lecture 6: primary transporters
Bacterial cells use both primary and secondary transporters. Primary transporters use ATP or other sources of energy directly to do transport while secondary transporters use energy gradients such as proton on sodium etc gradients.
A channel is a system where the channel is either open or closed. When it is open, specific ions move down the concentration gradient quickly. Transporters are slower and can pump molecules against the concentration gradient. They work by alternating axis mechanism where the pore only allows access from one side or the other depending on the concentration and changes due to conformational changes.
P type ATPases
These are important transporters that pump ions and lipids across cellular membranes. They are autophosphorylated and incluse Na+'/K+ ATPases which sets up the membrane potential and SERCA which is involved in calcium signalling in muscles. They are highly abundant
serca
SERCA gave the first structural information of a P type ATPase. Ca+ ions are key intracellular signalling ions and at rest, they are pumped out of the cytoplasm. When activated, Ca+ channels open to allow it back into the cytoplasm. Flow through these channels is quick however, pumping out of the cell is slower as it is done through transporters and not channels. This means that many ATPases are required to be able to pump CA+ out of the cell. For crystallography, they can be purified directly from muscle due to this.

SERCA1a transfers two CA+ per ATP hydrolysed in the forward direction and 2-3 H+ ions in the opposite direction.
transport cycle of SERCA
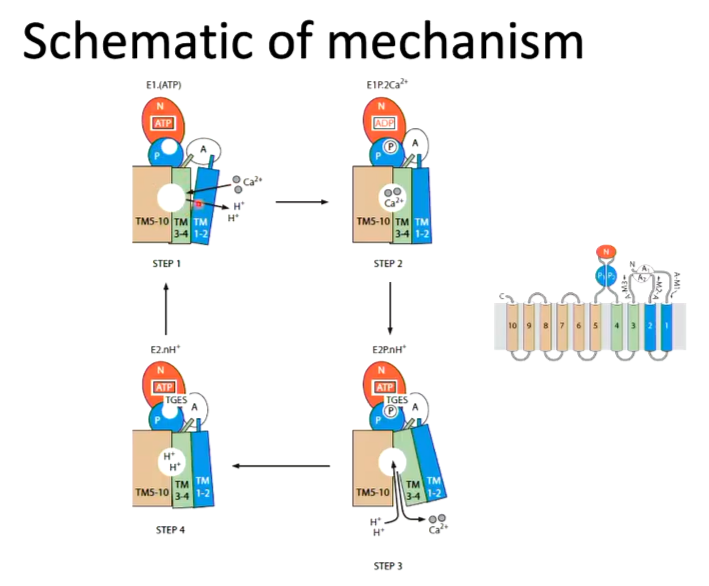
All P type ATPases work by the transport cycle where there are two states of the enzyme - a high affinity calcium binding state and a low affinity state. Calcium binds the high affinity state. ATP then binds and hydrolysed which causes a conformational change and switches state. Hydrolysis of the phosphate switches the enzyme back to the high affinity state
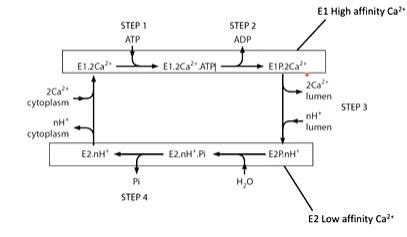
The first structure solved was the high affinity calcium state with calcium bound:
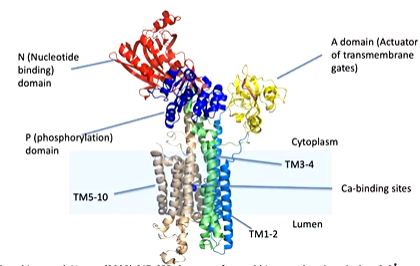
There is a transmembrane domain made up of 10 alpha helices donated TM1-2, TM3-4 and TM5-10. The calcium binding domain is located in the middle of the transmembrane domain. There is also cytoplasmic domains - an N domain (nucleotide binding) and an A domain (actuator of transmembrane gates) and P domain (phosphorylation).
Focusing on the transmembrane domains, the entrance to the calcium binding site is from the cytoplasm through TM1 and TM2. The binding site is well designed for calcium as there is positively charged calcium interacting with negatively charged glutamate as well as an unwound part of a helix which allows the calcium ion to interact with the carbonyl oxygens. There is also a negative dipole of the helix that can also interact with the positive charge of the ion
cytoplasmic domains
The cytoplasmic domains are the N domain, P domain and A domain. In the N domain, there is where the nucleotide should bins (in this case ATP). In the A domain, there is a TGES loop and the P domain which has a peptides from TM3-4 to the N domain which has an Asp 351 residue which is phosphorylated in the transport of calcium. The P domain has a rossmann fold structure.
The domains are arranged like:
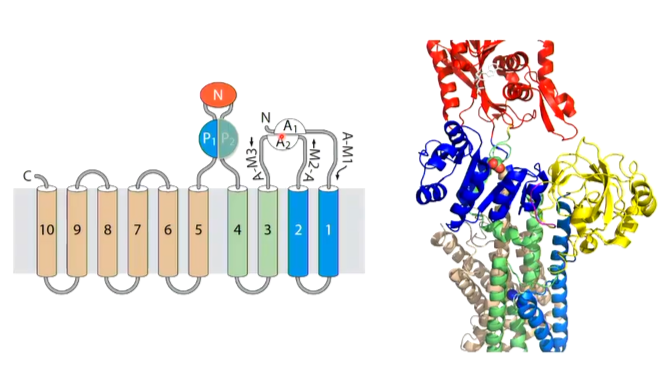
The domains are all interlinked and different sections complete different domains (eg TM2 completes the A domain and TM5 completes the P domain). These transporters work by conformational changes and so change to one of the domains will have a knock on affect and change the other domains as well
The binding of ATP will cause phosphorylation of ASP and the movement of calcium. There is 50 angstroms difference between these domains and so a conformational change is needed. The next structure worked on was where the aspartate was phosphorylated.
phosphorylation of aspartate
Upon the phosphorylation of aspartate, the N domain and A domain rotate and this means that the ATP site is next to the site of phosphorylation. This results a change in conformation in TM1 and 2.
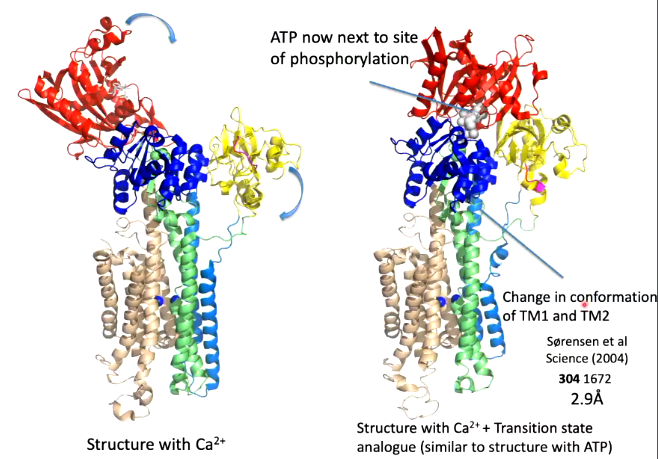
Instead of using ATP or another ATP analouge, ADP-aluminium fluoride was used as the shape of it is consistent with the transition state analouge of the reaction of the phosphorylation of aspartate. This transition state was well mimicked
Overlaying the two conformations before ATP binding and the transition state analouge, helices 1-4 all move but there is not much difference to the calcium binding site where the conformational changes moves the calcium binding site only closes but does not change position.
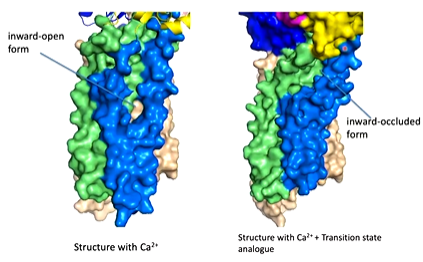
The next structure looked at was the low affinity calcium binding site where there should be a conformational change to the transporter
low affinity calcium binding state
They did a structure using the Asp 351 resembling a phosphorylated Asp. The TGES motif of the A domain replaces the ADP. There is also a shift in the conformation caused by the changes in N and A domains.
Looking at the surface representation of the structure, the movements of the N and A domains completely changes the calcium by dragging on the helices and opens it up so the calcium binding site is open to the outside of the protein. The calcium can come off and is replaced with proteins.
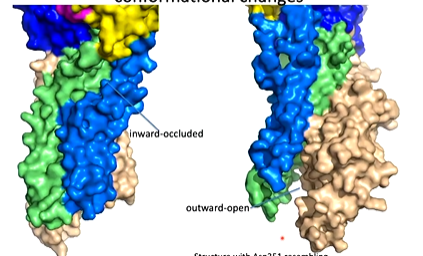
Beryllium fluoride was used as an analouge of phosphate and reacts with aspartate to do this. The TGES loop is made up of Thr 181, Gly 182, Glu 183 and Ser 184. tHIS IS REPLACED WHERETHE nucleotide binding domain was which has moved. The TGES domain has moved here and caused the conformational changes that allows the calcium to move out. The residues are interacting with residues on the phosphorylation domain. When you overlay the in and out conformations, you can see the movement of helices causing the Ca site to be disrupted.
Sodium potassium pump
This is a target for digitalis like compounds important for heart arrhythmia and is similar to SERCA but is more complex with an extra domain.
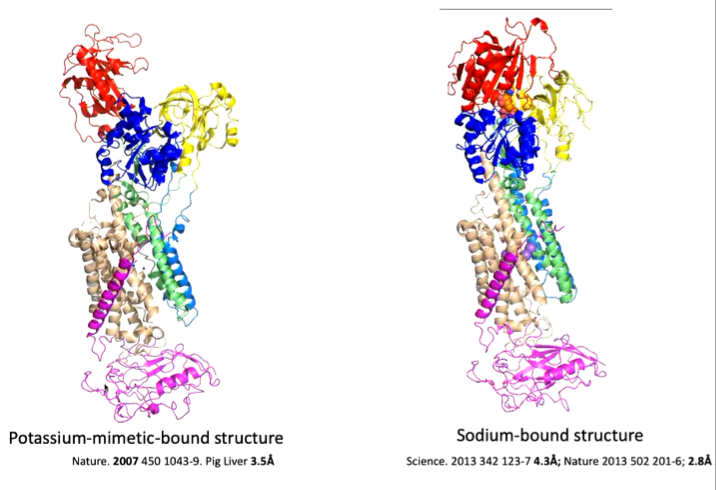
In the below schematic, it looks similar to SERCA (cytoplasm at the bottom) with extra domain.
Cryo-em
One paper gae 5 cryo-em structure of 5 different states of the protein and rather than being purified from source, it is purification of recombinant proteins produced in mammalian cells.
Morphing the 5 structures show the overall ideas are similar to SERCA, however the details are different as it is more of a rotation of domains rather than a translation. Each protein therefore is different as it has evolved differently
ABC transporters
These are ATP binding cassette transporters that contains both importers and exporters that transport large molecules. Importers are predominantly found in bacteria and exporters are ubiquitous with 44 distinct human ABC transporters. They contain nucleotide binding domains attached to a transporter domain:
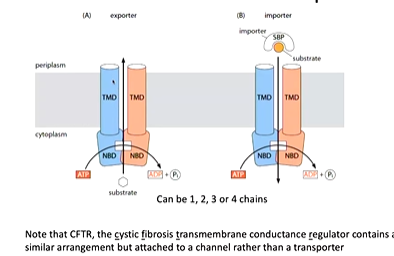
The cystic fibrosis transmembrane regulator also has this structure however is an ATP linked channel.
nucleotide binding domain dimer
hydrolysis happens here. It is a dimer that has a RECa domain (blue) and a alpha helix domain (green). ATP binds at the interface of the subunits not at the two domains of one subunit. There are therefore two binding sites.
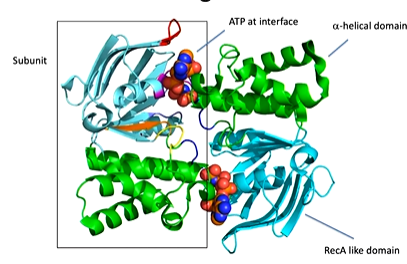
The protein has an A loop with a conserved aromatic residue interacting with the adenine of the ATP. There is also a P loop or walker A motif with a positively charged residue that can interact with the phosphate. There is also the helix dipole and the slight positive charge of it that can interact with the negative charge of the phosphate There is also a walker B motif (four hydrophobic residues, DE) with E thought to be the general base and D binding to Mg. There is also the ABC signature motif, Q loop and a D loop at dimer interface that affects the geometry of the AP site and an H loop or switch region. This all allows ATP to bind and be hydrolysed.
Exporters
Structures of bacterial proteins were first investigated using crystallography. The first structure of a mammalian exporter was of P glycoprotein that was low resolution. Cryo-em is giving many structures of human transporters and is often helped by technological improvements
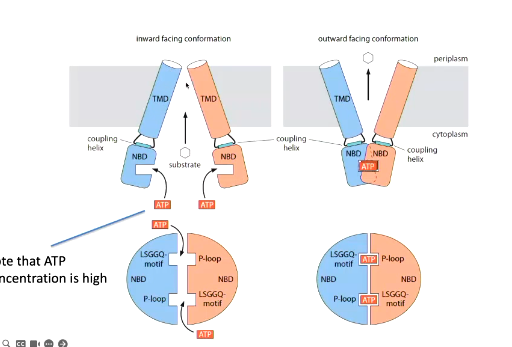
general mechanism of ABC exporters
The substrate binds from the inside where the membrane domain is open. The substrate causes a conformational change to the transmembrane domains that allows ATP to lock it in this confirmation. The two nucleotide domains are moved closer together through coupling regions and this pulls the transmembrane regions so the molecule can come off.
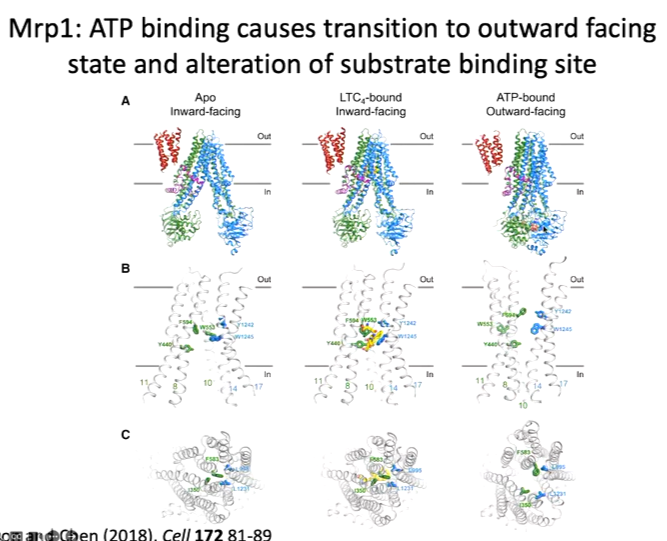
MRP1

It is one signal chain of protein with a few extra domains to the classical ABC domains. From the top side, it is closed off and there is no access to the substrate. The structure was solved with leukotriene bound at the centre of the protein. To fit molecules into the density pattern a lot of chemistry was used and there are interactions between the polar groups of the leukotriene and the polar groups of the protein. There are also polar pockets and hydrophobic pockets of the protein. The structure was compared with the Apo structure and overlaid it, there is a slight movement inward when the substrate is bound:

The next state is the ATP bound state to bring the two domains together. To get this structure, there is only one catalytic ATP site as the second one only contains an asp. They wanted to break both sites and prevent catalysis so that ATP bound bind but was not hydrolysed and so couldn’t switch back to the APO site. The mutation was E145Q and added the ATP and the substrate although the substrate is not seen in the structure:
importers
These are only found in prokaryotes excluding importers that also act as exporters.
There are different classes of importers and most of the initial ones have a substrate binding protein in the periplasm of gram negative bacteria. This is the part that bind the substrate (usually nutrients)
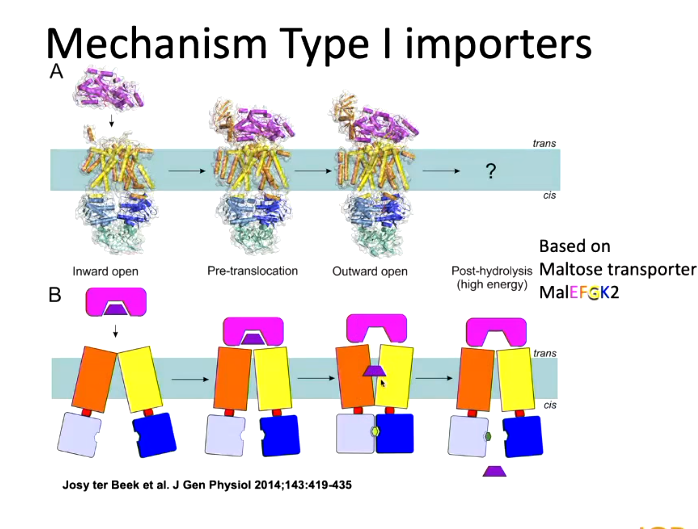
Type 1 importers have a substrate binding domain as well as another one. When a substrate binds, it binds in the middle and there is a large conformational change of the two domains to clasp together to grab the substrate.
There was a lot of work to arrive to this. The first substrate to be solved was MALef where maltose binds to the substrate binding domain specifically and then interacts with the APC transporter. The whole substrate binding domain mimics the binding of leukotriene to the inside of the ABS domain to cause a conformational change. ATP can the. bind and causes a conformational change so the substrate can enter the cell: