Kinetics
1/54
There's no tags or description
Looks like no tags are added yet.
Name | Mastery | Learn | Test | Matching | Spaced | Call with Kai |
|---|
No analytics yet
Send a link to your students to track their progress
55 Terms
what is absorption
transport of drug from the site of administration to systemic circulation
what drug specific factors affect absorption
solubility, pH, particle size, dissolution rate, route of administration
patient specific factors that affect absorption
age, blood flow at absorption site, GI content
conditions that affect blood flow
PAD, HF
is unionized or ionized drug better at absorbing?
unionized drug is more lipid soluble and uncharged so is better at crossing cell membranes
better at absorbing
passive diffusion
net movement of drug from an area of high to low concentration
most common
facilitated diffusions
carrier or channel mediated transport
does not require energy
cannot transport against concentration gradients
active transport
carrier or channel mediated
requires energy/ATP
enables movement from low to high drug concentrations
what does ficks law describe
how substances move (diffuse) from an area of high concentration to an area of low concentration
what are transporters
membrane bound proteins that facilitate movement of molecules in and out of the cell
influx transporter
transports drugs into the cell
bidirectional facilitated diffusion and do not require ATP
OATP, OAT, OCT, PEPT
efflux transporter
transports drugs out of the cell
require ATP to function
P-gp, MRP, BCRP, BSEP, MATE
bioavailability
fraction of the dose that reaches systemic circulation and ranges between 0 and 1
what are the three types of bioavailability
fraction absorbed
intestinal bioavailability
hepatic bioavailability
fraction absorbed
fraction that is absorbed intact in the intestinal membrane
intestinal bioavailability (Fg)
the fraction of the drug in the enterocytes that escapes metabolism
hepatic bioavailability (Fh)
the fraction that enters the liver and escapes first pass metabolism
dissolution
the process in which the oral dose dissolves into the GI fluid
determined by hydrophilicity, lipophilicity, crystalline form of the drug, particle size
pH partition hypothesis
a non-ionized species will more readily partition (preference) into a lipophilic solvent than an ionized species
whether a drug is charged depends on pKa and pH of its environment
what is the point of drugs being prepared into salts
improve wetting: how liquid spreads over drug and causes dissolution
leads to faster dissolution and increased rate of absorption
what does the noyes-whitney equation describe
the rate of dissolution of a drug (how fast a drug dissolves in a liquid)
higher concentration of drug inside the higher the rate of dissolution
the rate of drug absorption is controlled by
the slowest step
when dissolution rate controls absorption which equation describes the rate
noytes-whitney equation
when the membrane penetration controls drug absorption, the rate is approximated by
Fick’s law
dissolution and membrane penetration are what kind of process
follow first order kinetics
a weak acid or base partially ionizes into its ions in what type of solution
an aqueous solution
what is percent ionization
amount of acid or base that dissociates into its ions at a specific concentration
strong acids and bases dissociate into its ions at a specific concentration so their percent ionization is 100%
drug distribution
how rapid and to what extent drug in plasma gets up taken by tissue
cannot measure tissue concentration so plasma concentration is measured instead
effect only occurs when a drug reaches site of action
drug distribution is driven by what
passive diffusion of the unbound drug
distribution phase
after administration the plasma drug concentration is much higher than that in the tissues
drives diffusion of drug into the tissue
post-distribution phase
equilibrium is established between tissue and plasma, ratio between concentrations rises and falls in parallel
waht is volume of distribution
reflects how a drug will distribute throughout the body
Vd is constant for a drug and is the ratio of drug in body and the plasma concentration at a specific time
causes of low Vd
large
hydrophilic
plasma protein bound
impermeable membranes
what are some implications of low Vd
low dose to saturate plasma
fast clearance: if free drug
tissues receiving equal exposure (see same amount of drug everywhere
what are some causes of high Vd
small
hydrophobic
tissue protein bound (stronger affinity for proteins in tissues)
permeable membranes
implications of high Vd
allows targeting of tissues
can cause toxic buildup
concentrated in specific tissues
is drug binding to plasma proteins reversible or irreversible
reversible
examples of proteins involved in protein binding
albumin, AAG, and lipoproteins are primary plasma proteins involved in drug binding
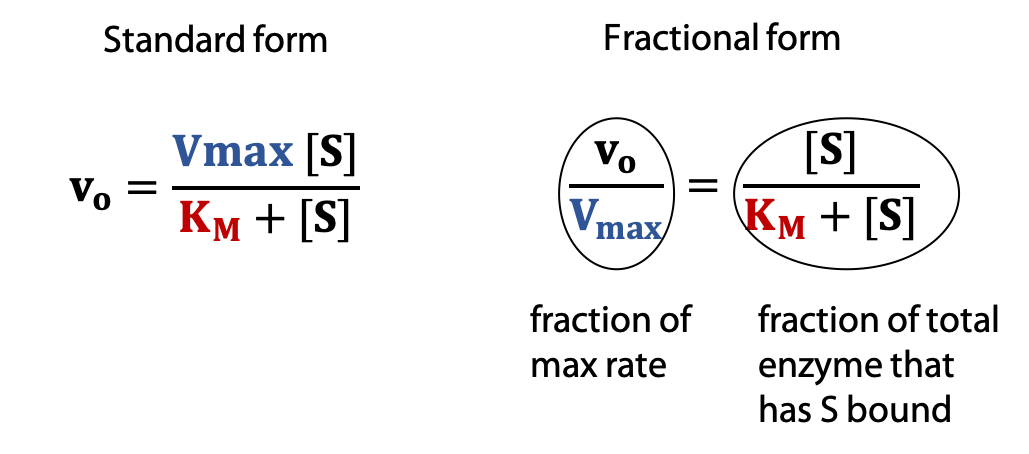
what does the Michaelis-mention equation describe
the velocity of metabolism (speed of metabolism)
when the velocity is half of Vmax, Chu is equal to
Km (Michaelis-Menton constant)
when Chu > km
rate is a zero order process
when Chu « km
rate approximates a first order process
renal excretion
parent drug is excreted out of the body via the urine
liver metabolism
parent drug is converted into metabolites by the liver
once the drug is converted to its metabolites, it is considered eliminated
rate of elimination and the rate of metabolism are what type of process
first order processes
clearance is dependent on what
liver and kidney function and how efficient they can extract drug from plasma
units of volume/time
total clearance is = to
renal clearance + hepatic clearance
clearance is constant but can be altered by
disease, medication, changes in organ function
large elimination rate constant means
short half life
low elimination rate constant equals
long half life
t1/2 =
0.693/K
first pass effect
removal of drug by GI tract and liver prior to entering systemic circulation
hepatic clearance is based on what model
well-stirred model
drug molecules are assumed to be distributed homogeneously
assumptions of the well stirred model
only unbound drug in the blood is subject to elimination
no membrane transport barrier
no concentration gradient of the drug within the liver
the concentration of the drug within the liver is equal to that in the venous blood
linear kinetics