Expression and analysis and human G6PD
Human glucose-6-phosphate dehydrogenase (G6PD)
at Xq28 region, 18 kb 13 exons
515 amino acids, 59.2 kDa
catalyses the first and rate limiting step in pentose phosphate pathway for the production of NADPH
protect cells from oxidative damage
G6PD deficiency
→ reduced production of NADPH selectively affect RBCs
they rely solely on pentose phosphate pathway for NADPH as they do not have mitochondria
oxidative stress may damage membrane protein of RBCs → hemolysis
G6PD deficiency patients: acute haemolytic anaemia when
injection of oxidant drugs
infections
ingestion of fava beans
~160 mutations reported
mutations may reduce activity or stability of G6PD
most G6PD-deficients are asymptomatic
HK: 4.3% male ; 0.5% female
Common G6PD variants in Chinese population
G6PD Canton
CGT → CTT
Arg459Leu
G6PD Kaiping
CGT → CAT
Arg463His
G6PD Gaohe
CAC → CGC
His32Arg
Recombinant DNA technology
Recombinant DNA
biologically active DNA
formed by the in vitro joining of segments of DNA from different sources
purpose
to clone genes
to make recombinant proteins when introduce to host cells
Traditional cloning process
vector preparation
insert preparation
ligation
transformation
colony screening
Restriction enzyme-based cloning
Restriction enzyme
restriction endonucleases
recognise and cut DNA at secific nucleotide sequences
Types of end cut
cohesive (sticky) end: fragments with short single-stranded overhanging ends
blunt ends: even-length ends from both strands
Cloning vector
a replicating DNA molecule that carries a foreign NDA fragment to be introduced into cell
e.g. plasmid DNA
properties
replicate independently of the host cells
unique restriction enzyme cleavage sites
✔︎ a reporter gene/ selectable marker to determine which host cells contain the recombinant DNA
elements contains
multiple cloning sites: for insertion of foreign DNA
origin of replication: allow recombinant DNA to replicate in bacteria
selectable marker: for identification of cells that have taken up the plasmid
e.g. antibiotic resistance gene
expression vector
cloning vectors that includes sequences needed for expression of the foreign DNA
for transcription and translation
composition
promoter
ribosome binding site
transcription termination
usage
foreign gene is inserted to to cloning site
i.e. between ribosome-binding sequence & terminator of transcription
promoter and other regulatory sequences from the host cell are also required
Procedures
cut DNA from different sources with the same restriction enzyme
→ produce DNA fragments with compatible ends
join DNA fragments with compatible ends by DNA ligase
recombinant DNA is created
Common strategies
double digest
using 2 enzymes with non-compatible ends
insert ligated to vector in one orientation only
single digest
insert ligated to vector in either orientation
using 2 enzymes with non-compatible ends or compatible ends or 1 enzyme only
Plasmid subcloning
prepare insert DNA containing coding sequence of G6PD
digest pGEM-T-G6PD DNA (parent vector) with NdeI and XhoI (restriction enzymes) to release the coding sequencing
prepare vector DNA for cloning G6PD
digest pET28a vector DNA with NdeI & XhoI → create DNA ends compatible for ligation to G6PD insert DNA
as the sticky ends produced by NdeI and XhoI are not compatible → vector DNA do not religate
construct G6PD expression vector
G6PD (1.5kb) DNA insert is ligated to pET28a (5.3 kb) vector DNA by DNA ligase
ligated in only one orientation → ensure proper expression using vector-encoded T7 promoter
coding sequence is fused in-frame to His-tag
His-tag is a tag made up of 6 consecutive histadine residues
insert DNA to vector DNA molar ratio= 3: 1 or 1: 1

recombinant expression plasmid is formed, containing
origin replication
antibiotic resistance gene
G6PD coding sequence
introduce the recombinant expression plasmid to bacteria——E. coli
characteristics of E. coli
most widely used as it has simple and well-understood genetic environment in which to isolate foreign
universal genetic code → accept foreign DNA derived from any organism
replicate very 22 min → amplify foreign DNA rapidly
each bacterium can carry up to several hundred copies of cloned gene
induce transformation by
pre-treatment with CaCl2 to render cells susceptible (competent cells) to take up exogenous DNA
electroporation (short electric pulse)
DNA uptake
DNA pass through channels formed at adhesion zone (where outer and inner cell membranes are used to form pores in the cell wall)
heat shock create a thermal imbalance on either side of membrane → help pump DNA via the adhesion zone
selection, identification and characterisation of the recombinant clone
transformation efficiency
a measurement of the quality of competent cells
defined as the number of colonies formed per μg of uncut plasmid DNA
10^5 per μg is adequate for simple cloning or subcloning
circular plasmid are more efficient for transformation than linear
bacteria can be transformed with
vector ligated to insert
self-ligated/uncut vector (w/o insert DNA)
uncut/nicked vector (w/o ligase)
identification methods
restriction enzyme analysis
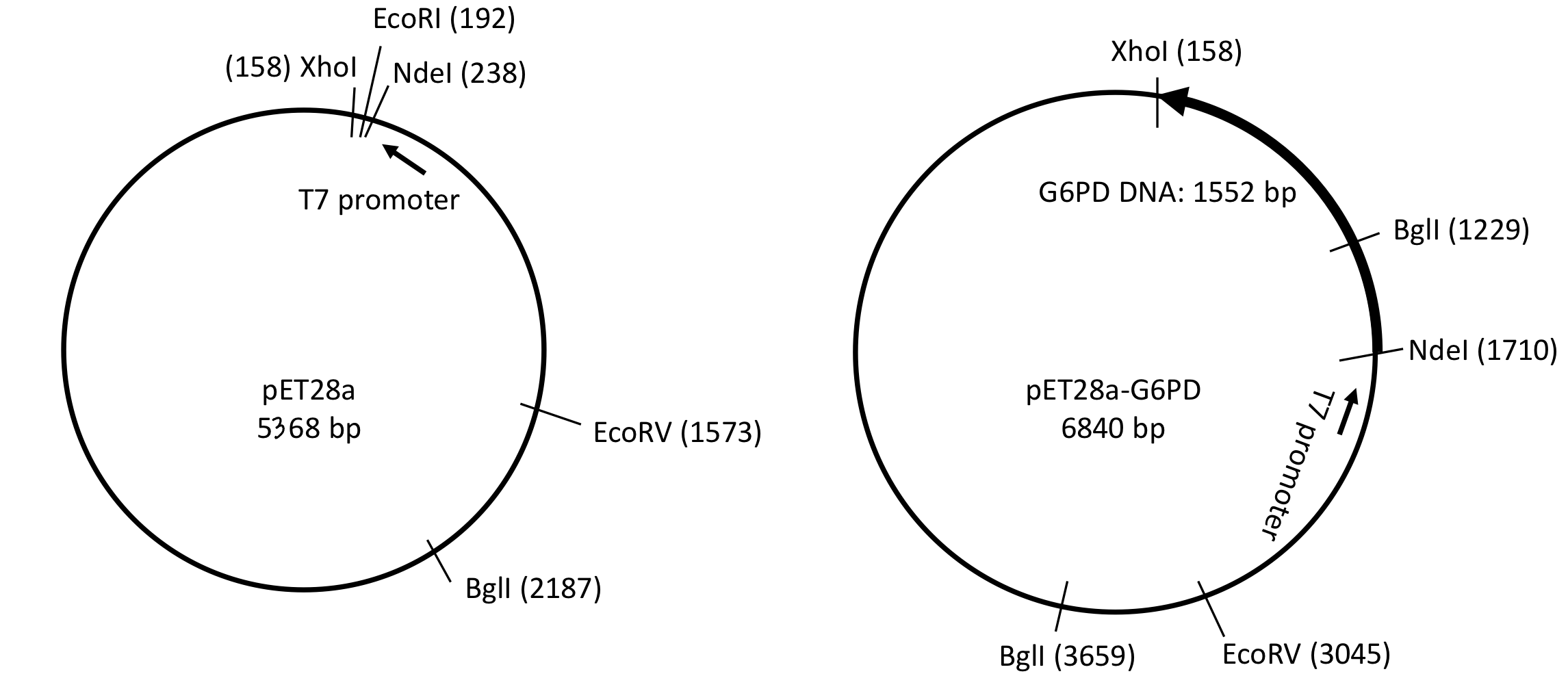
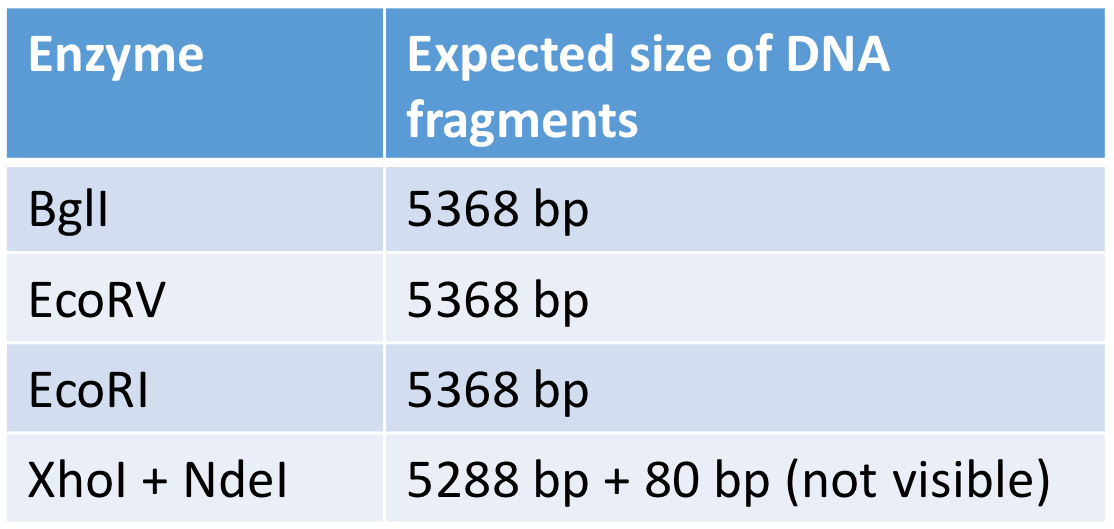
recognition sequence in pET28a
one BglI, one EcoRV and one EcoRI
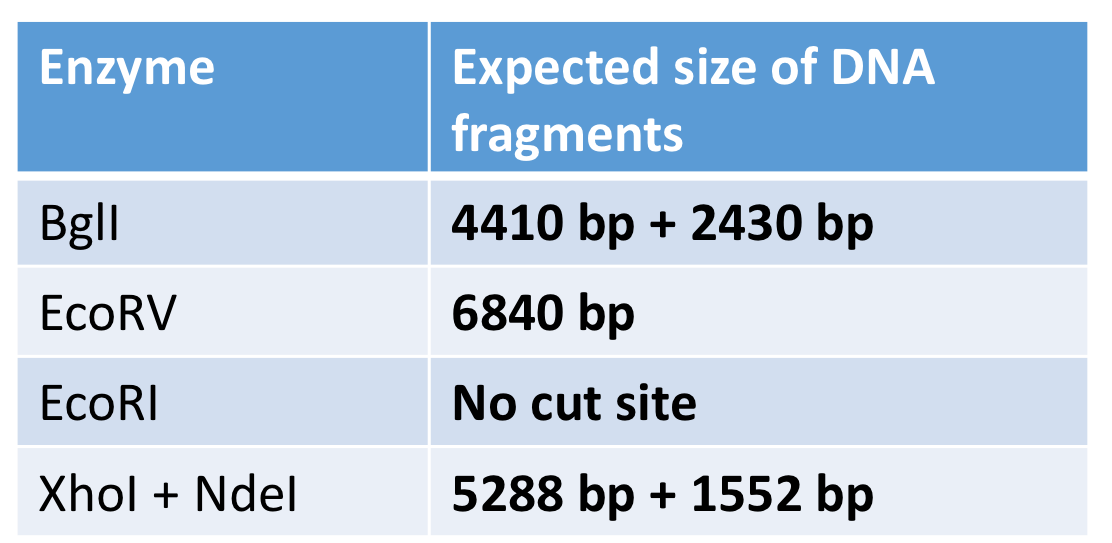
recognition sequence in pET28a-G6PD
two BglI and one EcoRV
colony PCR
insert-specific primer: positive applification confirms the insert’s presence
vector-specific primer: amplicon length determine the insert’s presence
insert- and vector-specific primer: PCR analysis of insert detection and amplicon length reveals cloning success
blue-white selection
insert DNA contains lacZ
product of lacZ turns X-gal to blue pigment
only bacteria that have taken up the plasmid grow in the presence of antibiotics
white colonies: bacteria that carry recombinant plasmid
blue colonies: bacteria that carry intact plasmid vector
DNA sequencing
positive clones should be confirmed by this method: ensure reading frame is correct and no extra mutations due to cloning process
pET expression system
Characteristics
high level of expression of recombinant protein → deleterious to cell
✔︎ inducible system:
expression of gene of interest is normally repressed
induced to express after cell culture has grown to appropriately high density
expression of cloned gene is controlled by T7 promoter: specific to T7 RNA polymerase but not E. coli RNA polymerase
require bacterial strain to express T7 RNA polymerase: cloned into bacterial chromosome
expression is controlled by the operator site in promoter
Inducible operon
structural genes (lacZ, lacY, lacA for metabolism of lactose): encoding proteins to be transcribed
promoter (lacP): for binding of RNA polymerase to initiate transcription
regulated by operator sequence
operator (lacO): regulate transcription, binding of repressor to operator blocks transcription → RNA polymerase cannot bind to the operator
Repressor protein (lacI)
has to binding sites: for operator and inducer (lactose/ ITPG)
╳ lactose → bind to operator → no transcription of operon
regulatory protein is synthesizd as an active repressor (constitutively expressed)
it becomes inactive when bind to inducer (lactose) → ✔︎ transcription of structural gene
Types of induction
ITPG induction
╳ ITPG: repressor protein is binding on the operator
no gene is transcribed
✔︎ ITPG: repressor is removed from the lac operator
induce gene expression
Auto induction
auto-induction medium
glucose:
preferentially used by bacteria → increase cells density
repress induction pof operator
lactose:
converted into allolactose in cell after glucose is depleted
auto induction of lac operon
glycerol:
support growth of cells
does not prevent induction
expression of recombinant protein in E. coli is induced when cells reached saturation
catabolite repression
presence of glucose lowers the cell concentration of cAMP
→ less CAP protein bind to promoter
less efficient transcription of operon
regulatory protein CAP (catabolite activator protein)
bind to cAMP (cyclic adenosine monophosphate) → complex
complex bind to DNA (upstream of promoter) → more efficient binding of RNA polymerase to promoter and activate transcription
availability of glucose represses the enzyme for lactose utilisation
Protein purification
Considerations
identify a suitable source of the desired protein or overexpress the the protein as recombinant protein (usually tagged) in a suitable host organism
Cell extraction preparation
collect cells by centrifugation
cell distruption
lyse the cells by using
detergent or enzyme (lysozyme)
sonication
mechanical homogenisation
use protease inhibitors and low temperatures to minimise proteolysis
centrifugation to remove cell debris
Selective precipitation (crude separation)
add ammonium sulphate
highly hydrated: reduce protein solubility by decreasing concentration of water available for protein-solvent interaction
more hydrophilic proteins precipitate at a higher concentration of ammonium sulphate
add water-miscible organic solvent
change solvent polarity → increase protein-protein electrostatic interaction
allow protein aggregation & facilitate precipitation
alter pH
affect ionisation of amino, carboxyl and side chain group of amino acid
protein aggregate and precipitate at isoelectric point
pH=pI (number of -ve & +ve charge is equal)→ protein net charge =0
pH > pI → protein is negatively charged
pH < pI → protein is positively charged
Chromatography
general principle: components are separated by the affinity to the stationary phase or distribution between stationary and mobile phase
affinity chromatography
specific interaction between protein and ligand on matrix
elution condition depends on specific protein and ligand
examples
glutathione (ligand) & GST fusion protein (target protein)→ elute when reduced glutathione & low ionic strength
Ni2+ (ligand) & His-tagged protein (target protein)→ elute if high ionic strength and imidazole
antigen (ligand) & antibody (target protein)→ elution condition depends
Purification of his-tagged protein by Ni-NTA agarose column
affinity medium (Ni-NTA) is equilibrated in binding buffer
Ni-NTA= Ni2+ coupled with nitrilotriacetic (NTA)→ ✔︎ couple to agarose resin
target protein (his-tagged G6PD) binds to column; unbound material eluted from column
target protein is recovered by changing the condition: adding imidazole to favour elution of the bound proteins
imidazole compete with histamine for binding to Ni2+
affinity medium is re-equilibrated with binding buffer
Ion-exchange chromatography
based on electrostatic interaction between protein & matrix
selective elution by changing pH (net charge) or increasing ionic strength of elution buffer to compete with protein in binding to matrix
hydrophobic interaction chromatography
separate proteins according to surface hydrophobicity
high salt: promote adsorption of hydrophobic protein surface to hydrophobic matrix
reduce solvation of sample solutes & expose the hydrophobic regions along the surface of protein molecule
elute by decreasing ionic strength of buffer
gel filtration
separate proteins by size
larger proteins do not enter matrix→ elute first
smaller proteins enter pores of matrix → retarded differentially
Dialysis or ultrafiltration
force sample across a semipermeable membrane by gas pressure or centrifugal force
purpose
remove salt
exchange buffer and concentration of protein
Biological activity assay
Purpose
to detect the protein
Types
Continuous assay
continuously monitor wither the disappearance of a substrate or appearance of product
measure directly by
change in absorbance
fluorescence
pH
Discontinuous assay
product measured after reaction manually stopped or quenched at different time
Coupled assay
for substrate-product pairs wit o spectroscopic properties
product from reaction catalysed by enzyme of interest: substrate in a second enzymatic reaction → give a compound that an be detected directly
example:
hexokinase converts D-glucose to DG6P
DG6P is converted to 6PGL and NADPH
measure hexokinase activity by measuring the increase in NADPH
Measuring G6PD activity by continuous assay

Principle
NADPH absorb radiation at 340 nm but not NADP
absorbance at 340nm increase when NADPH accumulate
determine inițial rate by determining the slope of the linear portion of the curve
dilute enzyme preparation if the initial rate is too high to accurately determined
Purification summary table
enzyme activity (U): (ΔA340/min)*reaction vol/(6.22*enzyme vol)
amount of enzyme that convert 1 μmole substrate to product in 1 min
specific activity (U/mg): activity per unit weight
indicate protein purity and quality
should increase after each purification step
overall yield or recovery= total target protein or activity in fraction / total target protein or activity in crude extract
purification factor= specific activity of fraction/ specific activity in crude extract
Protein concentration determination
UV spectromentry
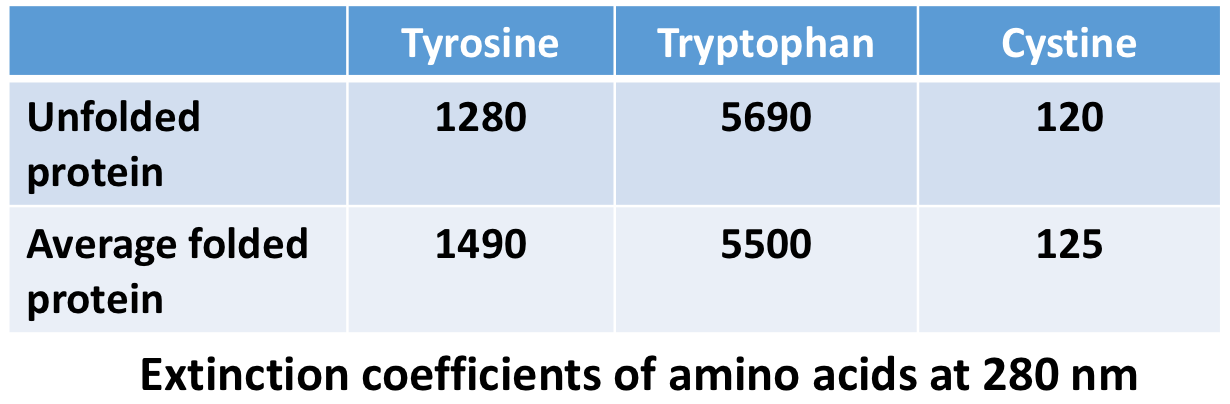
Absorbance at 280 nm by tryptophan and tyrosine and cystine
extinction coefficient of a protein at 280 nm (M^-1 cm^-1)= $ of tyrosine*ext(tyrosine) + # of tryptophan*ext(tryptophan) + # of cystine*ext(cystine)
disadvantage: can measure pure protein only
Dye-based method——Bradford method
protonated form of Coomassie Brilliant Blue G-250: orange or brown
dye becomes deprotonated when bind to side chain NH3+ group of protein→ blue
can be detected at 595nm
dye binds to proteins at Arg, His, Tyr, Trp, Phe
Disadvantage: affected by detergent in sample
sensitivity: 1-50 μg
Copper-based method
Lowry method
chelation(multiple bonds between organic molecules and metal) of Cu2+ by peptide bonds → reduction of Cu2+ to Cu+
Cu+ facilitate reduction of Folin reagent to a blue product → measured at 750nm
sensitivity: 5-100 μg
Bicinchoninic acid (BCA) method
Cu2+ in BCA reagent forms complexes with peptide bonds → reduce Cu2+ to Cu+
Cu+ interact with BCA to form a purple BCA-Cu+ complex → measured at 562nm
not compatible for samples with racing agent or copper chelating agents
sensitivity: 0.2-50 μg
SDS-PAGE
Purpose
check for purity of protein preparation
estimate protein molecular mass
for imumunodetection of protein
Procedures
Cast gel——stacking and resolving gel
sample preparation
determine sample protein concentration
pipet correct sample volume
add 2X or 4X loading buffer and boil sample for 5min
sample contains
DDT: reduce disulphate bridges
denature protein completely→ migration is not affected by protein secondary structure
protein subunits can separate independently
SDS
glycerol: add density to sample→ sink to bottom of wells
bromophenol blue: visualise samples in well & act as tracking dye
mount gel cassette into a vertical electrophoresis and apple gel running buffer
load the sample by electrophoresis
electrophoretic mobility of proteins in SDS-PAGE is inversely proportional to the logarithm of their mass
stain gel to visualise proteins
Discontinuous buffer system
buffer in tank is different from buffer in gel
different pH and % of acrylamide between stacking gel and resolving gel
stacking gel
concentrate all different seized proteins into a compact horizontal zone
proteins sandwiched between glycine molecules above and Cl- below
prevent protein bands from smearing and ensure good resolution
pH 6.8→ majority of glycine is zwitterionic (having both charges of functional groups) → lower mobility
mobility of ions: Cl- (completely ionised) > SDS > glycine
Cl- move away from Gly- to develop a voltage gradient → accelerate Gly-
→ all ionic spices move at the same speed
protein samples are stacked between the Cl- and Gly- into a very thin zone before separation in resolving gel
resolving gel
separate gel by size
at pH8.8: Gly- is predominantly -ve charged → increase mobility and overtake protein (move directly behind Cl-)
when Gly- move pat protein molecules: unstacking protein
formation of polyacrylamide gel
polymerisation of acrylamide monomer with cross-linking agent bis-acrylamide
reaction is initiated by APS and TEMED
average pore size is determined by the concentration of acrylamide and proportion of the cross-linking bis-acrylamide
Sodium dodecyl sulphate (SDS)
an anionic detergent
confers constant -ve charge : size ratio
SDS-protein is a uniform rod shape→ ✔︎migrate according to size
Immunoblot——Western blot
Purpose
confirm the identity of a target protein by
size (from SDS-PAGE)
binding to specific antibody
monitor column fractions during protein purification
compare protein expression in different tissue disease conditions or response to drug treatment
for diagnosis of diseases (e.g. HIV)
Procedures
separate proteins by electrophoresis
visualise protein separation by Coomassie blue staining or colloidal gold
transfer protein from gel to membrane (wet tank transfer or semi-dry transfer)
proteins are eluted from the gel and adsorb onto membrane
verify protein transfer
prestained markers transferred to blot
total protein detection on membranes
Ponceau S (100-1000ng)
anionic dye
can be removed after visualisation
blots can be used for immunodetection
Colloidal gold (100og-1ng)
interfere with subsequent immnuodetection of proteins on western blots
fluorescent protein stains (2-8 ng)
blocking unbound site on membrane
to prevent non-specific binding of antibodies→ reduce background signals
antibody binding (primary and secondary antibodies) and detection
visualise data
Detection method
Antibodies
primary antibody
against the target protein or specific epitope on protein
e.g. anti-His antibody, anti-G6PD
secondary antibody
recognise and bind primary antibody
unusually conjugated to enzyme (AP or HRP) and detected by enzyme-substrate reaction
can also be conjugated to biotin, fluorophere r radioisotope
Direct vs indirect detection
direct: primary antibody is labeled or tagged with a enzyme
no secondary antibody is required
indirect: unlabeled primary antibody against target protein is used
primary antibody is detected by secondary antibody
Detection of His-tagged G6PD
primary antibody: rabbit anti-G6PD polyclonal antibody is used → raise against a peptide corresponding to human-G6PD amino acid positions 50-100
secondary antibody: goat anti-rabbit polyclonal antibody conjugated with AP
insoluble purple NBT diformazan product is produced when substrate NBT/BCIP is incubated with AP
enzymatic dephosphosrylation of CSPD by AP → emission of light at a max wavelength of 477 nm
detected using CCD camera or exposing the blot to X-ray film