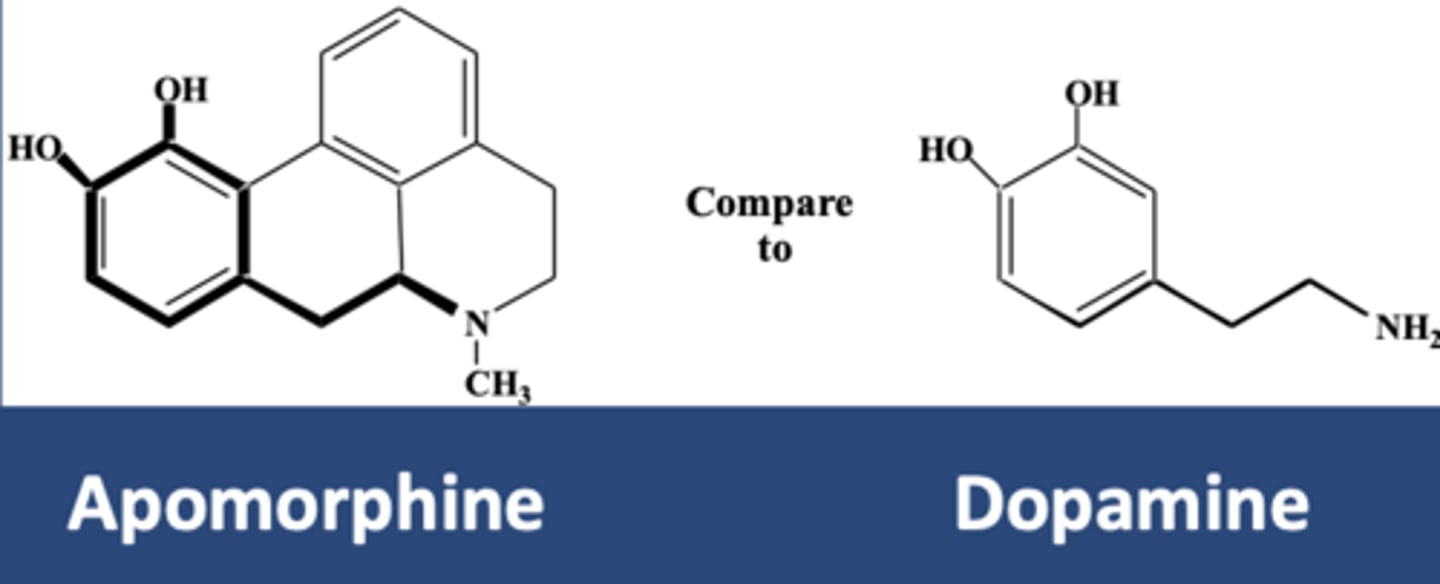
DA Agonists
-Apomorphine (APOKYN)
-Pramipexole
-Ropinirole
-Rotigotine
DA agonist efficacy
-Not a perfect treatment strategy
-There are dopamine systems in the brain outside the striatum (ADEs)
-Dopamine can act in peripheral tissues (ADEs)
-Loss of negative feedback of dopamine via circuits (no regulation)
-Normally, dopamine works phasically (brain releases it when we need it), so putting the same amount of an agonist in the body all the time isn't a perfect approach
Apomorphine (APOKYN)
-Dopamine agonist for PD
-Used in off periods of L-DOPA
Pramipexole, ropinirole, and rotigotine advantages
-Long DOA for less on/off phenomenon
-May be used early in course of the disease to delay L-DOPA therapy
-Oral or transdermal
Pramipexole, ropinirole, and rotigotine disadvantages
-N/V
-Hallucinations
-Hypotension
-Somnolence
-Compulsive behavior: gambling and punding (compulsive organization)
Enzyme inhibitor classes
-Carechol-O-Methyl Transferase (COMT) inhibitors
-MAOis
COMT inhibitor MOA
-COMT metabolizes L-DOPA and DA
-COMT inhibitors inhibit the metabolism
-Typically as adjuncts to reduce the off symptoms
COMT inhibitor drugs
-Tolcapone
-Entacapone
-Opicapone
Tolcapone
-Acts in periphery and CNS
-Giver q2-3days
-Hepatotoxic: requires liver function monitoring
-Reserved for refractory pts
Entacapone
-Acts only in periphery
-Given with each L-DOPA dose
Opicapone
-Acts only in periphery
-Given with each L-DOPA dose
MAOi MOA
-INhibition of MAO-A is associated with antidepressant activity
-Inhibition of MAO-B is helpful in PD
-Purpose is to decrease DA metabolism and decrease free-radical formation
MAOI drugs
-Selegiline
-Rasagline
MAOI pros/cons
-Therapeutic dose does not cause HTN crisis (no effect on MAO-A)
-Loses selectivity at high doses
-May interact with meperidine, TCAs, SSRIs, and tyramine
Amantadine MOA
-Block NMDA glutamate receptors
-Stimulates the release of DA and inhibits DA reuptake
-Used in mild cases or as L-DOPA adjunct
-Loses efficacy over a few months
Anticholinergics
-Specifically antimuscarinic (on GABAergic neurons)
-Used as adjuncts to address the DA/ACh imbalance in the striatum
-Most effective at reducing tremor, not as effective at reducing rigidity or bradykinesia
Anticholinergic drugs
-Benztropine
-Trihexyphenidyl
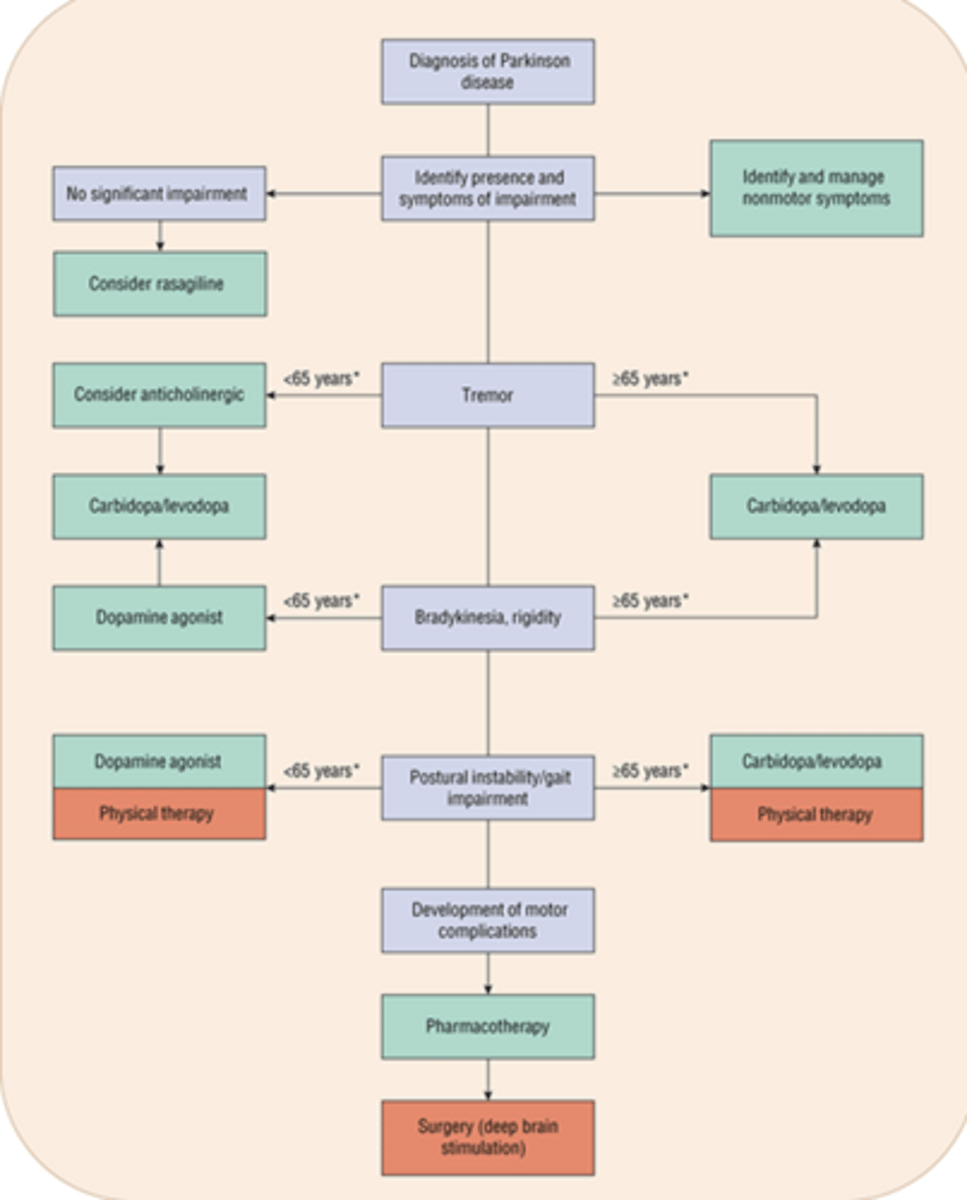
PD treatment summary figure
-Figure will be on exam
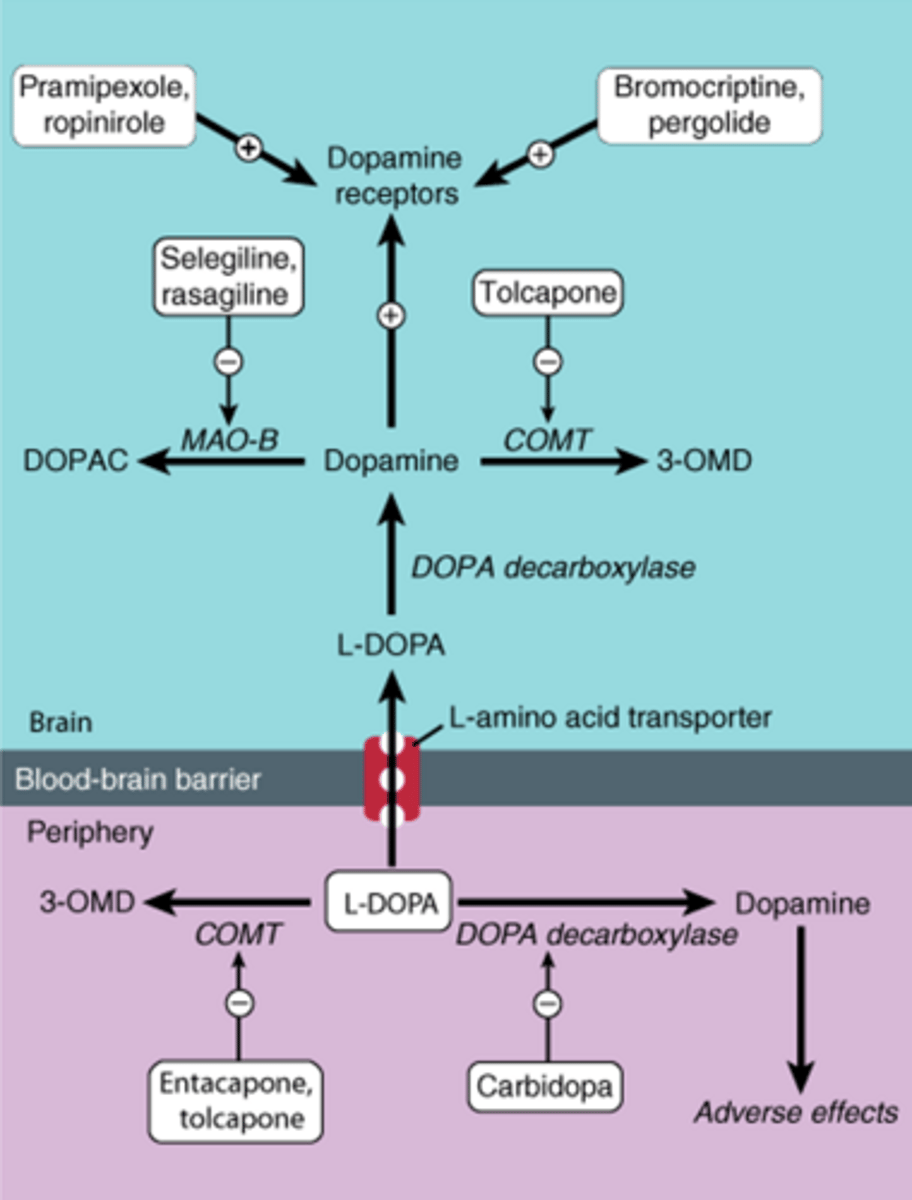
PD treatment algorithm