Section 8 Chapter 21 Recombinant DNA technology
1/17
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
18 Terms
What is recombinant DNA?
When the DNA of two different organisms has been combined and then transferred to another organism, such that the organism with the recombinant DNA is known as transgenic organism (Genetically modified organism, GMO)
The reason for no rejection is that the genetic code is universal, so proteins can still be synthesised.
Additionally, the mechanism for transcription and transulation is universal
However, organisms without introns cannot undergo splicing, so cannot form pre-mRNA similar to eukaryotic organisms
What are the three ways in making a DNA fragment?
Using reverse transcriptase on mRNA:
Scientists identify the organism that forms a lot of the protein needed (such as B cells from islets of langerhans that produce a lot of insulin), so this means these cells contain a lot of the mRNA required.
The reverse transcriptase binds to the mRNA strand and brings along complementary DNA nucleotides, forming cDNA (complementary DNA)
Then another enzyme removes the RNA bases, leaving only a single strand DNA nucleotides
Then DNA polymerase attaches to the single stranded DNA nucleotides, and brings over complemenetary DNA nucleotides and joins them together via condensation reaction forming the phosphodiester backbone.
Using restriction endonuclease:
The restriction endonuclease is complementary and specific to a specific base pairs on the DNA (recognition site), where there are two on the DNA base sequence, and this cuts the phosphidester bonds in the recognition site.
This then forms a DNA fragment
Using Gene machine:
Where a high tech machine identifies the primary structure of the protein, and then forms the mRNA base sequence and then identifies the DNA base sequence.
This DNA base sequence is then entered to the computer to check if a safe and non-harmful protein does not get formed.
The advantages and disadvantages of the three ways in forming the DNA fragment: EDIT THIS ONCE EXAM QUESTIONS FINISH
Using a gene machine it is faster, with greater accuracy, and the DNA formed is free of introns so can be transcribed and transulated by prokaryotic cells
What is the difference between in vivo and in vitro gene cloning:
In vivo gene cloning takes place inside the organism (called host cell)
In vitro takes place outside the organism (in glass)
Everything about in vivo gene cloning: Simple steps
The use of restriction endonuclease to form DNA fragment with sticky ends (not blunt) and same one for the Plasmid
And the DNA fragement should have additional bits, promoter and terminator region.
The DNA fragment should be added to the cut out plasmid via DNA ligase
Transformation: Plasmid entering the bacteria
Identifying which bacteria has the plasmid
For In vivo gene cloning the importance of sticky ends, and how they form recombinant DNA:
Using the restriction endonucleases, so that the sticky bits of the DNA hangs out, a similar enzyme is used to cut the plasmid, so that the DNA bases are complementary to each other. Then DNA ligase catalyses the reaction between the nucleotides, so that catalyses the reaction for phosphodiester backbone. The plasmid now has recombinant DNA.
Key point: there are other vectors that can be used, but plasmids are normally used, also plasmids almost always code for antibiotic resistance.
The problems with restriciton endonucleases used to cut DNA:
Since the same restriction endonucleases are used they form many overhanging DNA bases.
These bases are complementary to each other, and causes the nucelotides to join together, without being used in a plasmid, so cannot be used as a vector to transfer to host cells.
And the host cells will recognise that this is foreign DNA so will release an enzyme to break down the DNA fragements, so the required protein will not be transcripted and transulated
What extra bits do the DNA fragment require:
Additionally, the use of forming a DNA fragment, requires another extra lengths of DNA, such as a prototer region, allowing RNA polymerase and transcription factors to bind initiating transcription and transulation. Also for the release of RNA polymerase and transcription factors, terminator region required.
How can the plasmid enter the Bacteria?
Recombinant DNA enters host cells, through transformation: Plasmids are usually small, has a slightly negative charge, so they are polar, charged, hydrophillic and can’t pass through the phospholipid bilayer. They are mixed into a medium containing calcium ions (positively charged - so neutralised the electric repulsion) and heated to high temepratures, allowing more gaps between the phospholipids.
However, not all the plasmids enter the host cells: Some bacteria do not take in the plasmids, some of the DNA fragements join together and the host cells take this instead, and since bacteria recognises a foreign DNA it uses an enzyme to cut
How to identify which bacteria have the plasmid:
There are three scenarios that can happen:
The bacteria didn’t take plasmid
The bacteria took in plasmid, but not the plasmid with recombinant DNA
This is because, sometimes the cut plasmid reforms by itself, not having the DNA fragment
The bacteria took in plasmid, with the recombinant DNA
The first scenario: Did the bacteria even take the plasmid
So, most plasmids have antibiotic resistance, so add antibitic, will kill the bacteria without any plasmids
The second scenario: Use replica plating
In a petri dish, transfer the bacteria colonies in the exact positions. So, you have petri dish A and petri dish B
Then in petri dish A add antibiotic and some of the bacteria colonies with die, and identify the ones that die, they have the recombinant DNA
Another way is simplier: Fluroescent protein being easily seen OR produce an enzyme which action can be seen.
For fluroescent protein, the bacteria will glow and the bacteria that do not glow have the flourescent protein so has the recombinant plasmid
The recombinant plasmid will not turn the substrate blue as they cannot synthesise the protein.
The key idea: Bacteria with the recombinant DNA plasmid, has the DNA fragement added to the area where the plasmid is forming antibiotic resistance, so the bacteria with the recombinant DNA will die.
The steps of In vitro gene cloning:
Key point: the reaction inside the PCR can stop, if all the primers and the free DNA nucleotides run out.
Takes place outside the living organism, which is the polymerase chain reaction that takes place in the machine, which produces copies of the DNA fragement in a continuous cycle.
For new DNA fragments to be formed:
DNA nucleotides required
DNA polymerase (Taq polymerase)
Primers: Short sequences of single stranded DNA, that signal where the DNA polymerase should add nucleotides
Stage 1: 95 degrees
DNA fragment is heated to a high temperature, this breaks the hydrogen bonds between the nucleotides, causing the strands to separate
Stage 2: 55 degrees
The temperature is reduced, so primers can form complementary pairs at the end of each strand
Primers also prevent two complementary DNA strands from joining together (prevents annealing), and allow a starting sequence for DNA polymerase
Stage 3: 72 degrees
The temperature is increased, with the help of primers, taq polymerase adds complementary bases to each strands, forming two identical DNA strands
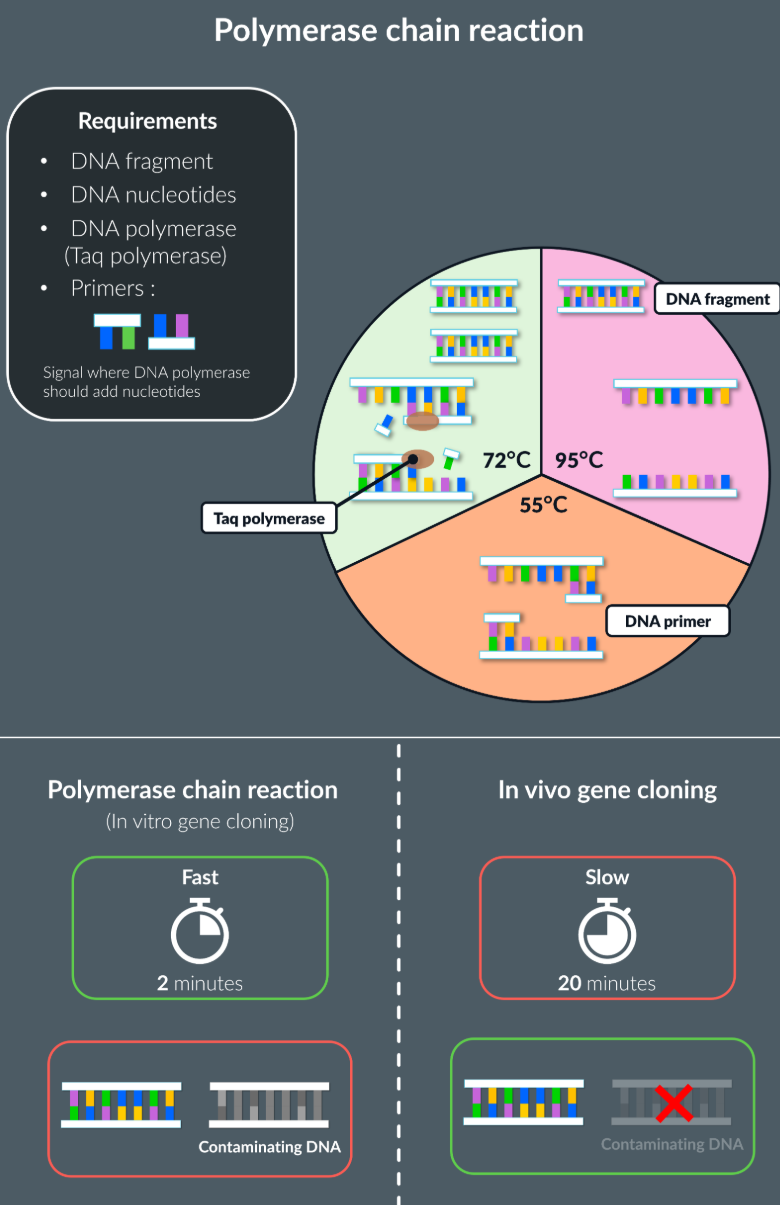
Comparing between in vivo and in vitro gene cloning:
Adv with in vitro:
Quicker
no living organism required, no complex culturing required
Adv with in vivo:
Usefull when wanting to introduce recombinant DNA to another organism such as humans (gene therapy)
No risk of contamination, as the gene is cut by the same restriction endonuclease, as only multiplies the DNA fragment that we want, as the in vivo means that any of the DNA fragment is replicated.
What are the two ways to label a DNA probe?
Radioactively label the probes, using nucleotides with isotopes of phosphate, these are radioactive so can be seen under a special scanner once hybridised with DNA base sequence
Fluorescently label probes, that emit light under special light, once DNA probe is hybridised with complementary DNA base sequence
The steps of locating genes (DNA hybridization):
DNA probe is made that has specific base sequence complementary to base sequence of a particular gene. It is made from replication of many DNA fragements from PCR (polymerase chain reaction), and is heated so that the hydrogen bonds are broken and only single stranded DNA probe is formed
The double stranded DNA is then heated, to separate the two DNA strands, and is mixed with the probe and cooled.
Then it is washed to remove any unhydridised DNA probe
Then identify the hydridised probe and DNA with special light for fluorescent and special scanner for radioactive hydribised probe and DNA
Why is it important to replicate many of the DNA probes from PCR (polymerase chain reaction)?
So that if only one DNA probe, then very unlikely to bind to complementary base pair on the DNA strand even though the locating gene DNA strand is present
Also once cooled there is a chance the DNA strands would just join via hydrogen bonding instead of with the DNA probe
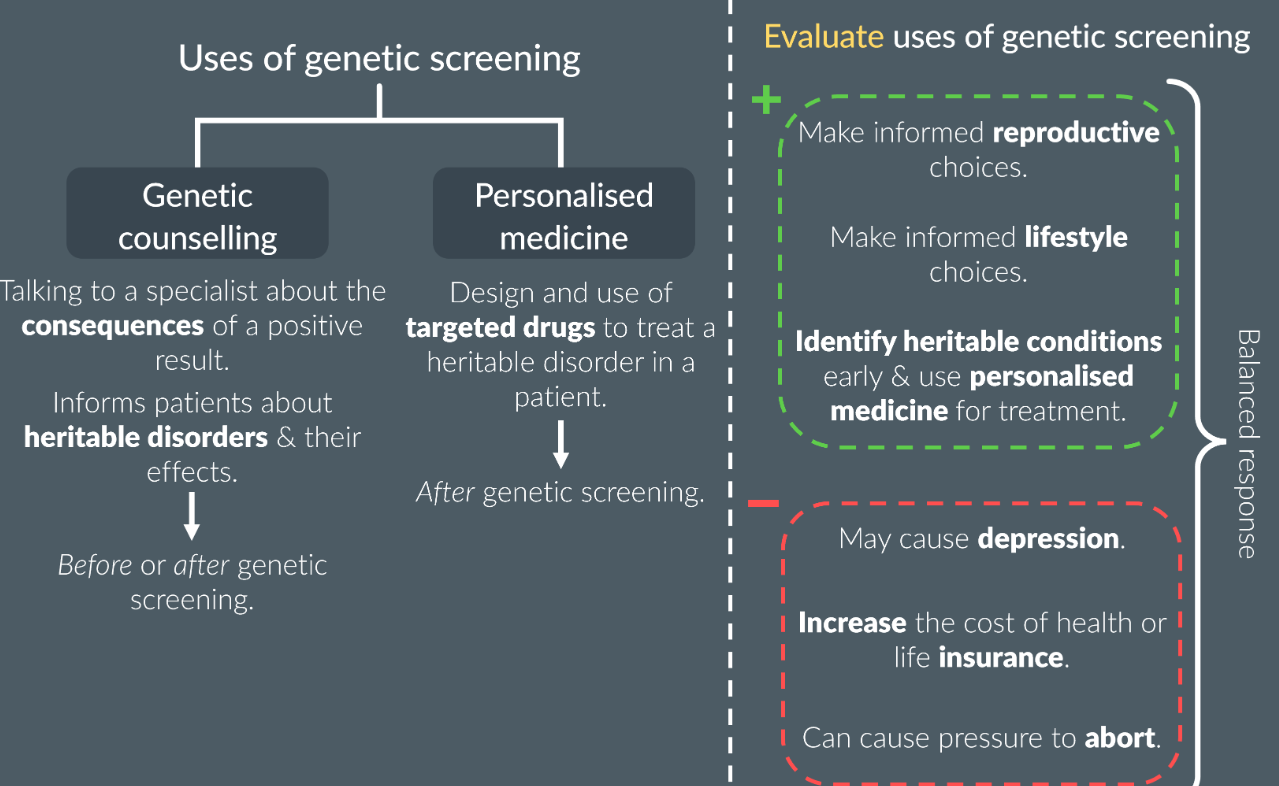
Personalised medicine and genetic counselling:
Everything about genetic fingerprinting:
Genetic fingerprinting is based on the number of non-coding regions (VNTRs - variable number tandem repeats), becuase non-coding regions don’t code for proteins, they do not effect organisms survival so there is great variation in the number of non-coding region repeats.
Gel Electrophoresis is used to separate DNA fragments based on their size, the DNA is negatively charged molecule, and with the smaller fragements they move faster towards the positive terminal, so they travel a greater distance.
Extraction, from a sample of blood or root hair, take out the DNA strand, then increase the quantity using PCR
Use restriction endonucleases to cut out specific DNA bases
Add sample to the sample well and add voltage, so can travel towards the positive terminal
Then use either radioactive or fluorescent DNA probes for hybridisation
Then take a picture using special light or special scanner

What method to find the base sequence of a gene?
Restriction mapping
Restriction endonucelase, cuts the DNA at various recognition sites, to form various fragments
They are separated and identified with gel electrophoresis
These results are scanned and interpreted by computer software
PCR cycles are used to speed up the process.