
Target Based Approaches and Phenotypic Screening
Definitions:
ADME = absorption distribution metabolism excretion
Medicinal chemistry = generation of small, druglike molecules with clinical activity through the application of synthetic organic chemistry
Drug-target interactions = key interactions which promote association between the ligand under development and the target protein
Notes:
Part 1: The Drug Discovery Pipeline: Target-based approaches (more traditional) and phenotypic screening (looking at key properties before we know what key proteins are)
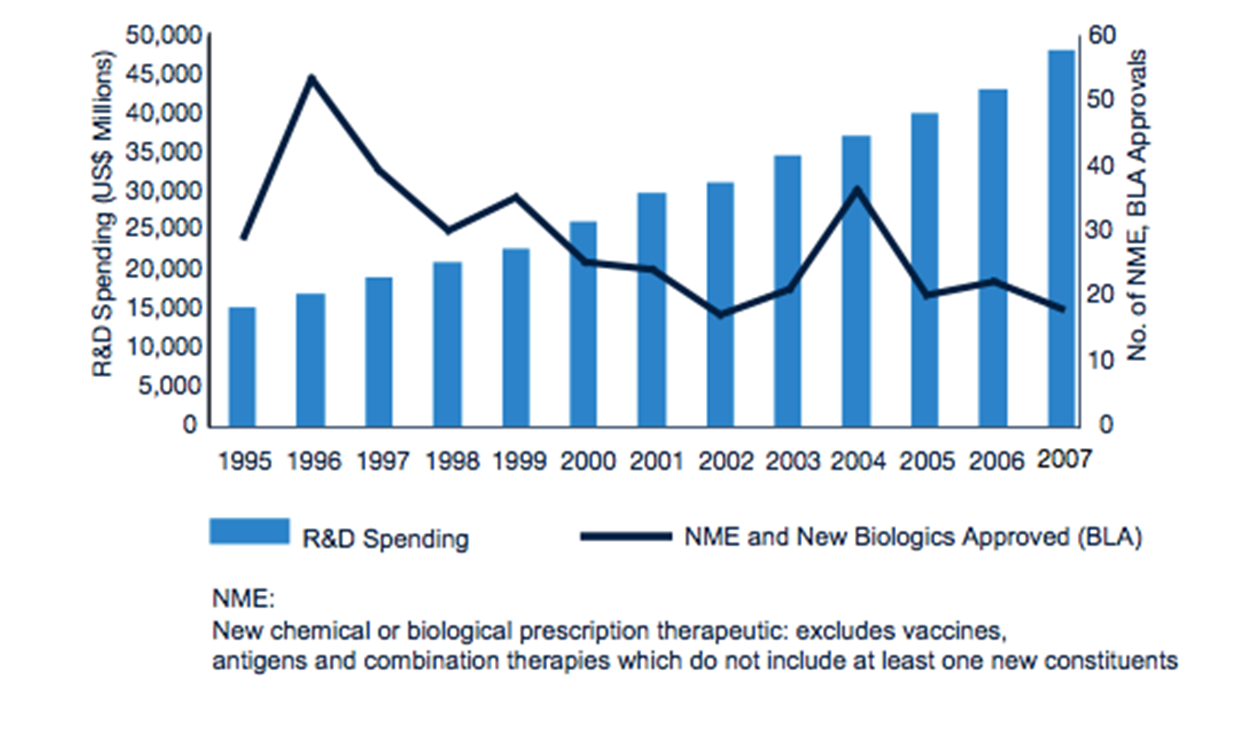
7: Drug discovery landscape - old data but gets message across. Less and less molecules making it to clinic but spending increasing
8: Finding a new drug against a chosen target for a particular disease usually involves high-throughput screening - large chemical libraries tested for their ability to modify target. Often robotically tested. Chosen drug targets modified to increase activity against target and reduce activity against unrelated targets. Drug likeness/ADME properties of molecule then improved.
9:
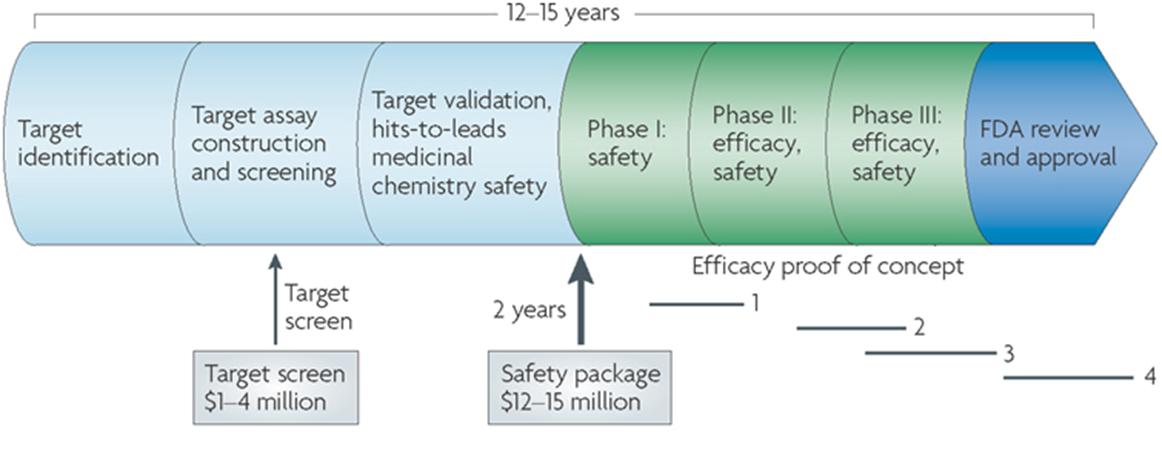
Light blue = preclinical, uses purified and isolated enzymes in vitro then goes onto animal testing
Interactions between medicinal chemists, pharmacologists, biochemists and pharmacists are required at every level for the drug development process. The chemists/pharmacologists/biochemists discover and optimise the drugs whereas pharmacists formulate and develop them. There’s very little point in investing between 100 million and 1 billion, and 10 to 15 years work discovering a drug and getting it to market only for the patient to misuse the drug and not receive any benefits from it.
10: The first step is target identification, where a target is identified which is associated with a desired clinical outcome eg:
ACE inhibition reduces blood pressure
blocking cardiac beta-adrenoreceptors can modify cardiac output and function
HIV protease inhibition halts viral replication
EGFR inhibition prevents cancer cell proliferation
11: Target selection:
New drug must have something to offer clinically and be capable of being highly profitable (big pharma view). From a regulator’s view, improvements on existing ‘gold standard’ drugs must be substantial. From a patient’s view, a drug is in demand if it avoids certain side effects of existing drugs. However a new drug may not be able to be afforded eg due to NHS budget or in developing countries where a particular issue is prevalent.
12: target examples and how a drug targeting them would work
Target | Mechanism |
Enzyme | Inhibitor - reversible/irreversible |
Receptor | agonist/antagonist |
Nucleic acid | intercalator (binder), modifier (alkylating agent) or substrate mimic |
Cell membrane ion channels | blockers or openers |
Transporters | uptake inhibitors |
13: A requirement of the drug development process is to validate that the selected protein is a robust target, which is confirmed using a number of molecular techniques to modify or remove protein of interest from a particular system eg:
siRNA based protein run-down
adenoviral/lentiviral delivery (transfection/infection method) into cells of genetic materials that encode for modified (eg site directed mutagenesis to make protein inactive) version of target → impacted cell responses/outcomes
genetic deletion (knockout) models - animals and cells prepared from them and compare to WT animal - check for fatal effects or if KO redundant
CRISPR - easily modifies genome allowing new scientific and therapeutic opportunities
14: Drug discovery and development is a challenging process. Challenges faced are having to produce a molecule with optimum pharmacodynamics (potency and selectivity for the target) while ensuring it has optimum pharmacokinetics (absorption into body with robust metabolic pathway).
15: The key to the success of a medicinal chemistry project is the ability to optimise molecules, making subtle changes to the molecule to make it a more successful drug.
Optimisation needs to account for potent and selective activity against the target protein associated with the clinical disease, and then achieving said activity at the desired concentrations in a physiological system (animal model and ultimately a human patient).
16: Put simply, pharmacodynamics are what the drug does to the body - the therapeutic effect, how it exerts its effect and how it interacts with the target. Small functional group changes can change the pharmacodynamic action of a drug eg beta agonist salbutamol and beta antagonist atenolol have for the most part very similar structures.


17: Pharmacokinetics is what the body does to the drug, entailing how well it’s absorbed, where the drug goes, how long it stays and how it’s metabolised.
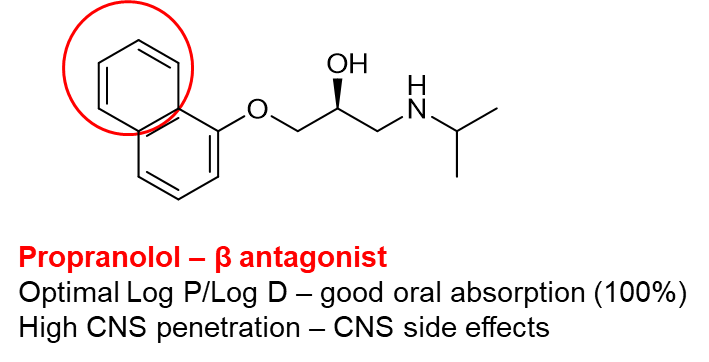
Like with pharmacodynamics, small functional group changes can change the pharmacokinetic action of a drug eg the only difference between atenolol which has poor oral absorption and low CNS penetration, and propranolol which has good oral absorption and high CNS penetration is a switch from amide group to an additional benzene ring.

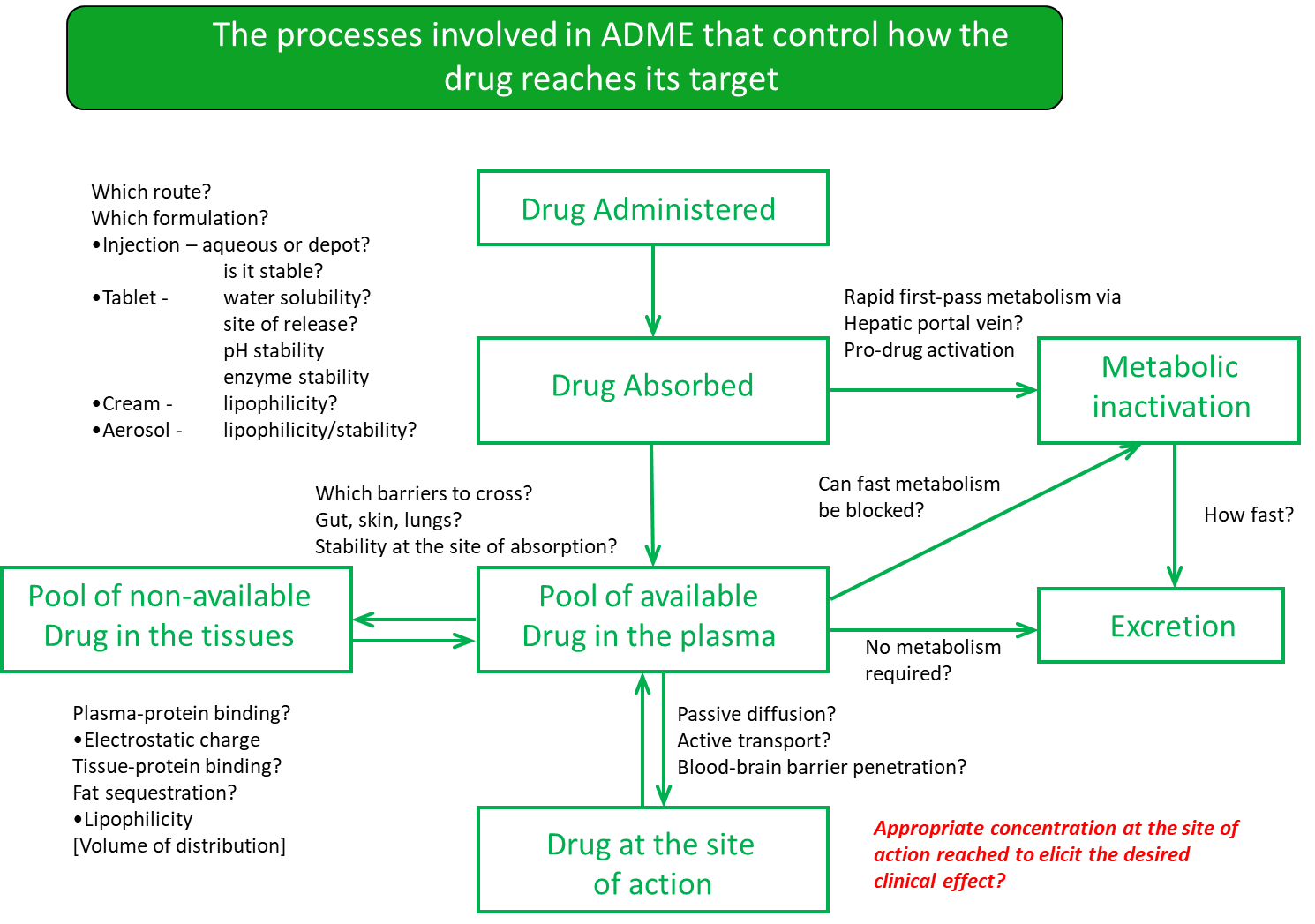
18: Pharmacokinetics usually assessed by absorption, distribution, metabolism, excretion and toxicology (ADMET)
19:
Part 2:
21: Compounds are sourced from natural products and synthetic molecules.
Natural products are best as purified molecules rather than extracts and are sourced from plants, sponges, bacteria and fungi etc eg taxol (anti cancer) derived from willow bark
Synthetic molecules typically used by big Pharma and are sourced from large scale libraries and are chemical building blocks of various molecular weights. These libraries are databases of compound chemical structure, purity, quantity and physiochemical characteristics. As much of the appropriate chemical space (?) is taken advantage of as possible to know what kind of chemical structure would be helpful
22: SAR = structure-activity relationship used for potency against target. Target may have different isoforms with different functions that don’t want targeting
usually start off with poor scaffold, scaffold hopping used to build more optimal drug
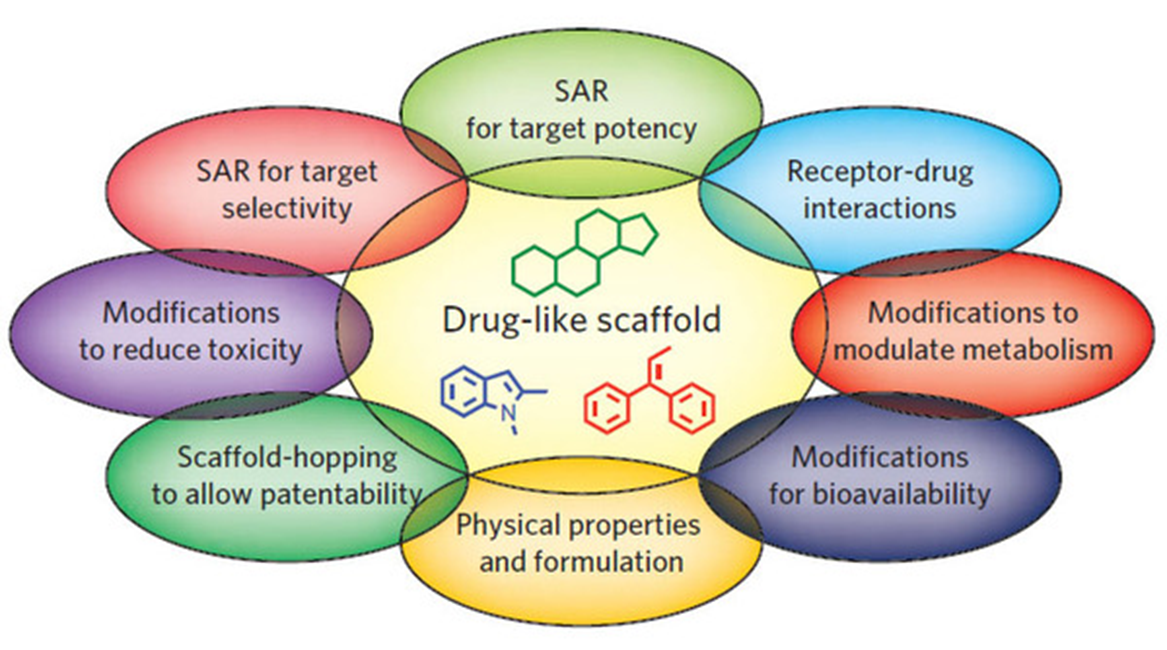
24: Drugs bind targets via their functional groups which allow them to interact with the target
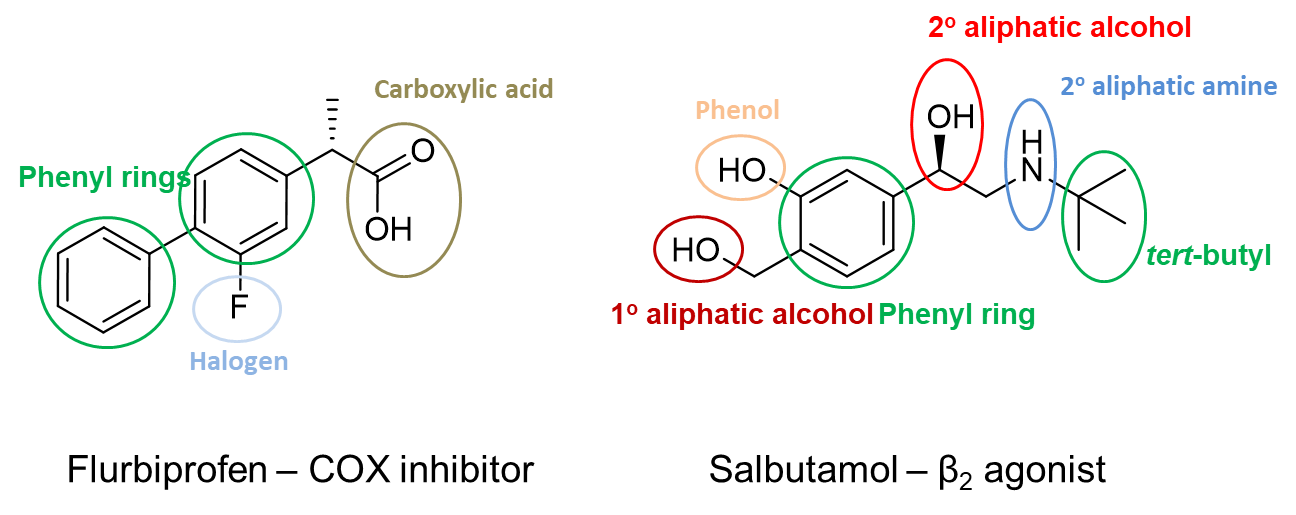
25: Drug functional groups interact with the different amino acids found in protein targets.
Drugs form short range van der Waals forces with non-polar, aliphatic side groups (glycine, alanine, valine, leucine, methionine and isoleucine) and aromatic side groups (phenylalanine, tyrosine and tryptophan)
Drugs form mid range hydrogen bonds with polar, uncharged side groups (serine, threonine, cysteine, proline, aspartate and glutamine).
Drugs form ionic bonds with positively charged (lysine, histidine and asparagine) and negatively charged side groups (glutamate and aspartate).
26:
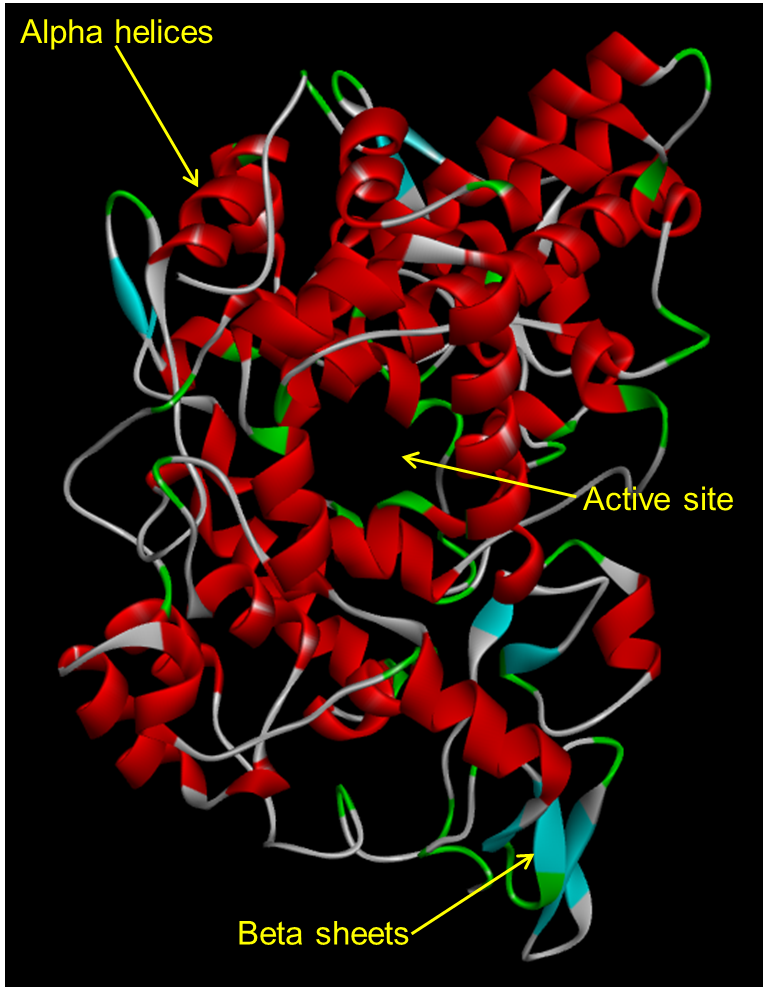
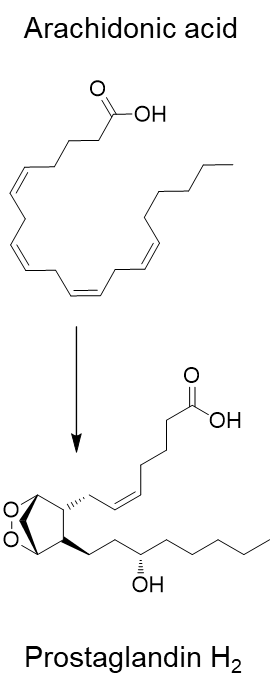
Above shows COX tertiary structure with the secondary structures displayed as a ribbon. COX’s tertiary structure allows it to perform its function by maintaining active site integrity for oxidation and cyclisation of arachidonic acid for prostaglandin synthesis.
29:
Enzymes globular tertiary structure has a surface which excludes small molecules from penetrating the interior. The active site is flexible and can expand and contract to allow substrates in and products out
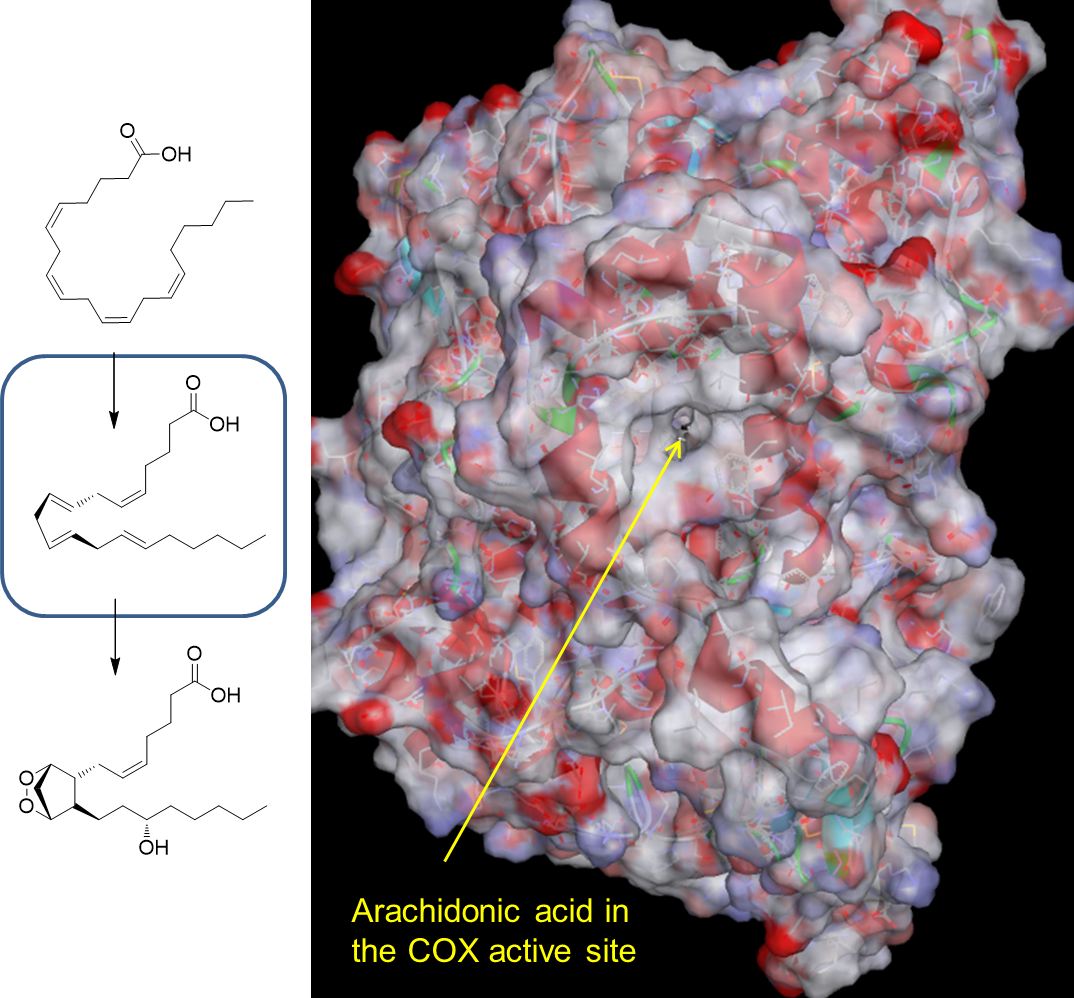
30: Interactions between amino acids bordering COX’s active site and arachidonic acid are a mixture of H bonds (directional, specific for substrate H bonding groups) and short range van der Waals between substrate surface and active site surface.
31: Flurbiprofen is a competitive COX inhibitor which binds in preference to arachidonic acid (has higher affinity) blocking prostaglandin synthesis therefore reducing inflammatory response.
For a compound to compete with the substrate it must have some structural resemblance to enable it to match the active site. To compete effectively it must form stronger interactions with the active site than the substrate does. Binding is dynamic meaning molecules associate and dissociate regularly so molecules need to remain bound to the active site for longer than the substrate to be effective.
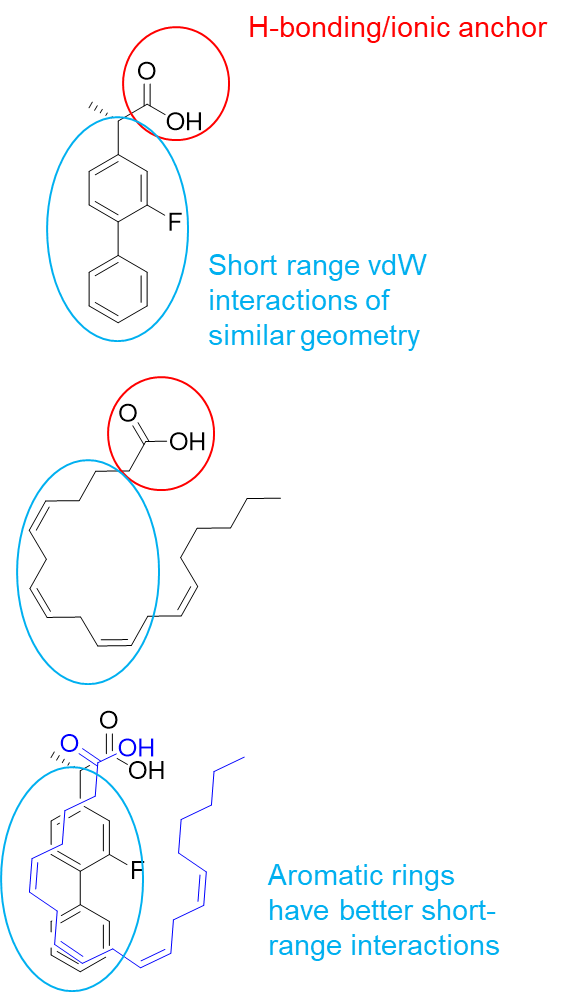
32: Driving potency and selectivity at the receptor level can be carried out via chain modification, ring substitution and modifications at different carbons.
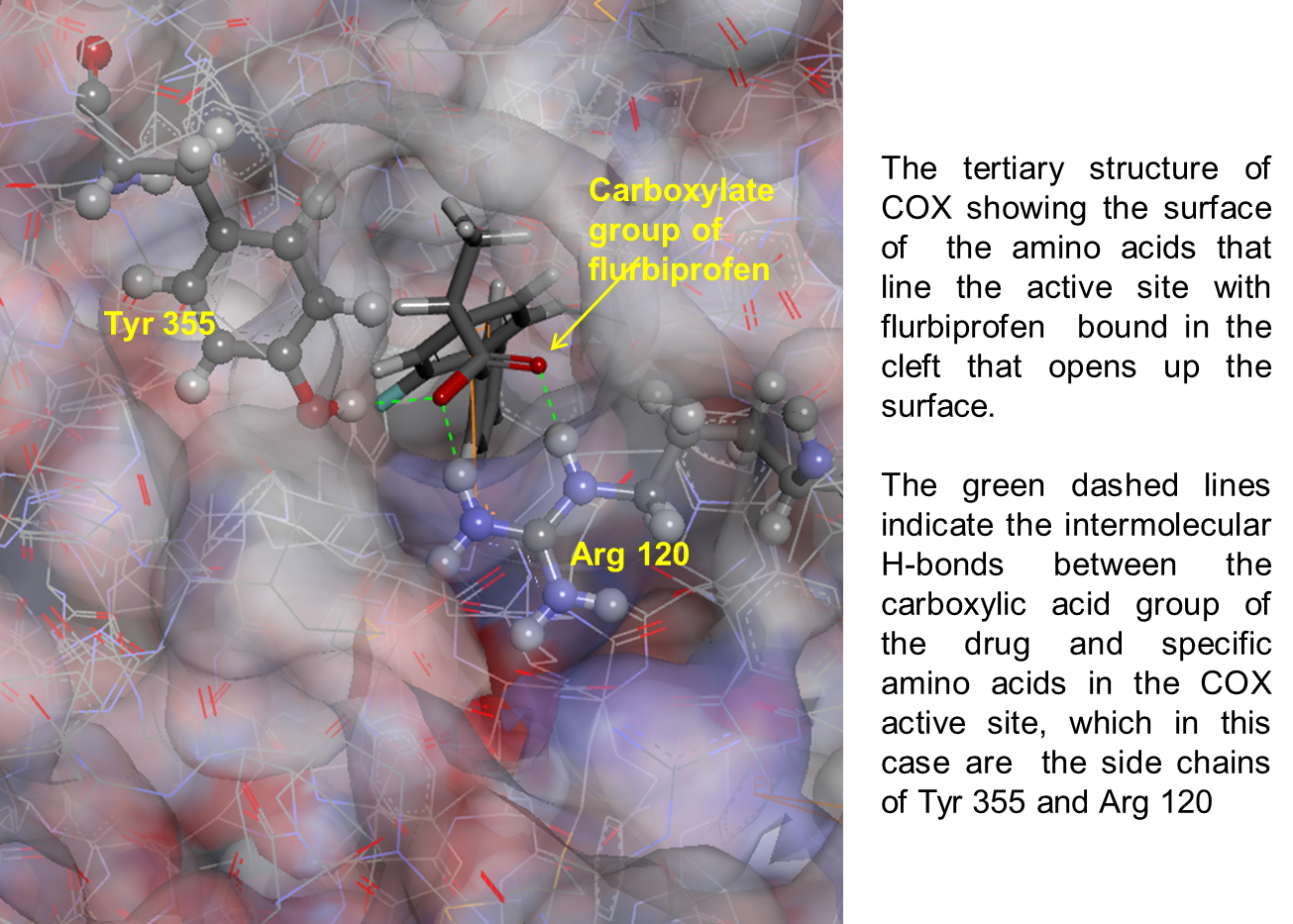
33: allows chemists to think if they can fill in any of the spaces
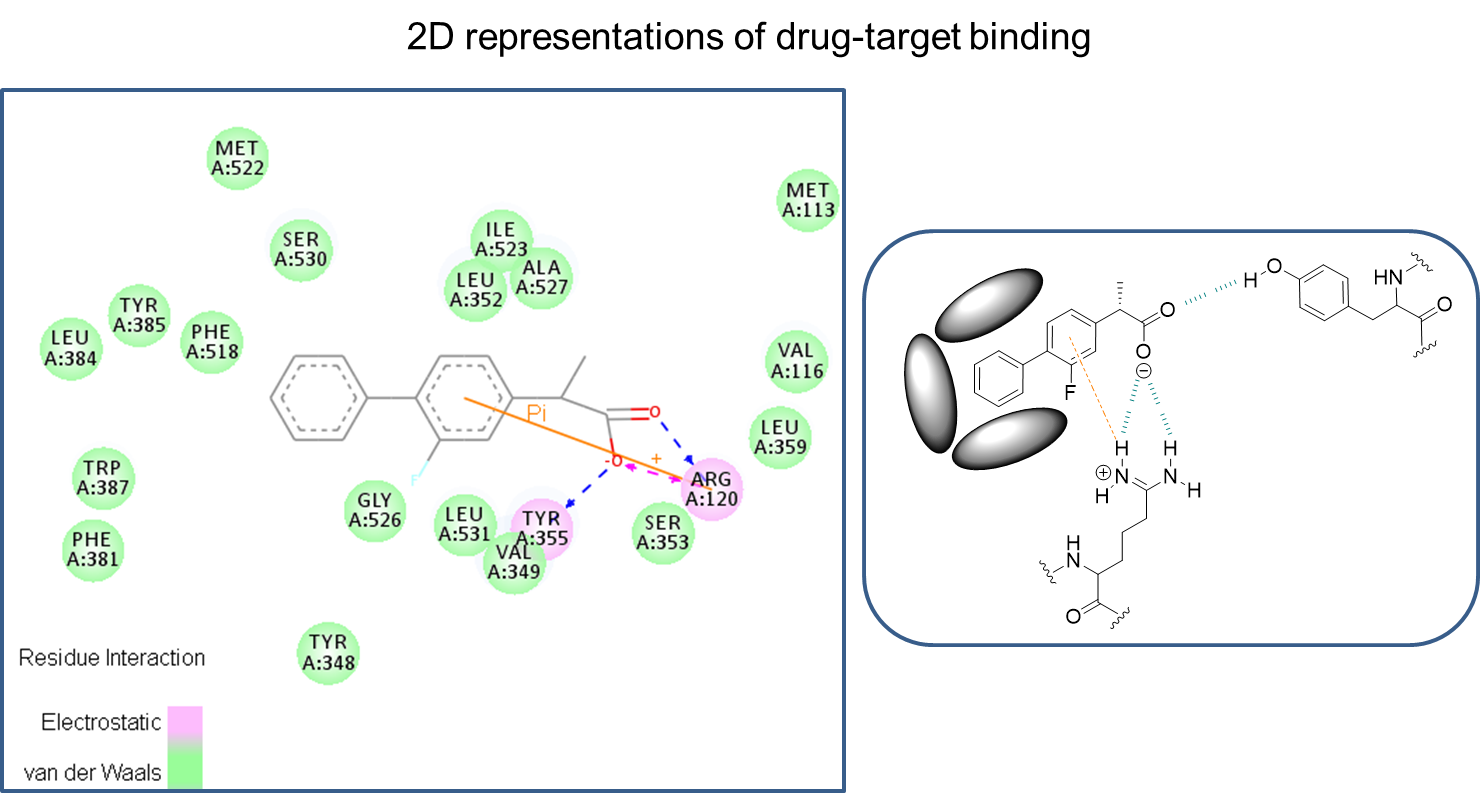
35: Driving potency and selectivity at the receptor level can be carried out via chain modification, ring substitution and modifications at different carbons to improve interactions for better potency and selectivity.
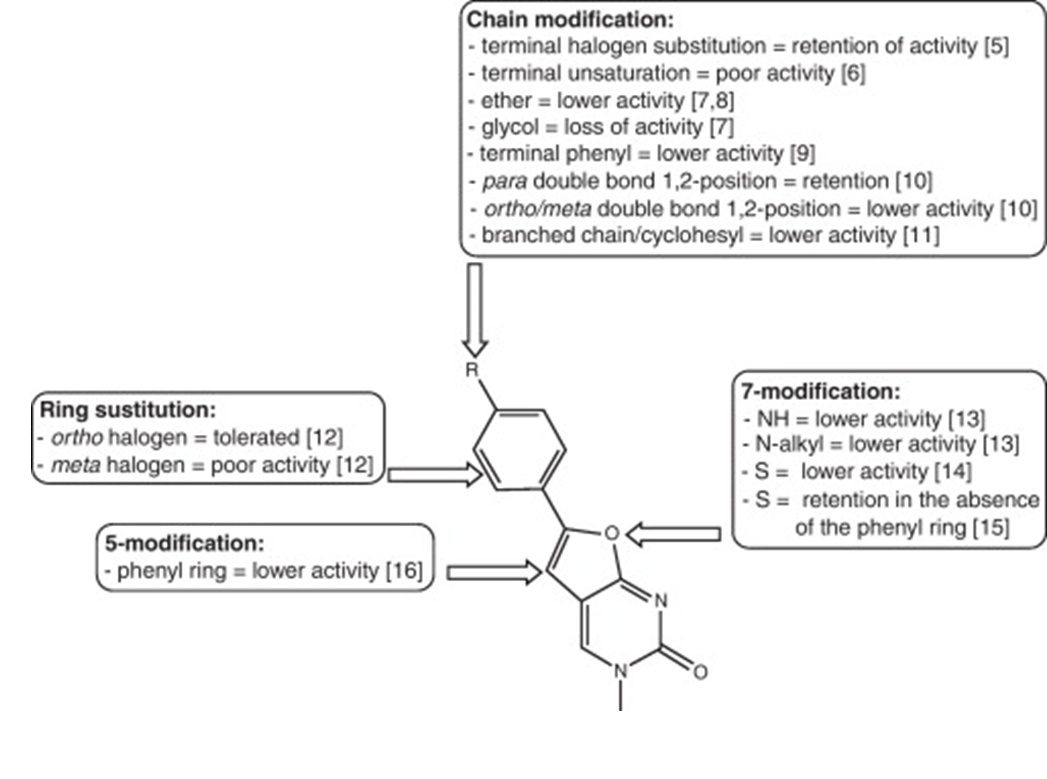
36: how can you improve molecule to make better/stronger/more interactions with amino acids in protein target
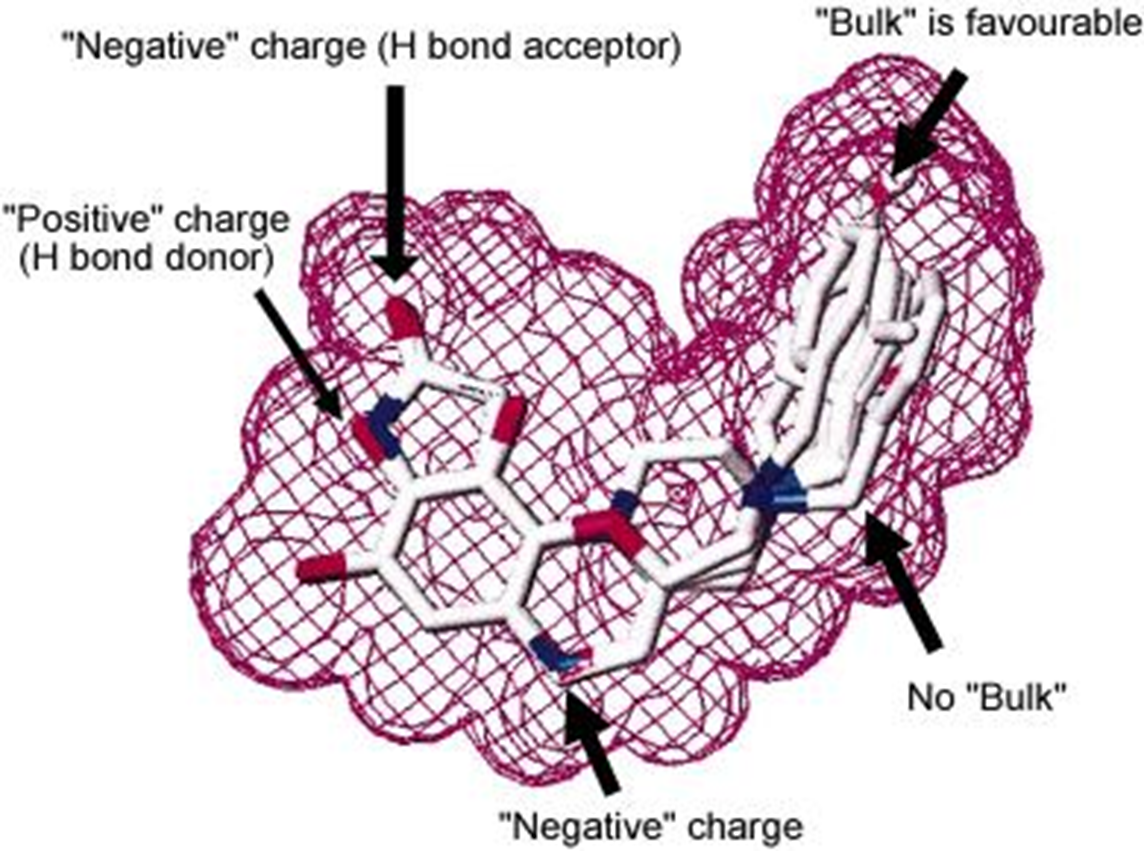
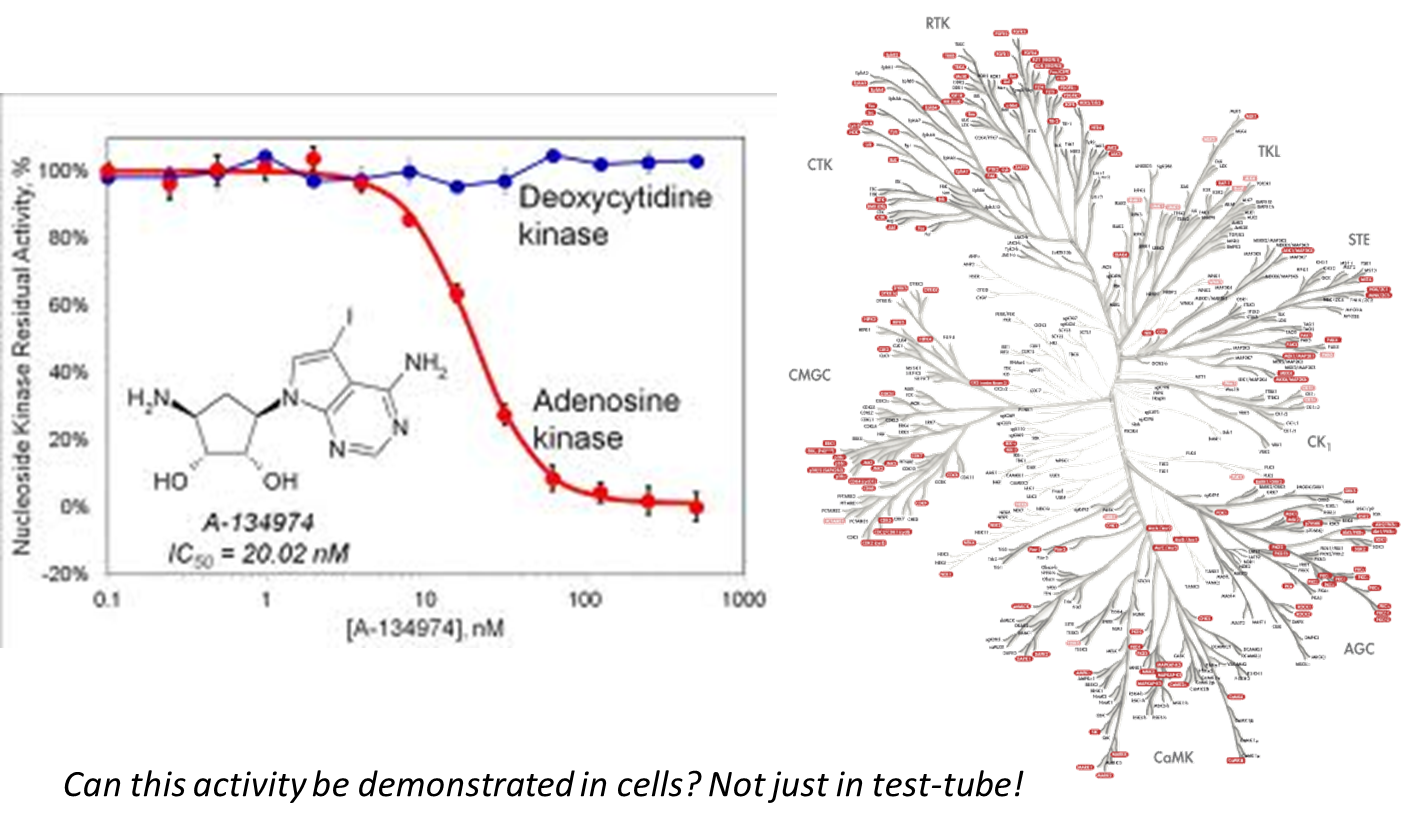
38: divergence between drug effects on two structurally related kinases - want to see divergence to know you’re driving towards inhibition etc of a particular target. These tests typically done with isolated enzymes in test tube/96 well plate but need to also be able to have same effect in a cellular environment - can it cross membranes and enter circulation
40: Pharmacokinetic considerations must parallel the optimisation of ADME properties.
Physiochemical properties must be modified to facilitate absorption via oral administration and structure must be modified to control metabolism and half life.
Can only call a molecule a drug once it has been approved for clinical use - before then it is just a druglike molecule
Chemical biology only considers pharmacodynamic properties, generating chemical tools, not leads, drug candidates or drugs.
41: Structural modification helps control metabolism and half life.
Structure-property relationship profiles cover solubility, membrane permeability, lipophilicity, pKa, stability, metabolite screening, CYP450 inhibition and plasma protein binding.
42: For drugs to be absorbed through the membranes of epithelial cells lining the small intestine they must diffuse through lipid membranes (lipophilic) and be single molecules, not aggregates. Need balance between hydrophilicity and lipophilicity
43: Target deconvolution - in phenotypic screening, need to go backwards to find key protein involved
44: Phenotypic screening identifies molecules with particular biological effects in cell based assays/animal models.
Simplest phenotypic screens use cell lines and monitor a single parameter eg cell death/protein production.
High content screening often used, where changes in expression of several proteins can be monitored simultaneously.
It can involve screening large libraries of compounds in automated high throughput cellular assays measuring levels of various proteins or effects on characteristics eg cell proliferation or responses such as apoptosis, migration and invasion.
45: Phenotypic screening requires target deconvolution (?) - phenotypic screens using fluorescence, allows chemical probe to be identified for target identification, optimisation then followed out
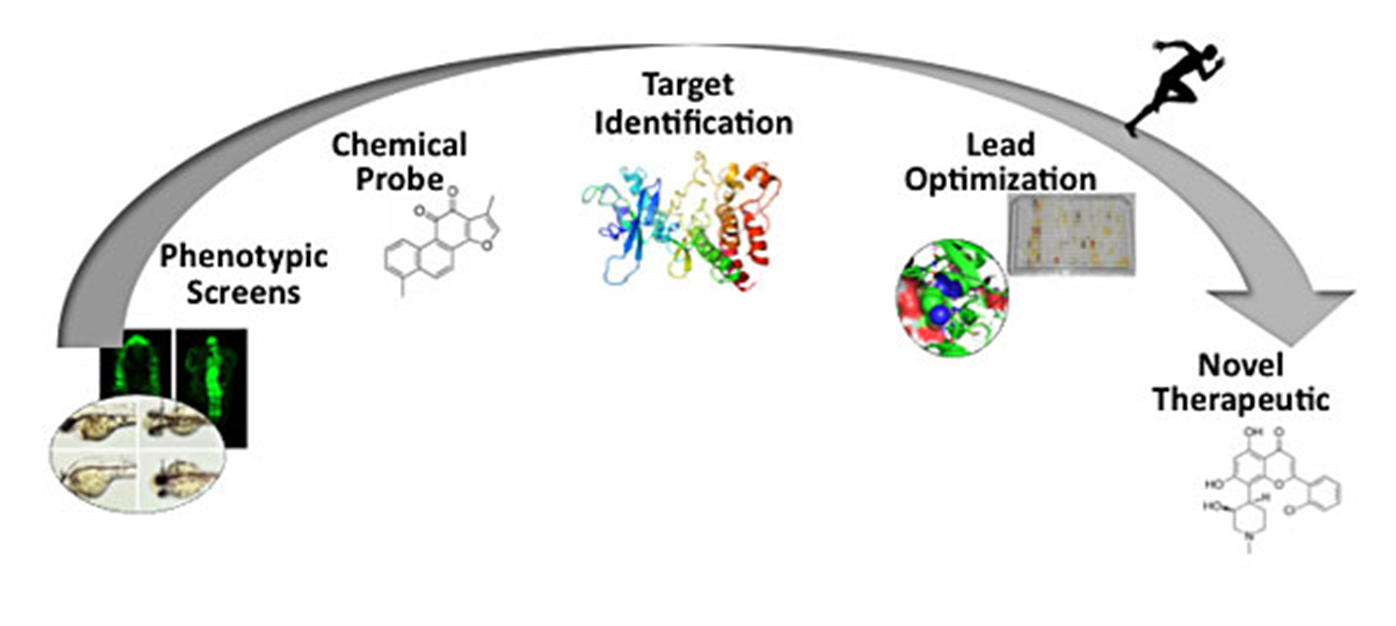
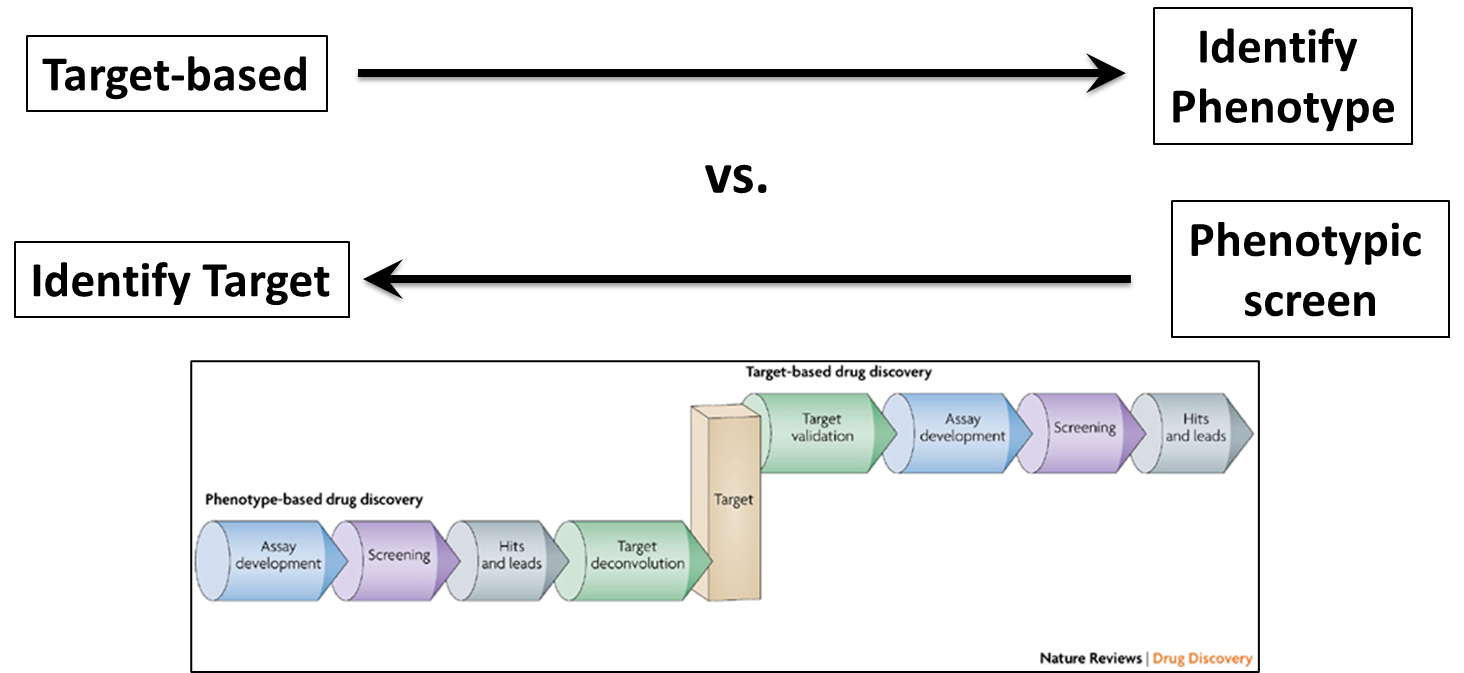
46: Target based approaches are used to identify the phenotype whereas phenotypic screens identify the target
Extra reading:
Biochemistry (Eighth Edition)
Berg, Tymoczko, Gatto Jr. & Stryer Chapter 36 Drug Development
An Introduction to Medicinal Chemistry (Fourth Edition) Patrick Chapter 2, 3 & 4 (aspects of)
Biochemistry Society – Policy document: Biochemistry Skills for Drug Discovery (https://www.biochemistry.org/Portals/0/SciencePolicy/Docs/Skills_for_Drug_Discovery_Position_Statement.pdf )
need to look over functional groups again lol and their interactions/bonds they form
Target Based Approaches and Phenotypic Screening
Definitions:
ADME = absorption distribution metabolism excretion
Medicinal chemistry = generation of small, druglike molecules with clinical activity through the application of synthetic organic chemistry
Drug-target interactions = key interactions which promote association between the ligand under development and the target protein
Notes:
Part 1: The Drug Discovery Pipeline: Target-based approaches (more traditional) and phenotypic screening (looking at key properties before we know what key proteins are)
7: Drug discovery landscape - old data but gets message across. Less and less molecules making it to clinic but spending increasing
8: Finding a new drug against a chosen target for a particular disease usually involves high-throughput screening - large chemical libraries tested for their ability to modify target. Often robotically tested. Chosen drug targets modified to increase activity against target and reduce activity against unrelated targets. Drug likeness/ADME properties of molecule then improved.
9:
Light blue = preclinical, uses purified and isolated enzymes in vitro then goes onto animal testing
Interactions between medicinal chemists, pharmacologists, biochemists and pharmacists are required at every level for the drug development process. The chemists/pharmacologists/biochemists discover and optimise the drugs whereas pharmacists formulate and develop them. There’s very little point in investing between 100 million and 1 billion, and 10 to 15 years work discovering a drug and getting it to market only for the patient to misuse the drug and not receive any benefits from it.
10: The first step is target identification, where a target is identified which is associated with a desired clinical outcome eg:
ACE inhibition reduces blood pressure
blocking cardiac beta-adrenoreceptors can modify cardiac output and function
HIV protease inhibition halts viral replication
EGFR inhibition prevents cancer cell proliferation
11: Target selection:
New drug must have something to offer clinically and be capable of being highly profitable (big pharma view). From a regulator’s view, improvements on existing ‘gold standard’ drugs must be substantial. From a patient’s view, a drug is in demand if it avoids certain side effects of existing drugs. However a new drug may not be able to be afforded eg due to NHS budget or in developing countries where a particular issue is prevalent.
12: target examples and how a drug targeting them would work
Target | Mechanism |
Enzyme | Inhibitor - reversible/irreversible |
Receptor | agonist/antagonist |
Nucleic acid | intercalator (binder), modifier (alkylating agent) or substrate mimic |
Cell membrane ion channels | blockers or openers |
Transporters | uptake inhibitors |
13: A requirement of the drug development process is to validate that the selected protein is a robust target, which is confirmed using a number of molecular techniques to modify or remove protein of interest from a particular system eg:
siRNA based protein run-down
adenoviral/lentiviral delivery (transfection/infection method) into cells of genetic materials that encode for modified (eg site directed mutagenesis to make protein inactive) version of target → impacted cell responses/outcomes
genetic deletion (knockout) models - animals and cells prepared from them and compare to WT animal - check for fatal effects or if KO redundant
CRISPR - easily modifies genome allowing new scientific and therapeutic opportunities
14: Drug discovery and development is a challenging process. Challenges faced are having to produce a molecule with optimum pharmacodynamics (potency and selectivity for the target) while ensuring it has optimum pharmacokinetics (absorption into body with robust metabolic pathway).
15: The key to the success of a medicinal chemistry project is the ability to optimise molecules, making subtle changes to the molecule to make it a more successful drug.
Optimisation needs to account for potent and selective activity against the target protein associated with the clinical disease, and then achieving said activity at the desired concentrations in a physiological system (animal model and ultimately a human patient).
16: Put simply, pharmacodynamics are what the drug does to the body - the therapeutic effect, how it exerts its effect and how it interacts with the target. Small functional group changes can change the pharmacodynamic action of a drug eg beta agonist salbutamol and beta antagonist atenolol have for the most part very similar structures.
17: Pharmacokinetics is what the body does to the drug, entailing how well it’s absorbed, where the drug goes, how long it stays and how it’s metabolised.
Like with pharmacodynamics, small functional group changes can change the pharmacokinetic action of a drug eg the only difference between atenolol which has poor oral absorption and low CNS penetration, and propranolol which has good oral absorption and high CNS penetration is a switch from amide group to an additional benzene ring.
18: Pharmacokinetics usually assessed by absorption, distribution, metabolism, excretion and toxicology (ADMET)
19:
Part 2:
21: Compounds are sourced from natural products and synthetic molecules.
Natural products are best as purified molecules rather than extracts and are sourced from plants, sponges, bacteria and fungi etc eg taxol (anti cancer) derived from willow bark
Synthetic molecules typically used by big Pharma and are sourced from large scale libraries and are chemical building blocks of various molecular weights. These libraries are databases of compound chemical structure, purity, quantity and physiochemical characteristics. As much of the appropriate chemical space (?) is taken advantage of as possible to know what kind of chemical structure would be helpful
22: SAR = structure-activity relationship used for potency against target. Target may have different isoforms with different functions that don’t want targeting
usually start off with poor scaffold, scaffold hopping used to build more optimal drug
24: Drugs bind targets via their functional groups which allow them to interact with the target
25: Drug functional groups interact with the different amino acids found in protein targets.
Drugs form short range van der Waals forces with non-polar, aliphatic side groups (glycine, alanine, valine, leucine, methionine and isoleucine) and aromatic side groups (phenylalanine, tyrosine and tryptophan)
Drugs form mid range hydrogen bonds with polar, uncharged side groups (serine, threonine, cysteine, proline, aspartate and glutamine).
Drugs form ionic bonds with positively charged (lysine, histidine and asparagine) and negatively charged side groups (glutamate and aspartate).
26:
Above shows COX tertiary structure with the secondary structures displayed as a ribbon. COX’s tertiary structure allows it to perform its function by maintaining active site integrity for oxidation and cyclisation of arachidonic acid for prostaglandin synthesis.
29:
Enzymes globular tertiary structure has a surface which excludes small molecules from penetrating the interior. The active site is flexible and can expand and contract to allow substrates in and products out
30: Interactions between amino acids bordering COX’s active site and arachidonic acid are a mixture of H bonds (directional, specific for substrate H bonding groups) and short range van der Waals between substrate surface and active site surface.
31: Flurbiprofen is a competitive COX inhibitor which binds in preference to arachidonic acid (has higher affinity) blocking prostaglandin synthesis therefore reducing inflammatory response.
For a compound to compete with the substrate it must have some structural resemblance to enable it to match the active site. To compete effectively it must form stronger interactions with the active site than the substrate does. Binding is dynamic meaning molecules associate and dissociate regularly so molecules need to remain bound to the active site for longer than the substrate to be effective.
32: Driving potency and selectivity at the receptor level can be carried out via chain modification, ring substitution and modifications at different carbons.
33: allows chemists to think if they can fill in any of the spaces
35: Driving potency and selectivity at the receptor level can be carried out via chain modification, ring substitution and modifications at different carbons to improve interactions for better potency and selectivity.
36: how can you improve molecule to make better/stronger/more interactions with amino acids in protein target
38: divergence between drug effects on two structurally related kinases - want to see divergence to know you’re driving towards inhibition etc of a particular target. These tests typically done with isolated enzymes in test tube/96 well plate but need to also be able to have same effect in a cellular environment - can it cross membranes and enter circulation
40: Pharmacokinetic considerations must parallel the optimisation of ADME properties.
Physiochemical properties must be modified to facilitate absorption via oral administration and structure must be modified to control metabolism and half life.
Can only call a molecule a drug once it has been approved for clinical use - before then it is just a druglike molecule
Chemical biology only considers pharmacodynamic properties, generating chemical tools, not leads, drug candidates or drugs.
41: Structural modification helps control metabolism and half life.
Structure-property relationship profiles cover solubility, membrane permeability, lipophilicity, pKa, stability, metabolite screening, CYP450 inhibition and plasma protein binding.
42: For drugs to be absorbed through the membranes of epithelial cells lining the small intestine they must diffuse through lipid membranes (lipophilic) and be single molecules, not aggregates. Need balance between hydrophilicity and lipophilicity
43: Target deconvolution - in phenotypic screening, need to go backwards to find key protein involved
44: Phenotypic screening identifies molecules with particular biological effects in cell based assays/animal models.
Simplest phenotypic screens use cell lines and monitor a single parameter eg cell death/protein production.
High content screening often used, where changes in expression of several proteins can be monitored simultaneously.
It can involve screening large libraries of compounds in automated high throughput cellular assays measuring levels of various proteins or effects on characteristics eg cell proliferation or responses such as apoptosis, migration and invasion.
45: Phenotypic screening requires target deconvolution (?) - phenotypic screens using fluorescence, allows chemical probe to be identified for target identification, optimisation then followed out
46: Target based approaches are used to identify the phenotype whereas phenotypic screens identify the target
Extra reading:
Biochemistry (Eighth Edition)
Berg, Tymoczko, Gatto Jr. & Stryer Chapter 36 Drug Development
An Introduction to Medicinal Chemistry (Fourth Edition) Patrick Chapter 2, 3 & 4 (aspects of)
Biochemistry Society – Policy document: Biochemistry Skills for Drug Discovery (https://www.biochemistry.org/Portals/0/SciencePolicy/Docs/Skills_for_Drug_Discovery_Position_Statement.pdf )
need to look over functional groups again lol and their interactions/bonds they form