Beta 2 Adrenergic Recptor
1/18
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
19 Terms
Comparing subtypes of adrenergic receptors
Valuable receptor in understanding how these classes of receptors function
2 subtypes of beta adrenergic receptors which have differential responses to agonists
Beta 1: isoprenaline > noradrenaline= Adrenaline
Beta 2: isoprenaline > adrenaline >> noradrenaline
Also have difference in response that they drive, through different pathways
Beta 1: ↑ [cAMP] → ↑ PKA —> ↑ muscle contraction
Beta 2 : ↑ MAPK ↑ cPLA2 → ↑ muscle contraction
Have some common motifs which can be evidenced by the agonists that they bind:
Aromatic ring with OH groups on the edge
OH and NH group
However, the variation in these molecules must be giving us this specificity
Alpha Receptor
Alpha 1: noradrenaline > adrenaline >> isoprenaline driving ↑ PLC —> ↑ IP3/DAG —> muscle contraction
Alpha 2: adrenaline > noradrenaline >> isoprenaline, driving ↓ [cAMP]
History
First characterised with radioligand binding studies, then cloned and expressed using cDNA libraries
Then structurally determined using crystal structures in 2007
Pivotal for acting as a model system to understanding the functioning of GPCRs which can help us to create drugs which target these
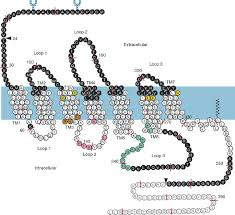
Primary structure
7 TM domains which anchor it to the lipid bilayer
N-terminal tail outside the cell which has glycosylation motifs which help to orientate the receptor in the membrane
Small EC loops and large IC loops and C terminal tail
Structurally these GPCRs look similar to the mAchR and so would expect some of the same features
Ligand binding domain in EC portion
G protein binding IC loop 3
TM domain 5 moving away for activation
NAdr Binding site
Hydroxyl groups and charged group which tell us about the binding
Aspartate 113 in TM3 involved in the binding of the positively charged residue, also have aspartate at 79 and 318 but when mutated show less of an effect —> suggests that Asp113 is involved in binding the charged NH3+ group
Series of compounds with varying hydroxyl groups substituted to observe the effect on affinity
Serine at 204 and 207 show high affinity binding but when we abolish either of these hydroxyl groups, binding affinity and receptor activity decreases by orders of magnitude showing that both of these OH groups are required for binding and activating the protein
Chimeric Proteins
Can create different chimeras by swapping out different TM domains of beta 1/beta2 to create a hybrid receptor
This allows us to link pharmacology to different domains in the protein
Experiment binding radioligand 125I-CYP (an antagonist) and displaced with salmeterol, a selective beta 2 agonist
Chimeras 1 and 2 have Ki in the nanomolar range, chimeras 3,4,5 and 6 have Kis which are much higher → can link certain parts of the receptor with high affinity binding
Binding site model
Charged interaction between aspartate 113 on TMD3 and NH3+ group
TMD5 has Serine 204 (and potentially serine 203) and 207 involved in OH group binding
Can we identify drugs which target one specific subtype?
Selective mutagenesis experiments which mutated single amino acids in the structure to identify subtype specificity
Tyrosine 308 mutated to an alanine in TM7
Usually when tyrosine interacts with salmeterol it base stacks with the aromatic molecules
This is what confers the selectivity to this subtype of receptor
G protein coupling
R—>R* state thought to be like an on/off switch activating the receptor, but this is not the case
All GPCRs have a basal level of activity so the receptor can activate the G protein even in the absence of an agonist (shown by a plot of cAMP against receptor level)
Two binding sites
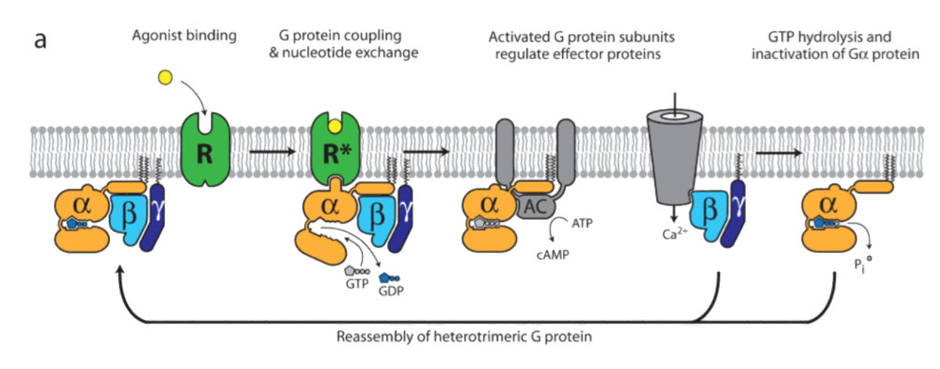
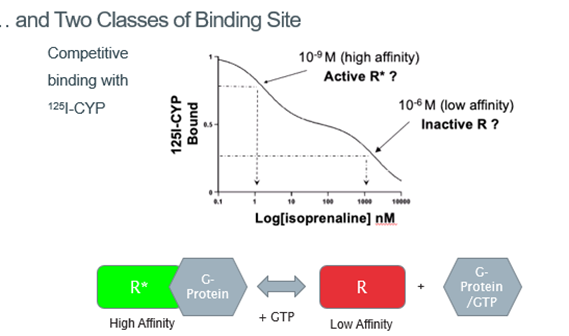
Radioligand diaplacement of 125I-CYP shows that there are 2 binding sites: one with low affinity binding, one with high affinity binding
Suggested that there are 2 different conformational states of the receptor: one which gives high affinity binding, one which gives low affinity binding
Suggested that we don’t have a sequential series of events, but instead have this equilibrium between the different conformational states which is shifted by the binding of an agonist
The other states are not unpopulated but are not highly populated
So at rest, the concentration of the inactive form of the receptor is higher than the active form, but when an agonist binds, the active form is more populated
Instead of acting as an on/off switch, altering the conformational equilibrium
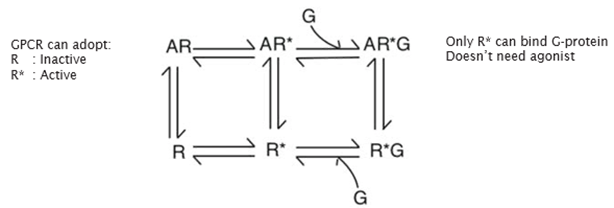
Model for receptor function
Absence of agonist, highly inactivated and high conc of activated form (basal)
Full agonist causes shift to a majority active state
Antagonist pushes eq to the inactive form
Inverse agonist pushes even more of receptors into the inactive state

Sequences responsible for receptor coupling
Can swap around the different intracellular loops to see how this effects downstream activation
If we delete the I3 loop and replace it with the one from alpha 2 we have reduced coupling so increased cAMP
Can swap the beta 1 and beta 2 IC loops and change the amount of cAMP produced
Can couple a GPCR to a G protein of our choice → the nature of these loops determines the interaction with the G protein
DRY motif
Stabilises the receptor in its inactive conformation
When you disturb this dry motif, observe an increase in basal levels of activity because you perturb the equilibrium and moved from a greater population of R→R* which interacts with the G protein
Basal level of activity strongly regulated by the DRY motif → if you want to activate the receptor, you need to disturb the interaction between the DRY motif
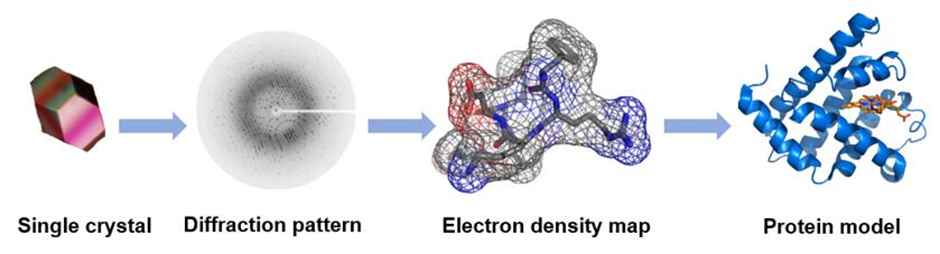
Structural Biology of GPCRs
In order to obtain a protein model, you need a crystal structure
But this required us to try and extract this from the lipid bilayer using detergents and lipids but we don’t know what lipids/detergents this protein will be stable in
We also have structural flexibility and have loop regions being highly disordered creates challenges when obtaining crystal structures
Ways around this:
T4 lysosozyme – clone in a protein which crystallises very well within the loop region and see if this promotes the formation of crystal structures
Can generate conformationally specific antibodies which bind particular loop regions and use this to drive the crystalisation of the G protein
Crystal structure eventually obtained with a T4 lysoszyme but was able to resolve a lot better structure on the IC side of the membrane compared to the EC (because the lysozyme locks the IC loops in place). But this still leaves the binding site partially unresolved.
In order to resolve the binding site, the structure of the IC surface must be disrupted, but this is the surface which is involved in binding G proteins. Shows how some of the structural information is lost in these studies
Binding of inverse agonists
Serine motifs involved in binding close to the inverse agonist (carazolol)
Polar interactions which are important, as well as hydrophobic interactions involved in binding
Beta 2 adrenergic binding site
Knowledge of GPCRs had come from rhodopsin, which had a chromophore molecule bound to it and therefore, the ligand didn’t need to come on and off the binding site
Relatively exposed binding site which is located between all the helices on the EC surface of the membrane
Then used this knowledge to map out how the molecule is going to bind to the receptor → integrated info from the site directed mutagenesis
Purification of the B2 adrenergic Gs complex
Lots of challenges and techniques required because not just trying to obtain a crystal structure of a protein, but also a protein couples to a G protein
Isolation of the active and inactive form confirmed
the TMD5 and TMD6 pushing away of the helical bundle
Change in the IC loop 2 (located near the DRY motif)
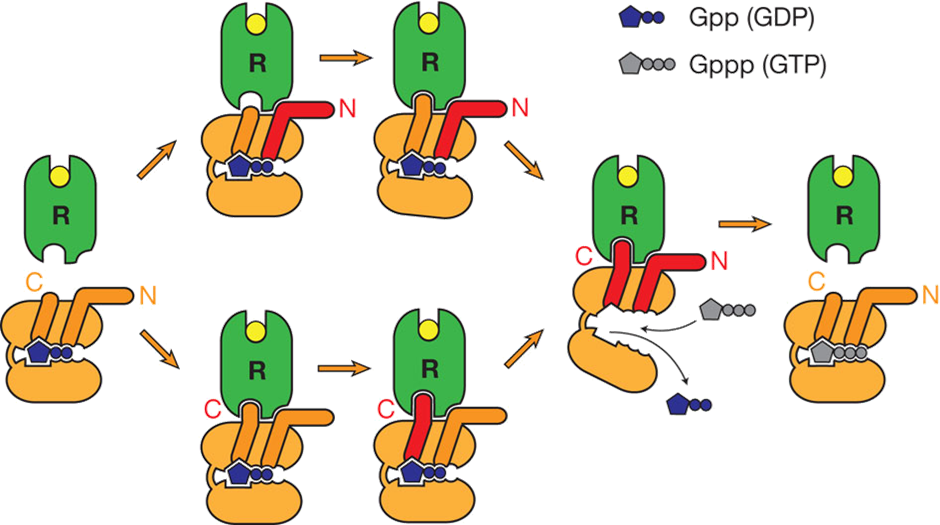
How doe we get binding of G protein to GPCR
Drug binding pulling the helical bundle opens up a hydrophobic cleft in the GPCR
Binding of the C-terminal helix to the hydrophobic cleft → centre of the helical bundle making hydrophobic interactions with TMD, but starts to form ionic interactions with the DRY motif
C-terminal (alpha 5) helix binding to the hydrophobic cleft which breaks apart the DRY motif and stabilises the active conformation of the receptor
C-terminal helix is a long helix and when it interacts with DRY motif of the GPCR, it causes it to be pulled away from a GDP binding site which breaks interactions with the GDP molecule → affinity for GDP decreases and released, allowing GTP to bind
Asp130/Arg131 salt bridge to Glu268 usually functions to hold receptor in inactive conformation but is disrupted
Instead Arg131 interacts with Tyr391 of Gas alpha 5 helix
G protein has propagated a conformational change in the alpha subunit, where it has reduced affinity for GDP and enhanced affinity for GTP
This causes conformational change in heterotrimeric G protein, causing it to dissociate away from the receptor
Model for activation of protein
Interaction of N and C terminal drives conformational change which drives the GDP—>GTP
Causes the disassociation which drives the activation of the G protein