BIMM 134: Discussion
1/89
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
90 Terms
[start wk.1] Cancer is the _____ leading cause of death in the US.
Cancer is the second leading cause of death in the US.
Note: All diseases show a statistically significant decrease in 2023 compared to 2022, except CANCER…
Lifetime risk of men/women getting cancer
1 in 3 (for men & women)
Top three incidences of cancer
Men:
Prostate (30%)
Lung & Bronchus (11%)
Colon & Rectum (8%)
Women:
Breast (32%)
Lung & Bronchus (12%)
Colon & Rectum (7%)
Top four cancer deaths
Men:
Lung & Bronchus (20%)
Prostate (11%)
Colon & Rectum (9%)
Pancreas (8%)
Women:
Lung & Bronchus (21%)
Breast (14%)
Pancreas (8%)
Colon & Rectum (8%)
Tobacco use
Lung cancer death rate: 90%
All cancer death rate: 1/3
At diagnosis, ____% of cancers have already metastasized
_____% of US Budget spent on cancer research (pre-Trump admin)
At diagnosis, 70% of cancers have already metastasized
~0.1% of US Budget spent on cancer research (pre-Trump admin)
What is cancer?
> 100 forms of the disease
Patient-specific mutations (inter-patient)
Genetically unstable
Variation within (intra) tumor
Variation between primary and metastasized (inter) tumors
Etc.
Main processes/pathways responsible of cancer
Cell division
Cell death
Cell differentiation
Metabolism
Multi-step process (4-7 mutations for malignant transformation)
But still requires at least one “renegade cell” to begin the development process
Classification - Naming, Grading, & Stage
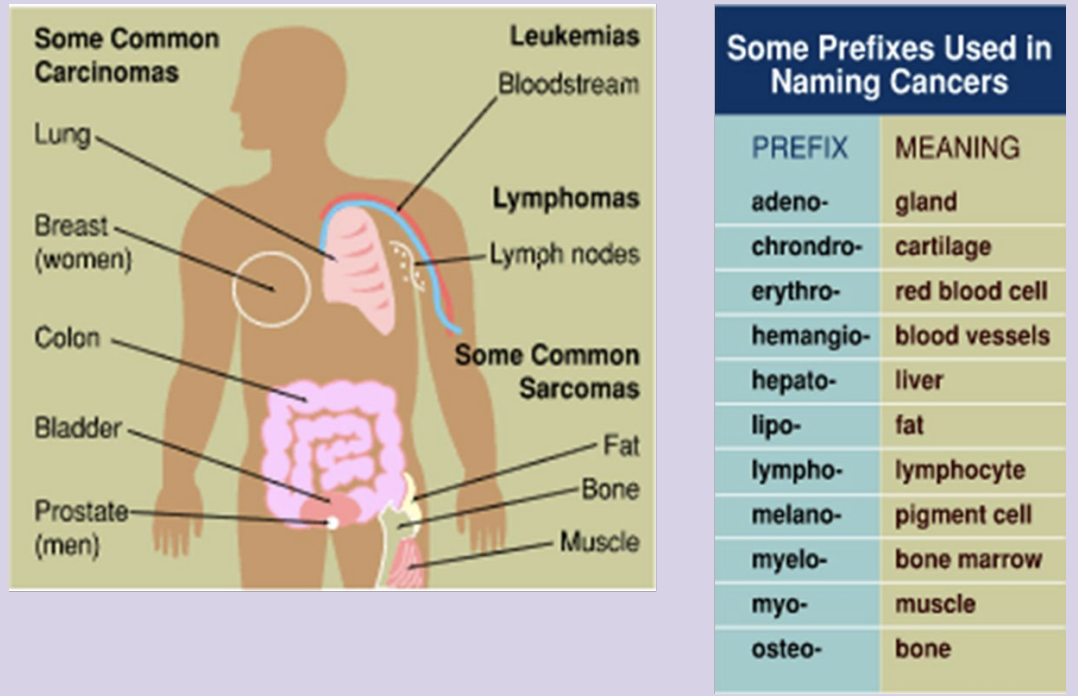
Prefix generally based on originating tissue
adeno-
gland
chrondro-
cartilage
erythro-
red blood cell
hemangio
blood vessels
hepato
liver
lipo
fat
lympho
lymphocyte
melano
pigment cell
myelo
bone marrow
myo
muscle
osteo
bone
Carcinoma
Epithelial cells lining body cavity, glands, skin
Squamous cell carcinoma
Derived from protective layer of cells over other underlying cells (ex: skin. cervix)
Adenocarcinoma
derived from cells lining secretory cells (ex: mucous producing cells within lung, colon, prostate)
Sarcoma
Derived from connective tissues (ex: bone, fat)
Hematopoietic
Blood producing cells
Lymphoma: from B- and T-cells (solid mass in lymph tissue)
Leukemia (circulating malignancy)
Grade: appearance of cells in biopsy
Low vs high
4 features to distinguish grade
Mitotic rate - how many/fast cells divide
Higher mitotic rate = more aggressive cancer (worse grade)
Nuclear grade - normal/abnormal nuclear shape
More abnormal nuclei = worse grade
Cellular differentiation - state of cell specialization
Less differentiated (more abnormal, less specialized) = worse grade
Surgical margins - proximity of tumor cells to surgical edge
Positive margins (cells at the edge) = higher chance cancer remains → worse prognosis
Tumor Stage
how far the cancer has spread
TNM Staging (I - IV)
T - primary Tumor (T0-4, x)
N - absence/presence in regional lymph Nodes (N0-3, x)
M - absence/presence of distant Metastasis (M0-1, X)

OG Hallmarks of Cancer
1. Gain-of-function oncogenes
2. Loss of function tumor suppressor
3. Loss of apoptosis
4. Gain telomerase activity
5. Gain blood supply
6. Spread/Growth at distant sites
Proto-oncogene vs Oncogene vs Tumor Suppressor
Proto-oncogene: promotes cell division under normal conditions
Ex: Ras and Myc
Oncogene: a proto-oncogene after a gain of function mutation
Only requires single mutation to promote cancer development (dominant)
Tumor suppressor: gene that normally prevents excessive cell division
Requires loss of function mutation in both alleles of gene to promote cancer development (recessive)
Ex: Rb and p53
Apoptosis: destruction of cell
Chromatin condensation
DNA fragmentation
Cell shrinkage
Cell protrusions “blebbing”
Necrotic cell death
Membrane disruption
Scattered cell debris
Decrease of apoptosis in cancer cells
Gain of anti-apoptotic proteins (ex. Bcl-2)
Loss of pro-apoptotic proteins (ex. BAX or p53)
Telomeres
repeat sequences at the ends of chromosomes
Normally protect from DNA damage, especially from the typical shortening of chromosomes after each DNA replication
Telomerase
protein that maintains telomere length in embryonic stem cells
After differentiation, telomerase activity is silenced in cells, which helps prevent uncontrolled cell division.
Differentiation = cells becoming specialized for a specific job.
Gain of Telomerase Activity
Cancer cells lose differentiation state → regain telomerase activity → unlimited lengthening of telomeres → cell immortality
Angiogenesis
Tumors are normally limited in growth to 1-2 mm
Require diffusion of oxygen/nutrients and removal of waste
Angiogenesis: formation of new blood vessels
Normally, the process is activated for new cell growth such as healing
Cancer cells initiate an angiogenic switch to promote cancer growth
Increases release of pro-angiogenic molecules (ex. VEGF)
Decreases release of anti-angiogenic molecules (ex. thrombospondin)
Allows invasion of cancer cells into the bloodstream → metastasis
Metastasis
Spread of tumor to distant sites
Up to 70% of patients with invasive cancers have metastasized
Metastasis leads to 95% of cancer related deaths
Example mutations:
Loss of E-cadherin
Gain of N-cadherin
Next Gen Hallmarks of Cancer
Reprogramming energy metabolism
Tumor-promoting inflammation
Avoiding immune destruction
Genome instability and mutation
Reprogramming energy metabolism - Warburg Effect
Cancer cells switch from oxphos to glycolysis even in the presence of oxygen
Faster than oxphos
Produces other intermediate biomolecules necessary for cell growth (ex. amino acids, lipids, nucleic acids)
Intermediates used in anabolic processes
Lactic acid creates an acidic tumor environment
Promotes tumor growth
Suppresses immune system
etc.
1.b. Reprogramming energy metabolism - Reverse Warburg effect
Reverse Warburg effect: cancer cells changing the behavior of surrounding normal cells
Cancer cells release ROS and other factors, which switch healthy stromal cells to aerobic glycolysis
Lactate produced by stromal cells is transported to cancer cells to use for energy and cell growth
Heterogeneity of tumor: Warburg and Reverse Warburg effect
2. Tumor-promoting inflammation
Inflammatory cells attracted to tumor
Factors from cancer cells induces release of carcinogen and promoters from inflammatory cells into the tumor microenvironment
3. Avoiding immune destruction
Disable immune response
Cancer cells evade immune response
4. Genome instability and mutation
Loss of DNA damage repair, detection and resistance mechanisms
Cell Signaling - Tyrosine Kinase Receptor
Ligand binding to surface receptor activates the tyrosine kinase → phosphorylates tyrosine residue on proteins → activating phosphorylation cascade
Various growth factors regulated by TKR
Important for cell growth and division regulation
Kinases/phosphatases in signal transduction pathways are mutated in cancer cells
Normal TKR Mechanism
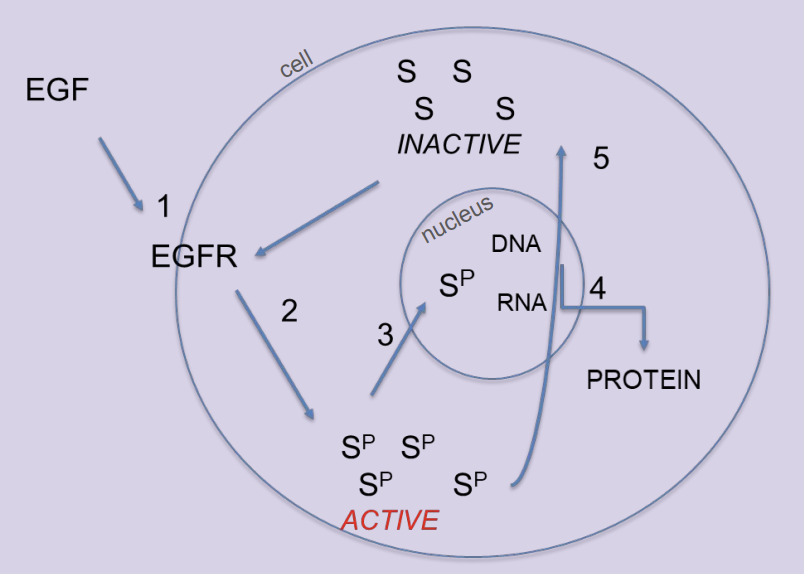
1st messenger (EGF) binds and activates TKR (EGF receptor)
TKR adds phosphates to various intracellular signal proteins (S) to activate them
This activates proteins in nuclear
Transcription factors → turning genes on/off for protein production
Proteins created for cell division
Phosphatases remove phosphates from signal proteins (S) → inactivate TKR (EGF receptor)
Cancer cells: proteins for cell division made w/o including growth signals
Types of Kinases
Tyrosine (Y) Kinase
adds phos to tyrosine residues
Serine(S)/Threonine(T) Kinase
adds phos to serine and/or threonine residues
Dual Kinase
has activity of both tyrosine and serine/threonine kinases
Lipid Kinase
adds phos to lipids
Tyrosine Kinase Receptors (TKR)
Many types of TKRs - share various protein domains between one another
Some ligands can activate various receptors
Some receptors can be activated by various ligands
The different receptor pathways can have some downstream pathway overlap
Ex: EGFR, HER2, etc.
EGF-RAS-MAPK Pathway (1 to 3)
EGF ligand binds to EGFR
Binding causes conformational change to enable dimerization
Dimerization initiates autophosphorylation of tyrosine residues on EGFR
EGFR is a tyrosine kinase
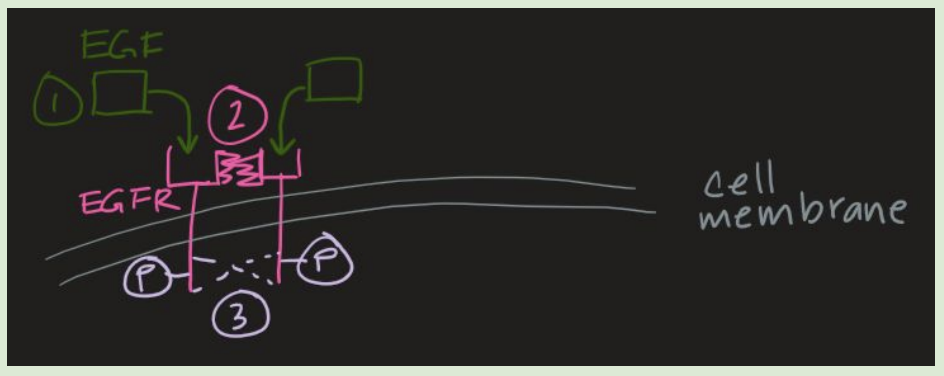
EGF-RAS-MAPK Pathway (4 to 5)
4. Autophosphorylation recruits GRB2 signal molecule to EGFR (interacting with SH2 domain of the signal molecule)
SH2: binding domain; key for messenger binding to TKS
SH2 Domain- allows activated receptors and GRB2 to bind
5. SH3 domain of GRB2 binds to SOS (intracellular messengers)
proteins bind to one another through SH3 domain
SH3: binding domain; key for messenger binding to TKS
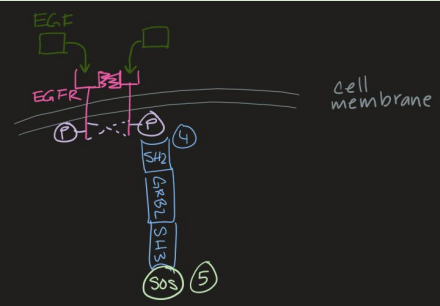
EGF-RAS-MAPK Pathway (6)
6. SOS recruitment activates RAS
SOS gets rid of GDP and allows GTP to bind so that RAS becomes active
SOS is a GEF protein
GEF proteins transfer GTP to other proteins
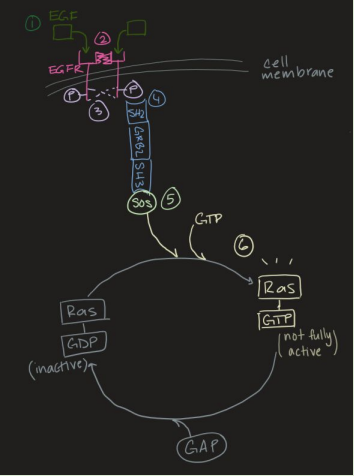
EGF-RAS-MAPK Pathway (7)
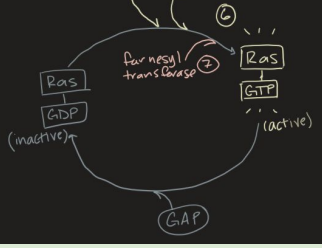
7. Farnesyl transferase adds farnesyl lipid and methylation to RAS-GTP and anchors it to the membrane = fully activating RAS
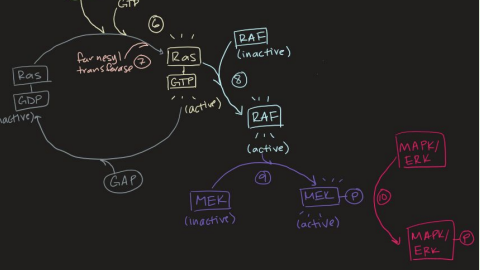
EGF-RAS-MAPK Pathway (8 to 10)
8. RAS activation recruits RAF
RAF = ser/thr kinase
9. Active RAF phosphorylates MEK
MEK = dual kinase
Dual kinases have tyrosine and serine/threonine kinase activity
10. Active MEK phosphorylates MAPK/ERK to activate it
MAPK = ser/thr kinases
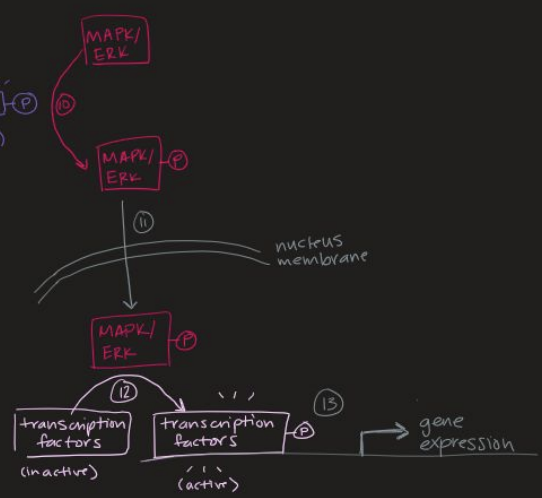
EGF-RAS-MAPK Pathway (11 to 13)
11. Activated MAPK/ERK is translocated to the nucleus
12. Once in the nucleus, MAPK/ERK activates various transcription factors — promoting cell cycle and division.
MAPK/ERK is Ser/Thr kinase
Ex: AP-1, Fos/Jun,
or Myc — transcription factor that helps change gene expression/ increases proliferation.
13. Active transcription factors will initiate transcription of various cell growth genes (ex: cyclins)
RAS-PI3K-AKT
Active RAS can also activate PI3K via direct binding
PI3K is lipid kinase
Active PI3K will phosphorylate PIP2, turning it into PIP3
PIP3 binds to both PDK1 and AKT
PDK1 and AKT is Ser/Thr kinase
PDK1 activates AKT via phosphorylation
Activated AKT activates mTOR
mTOR is a Ser/Thr kinase
mTOR
Increased survival
Increased proliferation
Increased motility
Angiogenesis
Decreased apoptosis
RAS-PI3K-AKT-mTOR
Mutations in PI3K pathway are most common in cancers
Loss of PTEN (phosphatase), a common tumor suppressor mutation
Converts PIP3 back to PIP2 → inhibiting the pathway
Oncogenic mutations
Point mutations
abnormal protein
Gene amplification
excess normal protein
Chromosomal translocation
When a piece of one chromosome breaks off and attaches to a different chromosome.
Local DNA rearrangements
Insertional mutagenesis
Oncogenic Mutations: Tyrosine Kinase Receptors
Point mutation/deletion
Ligand-independent dimerization and activation
Gene amplification
Overexpression of receptors
Ligand-independent dimerization and activation
Inducing conversion of paracrine to autocrine signals
Normally differentiated cells transition to paracrine signaling
Autocrine signals will activate TKRs → promoting continued growth
Ex: EGFR (HER1), HER2
Point Mutation - RAS
Single DNA base mutation
→ change 1 amino acid
→ abnormal protein
Normally, RAS is deactivated (GAP) after the signal pathway has been activated
Point mutation prevents its interaction with GAP
→ prevents hydrolysis of GTP
→ Ras stays activated
“Point mutations are prevalent in all three RAS isoforms (H-Ras, N-Ras, K-Ras)”
H-Ras (hematopoietic)
K-Ras most commonly mutated
(pancreatic, lung, and colon cancer)
N-Ras
(Neuroblastoma, Melanoma)
Isoforms have strong homology at the GAP binding site
Point Mutation - RAS
Most common mutations in codon 12, 13, 61
Glycine (G) to aspartic acid (D)/valine(V)/cysteine(C)
Adding bulky side groups that interfere with GAP-activation at the catalytic site on RAS
G12C - lung adenocarcinoma
Tobacco use
G12D - pancreatic and colorectal cancer
RAS mutation therapeutics
Originally considered undruggable bc
The GTP binding pocket is relatively inaccessible
High affinity for GTP
High levels of cytoplasmic GTP
Two main types of RAS mutation therapeutics
Off inhibitors: bind to inactive mutant
On inhibitors: bind to active mutant
RAS mutation therapeutics
Inhibitors target G12C mutation: Sotorasib & Adagrasib (RAS off)
Inhibitors target G12D mutation: RMC-6236 (RAS on)
Farnesyltransferase inhibitors
RAS mimic: Rigosertib
Inhibitors target G12C mutation: Sotorasib & Adagrasib (RAS off)
covalent binding to cysteine in the GTP binding pocket preventing/decreasing Ras activation
Don’t affect WT RAS or other RAS mutations
Eventual resistance to these drugs (inc RAS activation via other pathways and mutations)
Inhibitors target G12D mutation: RMC-6236 (RAS on)
Non-covalent binding to any G12 mutation
Potentially treat various RAS mutant driven cancers
Could avoid drug resistance seen with other RAS off therapeutics
Farnesyltransferase inhibitors
Prevents RAS localization to membrane
Not successful as stand alone therapeutic
RAS mimic: Rigosertib
Binds to normal RAS binding partners and inhibits their activity
RAF to inhibit downstream MAPK/ERK pathway
Also binds PI3K inhibiting downstream PI3K/AKT/mTOR pathway
Gene Amplification - Myc
Results in many copies of the gene (100-1000)
Excess protein is present in cells (protein itself is normal)
Amplified Myc is seen as extrachromosomal elements or intrachromosomal areas
Myc: transcription factor that form hetero dimer with the related TF Max
Binds 100s of gene promoters (esp those involved in cell cycle progression like cyclin D; CDK4/6)
Overexpression can activate Myc
Normal Myc expression is initiated when there is cellular stress, which subsequently activates Arf and p53 to pause cell cycle and induce apoptosis
For a full oncogenic effect, loss of p53 is also needed
Myc Therapeutic - OMOMYC
Small molecule inhibitor
Prevents Myc and Max from binding to DNA
→ blocks Myc turning proliferation genes on (ex: cyclin D; CDK4/6)
Chromosomal Translocation
A piece of a chromosome moves to another chromosome
Results in loss of normal protein control
Protein is made constantly instead of being initiated by a specific signal cascade
Chromosomal Translocation - BCR-ABL
ABL gene is normally part of chromosome 9
Translocation occurs on chromosome 22 downstream of the BCR gene
Resulting in the Philadelphia chromosome
→ BCR-ABL fusion protein
95% of chronic myelogenous leukemia cases have the Philadelphia chromosome
“The fusion protein contains the dimerization domain of BCR.”
Fusion of ABL to this dimerization domain promotes dimerization of ABL
This dimerization leaves ABL permanently activated
In normal conditions, ABL dimerization is controlled via a signal pathway
The tyrosine kinase domain of the fusion ABL will activate other growth pathways
RAS-MAPK and PIP3-AKT
Mutant protein is not transported to the nucleus (cytosolic)
→ increased activation of Ras/MAPK and PI3K pathways, and decreased normal apoptosis
BCR-ABL therapeutic: Gleevec (imatinib)
Gleevec targets the hybrid protein specifically, unlike other chemo and radiation therapies
Works by outcompeting ATP at the ATP binding site on the fusion protein
Prevents BCR-ABL from activating other proteins in signaling pathways
Anti-EGF receptor monoclonal antibodies
The antibody directly interacts with the receptor, blocking ligands from activating
Herceptin - Metastatic breast cancer with upregulated HER2
Binds and inactivates HER2
Erbitux - colorectal cancer with upregulation of EGFR (HER1)
These will be ineffective in patients with downstream oncogenic mutations (ex: RAS)
Intracellular tyrosine kinase inhibitors
Compete with ATP-binding site of kinase
TARCEVA and IRESSA (Gilotrif)
Developed to target common chemoresistance mutations in patients
mTOR inhibitor
Everolimus - inhibits downstream targets of mTOR
Inhibition of PI3K or tyrosine kinase pathways results in ________ of the other to increase cell survival and reduce ______
Inhibition of PI3K or tyrosine kinase pathways results in compensatory upregulation of the other to increase cell survival and reduce apoptosis
Breast cancer therapeutics
Tamoxifen
Herceptin
IBRANCE
LETROZOLE
Tamoxifen
Small molecule that inhibits estrogen/progesterone from binding to its receptor
Targets ER+ PR+ breast cancer
Blocking this slows growth of cancer cells
Herceptin
Antibody that binds and inactivates HER2
Used in patients who are HER2+++ (overexpression of HER2)
IBRANCE
Small molecule intracellular inhibitor of CDK proteins (control cell cycle/cell division)
Used in combination with anti-estrogen hormonal therapy
LETROZOLE
Hormone therapy - aromatase inhibitor
Increased survival compared to hormone therapy alone
Often used in combo with IBRANCE
Used to treat ER+ PR+ breast cancer
Three main mechanisms of Herceptin Resistance + Therapeutic Strats
Amplification or mutation of EGFR
Combine Herceptin or other EGFR Ab with a TK inhibitor like Iressa or Tarceva
Increase in alternative RAS activation
Combine Herceptin with RAS inhibitor (Rigosertib, FTI, Sotorasib, Adagrasib, etc)
Upregulation of PI3K/AKT/mTOR pathway
Combine Herceptin with PI3K/AKT/mTOR inhibitor (Everolimus)
RAS inhibitors
Sotorasib
Adagrasib
RMC-6236
Farnesyltransferase inhibitor (FTI)
Rigosertib
Myc inhibitor
OMOMYC
BCR-ABL inhibitor
Gleevec (imatinib)
Other kinase inhibitors
Erbitux (EGFR Antibody)
TARCEVA (intracellular TKR)
IRESSA (intracellular EGFR)
Everolimus (mTOR)
Vemurafenib (RAF)
Breast Cancer inhibitors
Tamoxifen
Herceptin
IBRANCE
LETROZOLE