CLÍNICA MÉDICA II
DIABETES MELLITUS TIPO 2
Critérios diagnósticos → mínimo de 2 alterados que podem ser na mesma amostra ou em amostras diferentes pois medem a glicose de maneira diferente e evitam o diagnóstico por hiperglicemia transitória, como em uso de corticoide ou infecção.
! critérios para Pré-DM necessita de APENAS 1 resultados alterado para diagnóstico !
*proposição para nomear Pré-DM para hiperglicemia intermediária
Glicemia Jejum
<99 = normal
100-125 = Pré-DM
>126 = DM
>200 + sintomatologia = DM sem necessidade de repetir exame.
TOTG de 2 horas → feito se apenas 1 dos exames estiver alterado e paciente tiver fatores de risco.
<140 = normal
140-199 = Pré-DM
>200 = DM
HbA1c
<5,7% = normal
5,7 - 6,4% = Pré-DM
>6,5% = DM
→ TOTG de 1h foi introduzido ao SBD em 2023 porém uso ainda é mínimo. Foi recomendado usar para pacientes de alto risco.
TOTG de 1 hora
<155 = normal
155 - 208 = Pré-DM
>209 = DM
Rastreio
Assintomáticos a partir dos 35 anos (3-3 anos)
Sobrepesos/obesos + FR independente da idade → individualiza periodicidade do rastreio
→ FR: HF, etnias (afrodescendentes, hispânicos, latinos), SOP, cardiopatas, HAS, HDL <35, TG >250, HIV, sedentarismo, acantose nigricans.
Continuidade
sem sintomas com 1 critério → semestral
Pré DM; exames normais e 3 FR; exames normias e FINDRISC alto/muito alto → anual
exames normais e < 3 FR; exames normais e FINDRISC baixo/moderado → trianual
Remissão
A possibilidade de remissão do diabetes acentua a importância do Pré-DM pois demonstra que é possível retornar ao normal com ou sem medidas farmacológicas e sempre com mudanças de hábitos de vida. É um “aviso”.
Remissão = glicose normal por no mínimo 3 meses sem uso de hipoglicemiantes.
Complicações Macrovasculares → estão relacionadas à doença aterosclerótica
Doença aterosclerótica coronariana
Doença cerebrovascular
Doença arterial periférica
Complicações Microvasculares → pesquisa desde 1a consulta
Retinopatia → encaminha para oftalmologia
Nefropatia → creatinina, TFG, urina rotina (proteinúria)
Neuropatia
Comorbidades
HAS
IC
Obesidade
Dislipidemia
Doença Hepática Metabólica
Sedentarismo
Tabagismo
Complicações crônicas
Doença ocular diabética
Nefropatia
Neuropatia
Osteodistrofia diabética
Doença Cardiovascular (coronariana, cerebrovascular, periférica)
TRATAMENTO DE PRÉ-DM
Objetivo → PREVINIR a DM2, não é normalizar a glicose. Embora a normalização possa acontecer como consequência de uma boa terapêutica e adesão.
! o tratamento não-farmacológico é a BASE de todos tratamentos de DM2 e sempre deve ser indicado → consegue diminuir 1Ac em até 2% !
→ a diminuição da Hb1Ac age apenas na melhora de desfechos microvascualres, já os macrovasculares precisam de outras ações (ex: mudança de hábito de vida). LOGO, metformina não substitui tratamento não-farmacológico.
→ Quando começar tratamento farmacológico:
25-69 anos
IMC > 35
GJ > 110
Hb1Ac > 6%
DMG prévio
→ Apenas 30% das Pré-DM se tornam DM2
→ A meta para Pré-DM é no máximo 6%, abaixo disso há alto risco de hipoglicemia.
Efeito Legado (para DCV) → Quanto mais rápido o tratamento intenso é iniciado, melhor o desfecho da doença para o RESTO DA VIDA do paciente. Dessa maneira, é preciso equilibrar a intervenção e a qualidade de vida.
TIR [Time In Range] → A medida de tempo em que a glicose fica dentro da faixa-alvo ideal. É 70-180mg/dL. Usado quando há monitorização contínua.
→ A variabilidade de glicose também demonstrou ter alto efeito lesivo na camada endotelial vascular.
! As complicações macrovasculares já começam na Pré-DM, enquanto as microvasculares são EXCLUSIVAS da diabetes !
Hipoglicemia
N1 → <70
N2 → <54
N3 → alteração de estado mental, precisa de ajuda de terceiros para se recuperar.
TRATAMENTO DE DM2
Metas
Hb1Ac → <7% (porém pode variar conforme o paciente e seu risco de hipoglicemia, ex: idoso frágil é <8%).
6,5% → paciente de baixo risco, boa saúde, funcionalidade e poucas ou nenhuma comorbidade
7% → Maioria dos pacientes adultos
7,5% → Maioria dos pacientes idosos
8% → Idosos com problemas complexos ou saúde intermediária
Sem meta → Idosos com saúde complexa, saúde baixa OU pacientes com expectativa de vida limitada.
ADA 2025
Glicemia pré-prandial → <100
Glicemia pós-prandial → <160
TIR → 70-180mg/dL
→ A meta da gestante é a unica Hb1Ac <6% pois melhora desfecho materno-fetal, outros testes tem como meta os valores de normalidade.
→ Hb1Ac = glicemia média de 154. Logo resultados de diário de glicemia tem que bater com a média.
Tratamento Não Farmacológico (TODOS)
Mudança de estilo de vida
Dieta → tem uma dieta padrão para início.
Exercício Físico → 150 minutos por semana, nunca menos de 30 minutos por sessão e tentar não passar mais de 2 dias sem fazer.
Higiene do Sono
! TODOS PACIENTES COM DM2 DEVEM USAR METFORMINA !
Escolha do Medicamento
→ 6 Ps: Patogênese (resistência a insulina ou queda da secreção de insulina); Potência (impacto na glicose, peso e proteção cardiorrenal), Precauções (contra-indicações e efeitos adversos); Plus (impacto no peso, ACV, função renal, fígado e ossos), Praticidade (modo de uso), Preço.
Secretagogos→ têm alto risco de hipoglicemia, primeira escolha deve ser a Glicazida MP (menor risco de hipoglicemia).
CI: IR, IC, acidose, IH, IP
Incretínicos → IDPP4 e A-GLP-1 [A vantagem dos incretínicos é que eles são glicose dependentes, logo só liberam insulina se há hiperglicemia o que impede quedas hipoglicêmicas.
Semaglutida - Ozempic
Dulaglutida - Trulicity
Liraglurida - Victoza
*Agonista de GLP1/GIP → Tirzepatida (Mounjaro), ainda não disponível no Brasil
Metformina (biguanidas) → diminui produção de glicose pelo fígado (faz hiperglicemia de jejum) e diminui resistência a insulina periférica
Metformina XR → prolonga a liveração por capsula que sai nas fezes, diminuindo os principais efeitos adversos (TGI)
Deve-se fazer controle anual da Vit. B12 pois medicação afeta a absorção.
ISGLT-2 → reduz reabsorção renal da glicose, provoca glicosúria.
EA: aumento de infecções genitourinárias, aumento do risco de cetoacidose diabética em DM1
Dapaglifozina - Forxiga
Glitazonas → reduzem resistência à insulina
Pioglitazona EA → IC III/IV aumenta taxa de internação, aumento de taxa de fraturas.
Não devem ser associados → Insulina + Secretagogos ; IDPP4 + AGLP-1
Potência: [mais] A-GLP-1 ; ISGLT-2 ; Metformina ; Glitaozna ; Sulfoniureia ; IDPP4 [menos]
Plus
Obesidade → A-GLP-1 ; A-GLP-1 + GIP ; ISGLT-2 e Metformina (baixa evidência)
HAS, IC e DVC → A-GLP-1 e ISGLT-2
Doença Hepática Metabólica → Pioglitazona ; A-GLP-1 ; A-GLP-1 + GIP ; ISGLT-2
→ DM2 + Doença Cardiovascular Aterosclerótica
Iniciar já com terapia dupla → Metformina + AGLP1 ou ISGLT2
Se meta não é cumprida, inciar terapia tripla com esses três
→ DM2 + Insucificiência Cardíaca
Iniciar com terapia dupla → Metformina + ISGLT2
Se meta não é cumprida, iniciar terapia tripla → Merformina, ISGLT2 + A-AGLP-1
CI → Pioglitazona e Saxagliptina não devem ser dadas a IC
Exceções: A metformina não edve ser usada em pacientes instáveis, logo é CI se TFG <30, ou se IC é aguda ou se IC IV.
→ DM2 + Doença Renal
TODOS devem usar IECA ou Bra → suspender se Hiper K ou TFG cair >30%
TFG < 60 + RAC >30 → ISGLT2 + IECA/Bra + Metformina
Metformina só é indicada até TFG de 30, abaixo disso recomenda-se IDPP-4 ou Insulina.
Insulina para DM2
Sintomatologia (poliúria, polidpsia, perda de peso, polifagia)
HbA1c > 9% ou GJ >250
→ O uso pode ser transitório!
Tratemento Cirúrgico
→ Cirurgia bariátrica: para tratar obesidade
→ Cirurgia metabólica: para tratar síndrome metabólica (são o mesmo procedimento)
INSULINOTERAPIA
FISIOLOGIA
→ Em jejum, a glicose tem produção predominantemente hepática e levemente renal.
→ Insulina tem ação anabólica
Fígado: inibe glicogenólise e gliconeogênese
Músculos: promove captação da glicose
T. Adiposo: Inibe a lipólise.
EFEITOS ADVERSOS
Hipoglicemia
Lipodistrofias
Ganho de peso
Reações alérgicas
METAS DM1
< 6,5% se:
Baixo risco de hipoglicemia
Não piora qualidade de vida
Não sobrecarrega o cuidado da diabetes
Em fase de remissão (Lua de Mel)
Fase de Lua de Mel: período aproximadamente 3 meses após início da insulinoterapia que há remissão parcial da DM1, ou seja, normalidade dos níveis glicêmicos com baixa quantidade de insulina terapêutica. Tem duração média de 10 meses.
DT: <0.5 UI/kg/dia
< 7,5% se:
Hipoglicemia assintomática ou incapacidade de perceber hipoglicemia
Histórico de hipoglicemia grave
Falta de acesso aos análogos de insulina
Falta de acesso a sistemas avançados de liberação de insulina
Impossibilidade de monitorização glicêmica regular
Impossibilidade de monitorização contínua de glicose
Alvos glicêmicos menos rígidos se:
Menor expectativa de vida
Comorbidades limitantes → Neoplasia avançada, doença cardiovascular, doença renal do diabetes avançada.
Risco de hipoglicemia grave
Função cognitiva comprometida
Capacidade funcional comprometida
[ambas visam o menor risco de hipoglicemia]
INSULINA BASAL IDEAL: longa duração sem pico. (faz cair antes de dormir até acordar de 40-60mg)
INSULINA BÓLUS IDEAL: ação rápida e término rápido
FARMACOCINÉTICA
→ possíveis modos de ação: Ultralenta, Lenta, Intermediária, Rápida, Ultrarrápida, *Ultra-Ultrarrápida
Ultralentas
Degludeca → Início em 30-90 minutos, não tem pico, dura entre 36-42h e pode ser usada a partir de 1 ano de vida
Glargina U-300 → Início em 2h, não tem pico e dura 36 horas. Pode ser usada a partir de 18 anos.
Lentas
Glargina U-100 → Início em 2h, não tem pico e dura 20-24h. Pode ser usada a partir dos 2 anos.
Orientação aplicar sempre no mesmo horário.
Detemir → Início em 3-4h, não tem pico e dura 13-20h. Pode ser usada a partir de 2 anos e em gestantes.
Intermediária
NPH → Início em 2-4h, pico em 6-8h e dura 12-18h. É usada como insulina basal, sendo aplicada de 2-4x/dia.
Orientação é aplicar logo antes do café da manhã.
Rápida
Regular → Início em 30 minutos (5 se EV), pico em 2-3h e dura 4-6h. Sua administração é endovenosa.
Ultra-Rápidas
Lispro, Asparte e Glulisina → têm início em 5-15 minutos, pico em 30-90 minutos e duram 4-6h. Podem ser usadas em crianças e gestantes (exceto Glulisina)
(Lispro e Asparte podem ser administradas com canetas com meia unidade e todas ultra-rápidas tem administração endovenosa e em bombas)
O melhor tempo de aplicar essas insulinas é 10-15 minutos antes da refeição.
Ultra-Ultrarrápida (É a que mais se aproxima da insulina endógena)
FIASP [Asparte UR] → Início em 2-5 minutos, pico em 1-3h e dura 5 horas. Sua utilização pode ser PÓS-PRANDIAL ou DURANTE.
→ Para a segurança de idosos e nenéms, a insulina Pós-Prandial evita riscos de hipoglicemia caso haja recusa alimentar.
PLANOS DE INSULINOTERAPIA INJETÁVEL
Preferível: Múltiplas doses de insulina com análogos de ação longa + análogos de ação rápida ou ultra-ráida (Problema: $$$)
Outros: Múltiplas doses de insulina com NPH [preferível aplicar 3x, metade antes do café, ¼ meio dia e ¼ antes de dormir] + análogos de ação rápida ou ultra-rápida OU Múltiplas doses de insulina NPH + Regular OU pré-misturadas (Problema: risco de hipoglicemia)
BOMBAS DE INSULINA
→ Infusão subcutânea de insulinas ultra-rápidas (Lispro, Asparte e Glulisina) e Fiasp.
→ Indicações:
Hipoglicemias
Fenomêno do Alvorecer → aumento fisiológico de cortisol e GH aumentam glicemia.
Gestantes
Gastroparesia
Crianças
Estilo de vida “sem rotina”.
DOSE DIÁRIA TOTAL: 0,5UI/kg/dia (50% basal e 50% bólus → 10-20% por refeição)
Aumenta a necessidade de insulina: puberdade, final de gestação, medicamentos (corticóide), doenças intercorrentes, hipertireoidismo, Cushing, estresse.
Diminui a necessidade de insulina: DRC, Fase de Lua de Mel, Hipotireoidismo, Má absorção intestinal, insuficiência adrenal
DM2 → 0,1-0,2UI/kg/dia
→ Considerações para Bólus Prandiais
Qual a glicemia pré-prandial? → Correção de bólus
O que será ingerido? → Contagem de carboidratos ou dieta pouco variável.
Quantos UI de insulina cobrem Xgr de CHO? → Contagem de carboidratos
Quantos UI de insulina fazem glicemia cair? → Correção de bólus.
CORREÇÃO DE BÓLUS
A correção de bólus leva em conta o Fator de Sensibilidade (quanto 1UI de insulina abaixa na glicemia), a glicemia medida pré-prandial e a glicemia alvo (costuma ser 100).
Calcular Fator de Sensibilidade:
1800/Dose Diária Total de Insulina (0,5UI/kg/dia)
Calcular Correção:
Glicemia medida - Glicemia Alvo / Fator de Sensibilidade
Ex: Paciente de 70kg com glicemia pré-prandial de 170, alvo é de 120.
1800/35 = 50
170-120/50 = 0.9
BÓLUS ALIMENTAÇÃO
Regra Geral: a proporção de UI:CHO é 1:15g.
400/DDT é a contagem mais fidedigna.
Existem tabelas com quantidade de CHO em alimentos comuns, como 1 copo de leite (12g) e pão integral (12g)
Ex: Paciente de 70kg com glicemia pré-prandial de 170, alvo é de 120. Irá comer 1 copo de leite e 2 fatias de pão integral.
1800/35 = 51
170-120/51 = 0.98 → Bólus correção.
Bólus alimentação: 1UI:15g → 12+24 = 36g CHO → 36/15 = 2,4UI
Correção + Alimentação: 2,4 + 0,98 = 3,38 UI
Arredonda para baixo → 3UI.
! Se Glicemia pré-prandial for menor que a glicemia alvo, o valor será negativo e deve-se SUBTRAIR do valor do bólus alimentação !
Regra Geral Adultos → 1 UI para que cada 50mg acima da glicemia alvo (100mg)
→ subir 40-60mg após alimentação está adequado.
TRATAMENTO NÃO INSULÍNICO PARA DM1
→ Não existe evidência científica
→ ISGLT2 são contra-indicados para DM1 pelo alto risco de cetoacidose
TRANSPLANTE EM DM1
Transplante de ilhotas
Transplante de Pâncreas
Transplante duplo: Pânceas e Rim → É o mais usado.
INSULINOTERAPIA EM DM2
Início é apenas com basal 0,1-0,2UI/kg/dia ASSOCIADO ao tratamento não-insulínico (exceto sulfoniureias)
Se a dose precisar ser aumentada e chegar a 0,5UI/kg/dia deve-se considerar associar A-GLP-1 antes de começar bólus.
Degludeca + Liraglutida
Glargina U100 + Lixisenatida
Se A-GLP-1 não forem possíveis ou ineficazes, iniciar tratamento basa/bólus.
→ Indicações para insulinoterapia em DM2
Falência de Células Beta → emagracimento, 4ps (poliúria, polidipsia, polifagia, perda de peso)
Descompensações agudas
Internações
Cirurgias
Gravidez
Insuficiência Renal
Insuficiência Hepática
Sintomático ao diagnóstico + GJ > 300 e/ou A1c >10% e/ou cetonúria/cetonemia
Sintomático + GJ>250 e A1c >9%
NÓDULOS TIREOIDEANOS
PREVALÊNCIA
3-7% das pessoas tem nódulo palpável, enquanto 20-76% tem nódulo visível em USG → a recomendação é fazer USG em quem possui nódulo palpável
Cerca de 90% dos nódulos são benignos, intuito do USG é identificar malignidade
ETIOLOGIA
Doença nodular folicular (bócio colóide ou adenomatoso)
Cistos simples ou secundários a outras lesões da tireóide
Tireoidites
Doenças granulomatosas → Sarcoidoses
Neoplasias: adenomas, neoplasias de baixo risco, carcinomas, linfomas, tumores raros e lesões metastáticas
SUGESTIVO DE MALIGNIDADE
Características pessoais:
<30 anos
>70 anos
Homens
Radiação prévia de cabeça ou pescoço
HF de Ca. medular ou NEM → Neoplasias Endócrinas Múltiplas
Carcaterísticas do nódulo
Crescimento rápido → 20 % de aumento em pelo menos 2 dimensões (no mínimo 2mm) ou 50% do volume
>4cm
Lindafenomegalia → Ca. tireodeano mais comum é o papilífero e costuma metastizar para linfonodos
Endurecido, aderido
Disfonia, compressão
AVALIAÇÃO LABORATORIAL
→ Caso haja disfunção tireoideana, hormônios costuma subir pois nódulos são autonômos
TSH e T4 livre → função tireoideana costuma ser normal, se hipotireoidismo não exclui malignidade e se hipertireoidismo indica pouca probabilidade de câncer.
Calcitonina → suspeita de carcinoma medular, porém ainda controverso como rotina
EXAMES COMPLEMENTARES
USG é o padrão-ouro
Cintilografia é indicada em caso de hipertireoidismo (TSH baixo)
CINTILOGRAFIA → indicada apenas se TSH baixo
[ tem pouco valor na distinção de malignidade e benignidade ]
Maioria dos nódulos são hipo ou normocaptantes → se for o caso, indicação é seguir com PAAF
<15% dos hipocaptantes são malignos
1-2% dos hipercaptantes são malignos
ULTRASSONOGRAFIA
→ Tireoide normal: hiperecogênica em relação aos tecidos vizinhos. Volume 10-15 cm3
Composição: sólido, cístico ou misto → Sólido tem mais risco de malignidade
Ecogenicidade: hipo, iso e hiperecogênico → quanto mais hipoecoido maior risco de malignidade (50-60%)
Margem/halo: regular, irregulares, infiltração
Forma: altura e largura
Focos ecogênicos: calcificações periféricas (casca de ovo), macro e microcalcificações → todos os focos ecogênicos aumentam risco de malignidade, principalmente microcalcificações
Características linfonodais
→ Nódulos císticos anecoidos são sempre benignos
→ Nódulos espongiformes: Agregação de múltiplos componentes microcísticos em mais de 50% do volume do nódulo [aspecto de colmeia]; Maioria é benigno
→ Colóide espesso é visto como “cauda de cometa” e pode ser diferenciado das calcificações pois essas fazem sombra acústica
DESCRIÇÃO DO USG
Medida das 3 dimensões acuradas
Localização do nódulo
Descrição de no máximo 4 nódulos [escolhe-se os com maiores TI-RADS, independente do tamnho]
Características do nódulo
ACOMPANHAMENTO COM USG (Nódulos sem critérios para PAAF)
6-12 meses: < 1cm com características suspeitas
12-24 meses: Nódulos de baixa ou intermediária suspeição
2-3 anos: Nódulos de muito baixo risco
CARACTERÍSTICAS SUSPEITAS DE NÓDULOS SÓLIDOS (SSF)
Margem ou halo irregular
Altura maior que largura
Focos ecogênicos puntiformes
Macrocalcificação
PADRÃO DE RISCO USG ATA 2025 → utiliza SSF + ecogenicidade
SFF | MUITO HIPOECOICO | HIPOECOICO | ISO/ HIPERECOICO |
2 ou + | Alto | Alto | Intermediário |
1 | Alto | Intermediário | Intermediário |
0 | Intermediário | Baixo | Baixo |
→ Baixo risco: PAAF se >1,5cm
→ Risco intermediário: PAAF se >1cm
→ Alto risco: PAAF se >1cm
TI-RADS
Composição:
sólido - 2
cístico - 0
misto - 1
Ecogenicidade:
muito hipoecoido - 3
hipoecoido - 2
isoecoico/hiperecoico - 1
anecoico - 0
Forma
altura maior que largura - 3
lagura maior que altura - 0
Margem/halo
lisa - 0
regular - 0
irregulares - 2
infiltração - 3
Focos ecogênicos:
cauda de cometa - 0
calcificações periféricas (casca de ovo) - 2
macrocalcificações - 1
microcalcificações - 3
→ TI-RADS 1: 0 pontos; sem seguimento
→ TI-RADS 2: 2 pontos; sem seguimento
→ TI-RADS 3: 3 pontos
>2,5cm → PAAF
>1,5cm → seguimento 3-5 anos
→ TI-RADS 4: 4-6 pontos
>1,5cm → PAAF
>1cm → seguimento 1, 2, 3 e 5 ano.
→ TI-RADS 5: >6 pontos
>1cm → PAAF
>0,5cm → seguimento anual por 5 anos
PAAF
Punciona no máximo 2 nódulos (com maior TI-RADS)
Melhor exame para distinguir entre lesões malignas e benignas
Limitação: diferenciação entre adenoma e carcinoma folicular [tem que ver invasão de cápsula]
Deve-se repetir a PAAF sempre que houver mudança no padrão do USG
Repetir após 3 meses se: BI, BIII ou BIV
BETHESDA → Classificação para análise da citologia
I → inconclusivo; repete PAAF [nova PAAF dará o diagnóstico em 60- 80 % dos casos]
II → benigno; acompanhamento clínico com USG
Repete USG 6-24 meses após PAAF inicial, e repete PAAF se:
se aumento > 20% em pelo menos 2 dimensões
Se aumento > 50% volume
Se surgirem características de malignidade
III → atipia de significado indeterminado; repete PAAF + teste molecular (pode fazer lobectomia diagnóstica ou vigilância)
Normalmente se PAAF repetida persiste em BIII, faz cirurgia e decide acompanhamento
IV → Neoplasia folicular; teste molecular e/ou lobectomia diagnóstica
V → suspeita de malignidade; teste molecular + lobectomia ou tireodectomia subtotal ou total [normalmente total]
VI → Maligno; lobectomia ou tireodectomia subtotal ou total [normalmente total]
PAINEL MOLECULAR
→ Faz para Bethesda III e IV
BRAFV600E → carcinoma papilíferos menos diferenciados e anaplásicos
RET/PTC → metástase linfonodal; também em tumores benignos
RAS → carcinoma folicular; também em adenoma folicular
PAX8/PPARᵧ → carcinoma folicular
TERT → indica maior agressividade
TRATAMENTO
Decisão entre acompanhemnto e cirurgia de acordo com risco de malignidade
Disfunção tireoidiana (hipertireoidismo) e sintomas compressivos dizem a favor de intervenção cirúrgica
Atualmente, alcoolização e radioablação so usados apenas para tumores benignos.
OBESIDADE
→ É uma doença poligênica com formas monogênicas raras.
CONCEITO: Acúmulo anormal ou excessivo de gordura corporal que pode atingir graus capazes de afetar a saúde (OMS).
CLASSIFICAÇÃO PELO IMC
Abaixo do peso ideal | <18.5 |
Peso ideal | 18.5 - 24.9 |
Sobrepeso | 25 - 29.9 |
Obesidade Grau I | 30 - 34.9 |
Obesidade Grau II | 35 - 39.9 |
Obesidade Grau III | >40 |
→ Limitações: não demonstra o percentual de gordura e distribuição corporal e não determina quando o excesso de adiposidade é um problema de saúde.
ALÉM DO IMC
→ 3 medidas corpotais que pode ser usadas para confirmar excesso de gordura corporal (se IMC >40 não há necessidade )
Circunferência cintura: >102 para homens e >88 para mulheres
Relação cintura-quadril: >0.90 homens e >0.85 mulheres
Relação cintura-altura: >0.5 para todos
Tomografia computadorizada
→ Nova proposta de 2025 coloca obesidade como IMC + outra avaliação com excesso de godura (TC ou medida) OU medida corporal + TC.
EDMONTON → classificação da obesidade de acordo com acometimento fisiológico, mental e funcional.
0 - ausência de fatores de risco, sintomas físicos, psicopatologias e acometimento funcional
1 - fatores de risco subclínicos, acomentimento físico, psicológico e funcional leve
2 - presença de doença crônica relacionada à obesidade e limitação moderada nas atividades diárias
3 - Dano de orgão-alvo estabelecido, osteoartrite incapacitante, acometimento psicológico e funcional importante
4 - Injúrias severas, acometimento geral severo
FISIOPATOLOGIA
Epigenética → interação fatores genéticos e ambientais
Desequilíbrio positivo entre consumo e gasto energético
BALANÇO ENERGÉTICO
Controlado no hipotalámo e por um grande número de peptídeos secretados pelo intestino e tecido adiposo
Na obesidade há uma alteração no “sensor” do SNC de fome/saciedade e do gasto de energia → aumento da apetite e redução da termogênese
TECIDO ADIPOSO
Hiperplasia e hipertrofia do tecido adiposo → hipoperfusão → infiltração de macrofágos no tecido adiposo → secretam fatores inflamatórios (TNF-alfa e IL-6) → atuam na resistência à insulina → inflamação crônica de baixo grau
COMORBIDADES
Sistema Cardiovascular → HAS, doença coronariana, acidente vascular cerebral, IC
Metabólicas → DM2, DMG, dislipidemia, hiperuricemia, gota, doença hepática metabólica
Sistema reprodutivo/urinário → Infertilidade, hipogonadismo, SOP, ITU
Sistema gastrointestinal → Refluxo gastroesofágico, doença hepática metabólica
Neoplasias → Mama, colon, endométrio, vesícula, esôfago, fígado, próstata
Psiquiátrica → Depressão, demência
Sistema respiratório → Apneia do sono, asma.
Sistema osseo → osteoratrites
Alterações cutâneuas → lipedema
TRANSTORNOS ALIMENTARES
Estão presentes em 20-30%
Dentre eles: compulsão alimentar; purgação; ausência de saciedade; hábito do comedor noturno; bulimia
TRATAMENTO
→ Redução de 5-10% do peso melhora: apneia do sono, risco de DM2, qualidade de vida, perfil lipídico, risco cardiovascular, níveis pressóricos.
→ Nova proposta: obesidade controlada → considera peso máximo atingido durante a vida (18-85 anos) e olha trajetória:
Obesidade inalterada → perda de <5%
Obesidade reduzida → perda de 5-10%
Obesidade controlada → perda de >10%
Tratamento não medicamentoso (BASE!)
Reeducação alimentar → terapia nutricional: alimentação saudável + relação saudável com comida
Atividade física → preferência por atividade aeróbica por 30-60 minutos de média/alta intensidade
Tratamento Medicamentoso → Revisar e ajustar o tratamento no mínimo a cada 03 meses
Considerar quando:
IMC >30
IMC >27 + comorbidade (HAS, DM2, hiperlipidemia, apneia do sono)
Fármacos:
Sibutramina → sacietógeno que foi retirado do mercado americano e europeu pelo risco de desfechos cardiovasculares (16% RR, scout trial).
No Brasil é CI para HAS, DCV, DM2 + FR, > 65 anos, crianças e adolescentes
EA: boca seca, cefaleia, taquicardia, elevação de PA, constipação intestinal, agitação, nervosismo
Orlistate → inibe 30% da absorção de gordura pelo TGI
EA: : esteatorréia, flatulência, descarga de gordura, urgência fecal, deficiência de vitaminas lipossolúveis ( mais raro)
Liraglutida → Retarda esvaziamento gástrico + Efeito incretínico → Secreção de insulina e Inibe glucagon + Ação hipotalâmica em vias anorexigênicas/apetite hedônico
Semaglutida (para tratamento de obesidade dose deve chegar a 2mg)
Bupropiona/naltrexona → compulsão, diminui apetite
Lisdexanfetamina → Transtorno alimentar
Tirzepatida → aprovada FDA, ainda não comercializada no Brasil, aqui aprovada para DM
Off-label:
fluoxetina, sertralina → compulsividade
semaglutida oral
topiramato → transtorno alimentar, hipotimia
CIRÚRGIA BARIÁTRICA
Mecanismo:
Redução de apetite → restrição, mal-absorção
Alteração dos sinais periféricos da regulação do peso corporal (comunicação intestino-cérebro) → Aumento secreção de hormônios da saciedade (GLP-1 e PYY) + Redução secreção de grelina
Indicações:
IMC ≥ 50
IMC ≥ 40 com ou sem comorbidades e tto por no mínimo 02 anos
IMC entre 35-40 com comorbidades sem sucesso com tratamento clinico por no mínimo 02 anos
IMC 30-35 → *Cirurgia metabólica: visa controle da síndrome metabólica
Contra-indicações:
Limitação intelectual significativa
Transtorno psiquiátrico não controlado
Doença cardiopulmonar descompensada
Doença que predispõe sangramento gastrointestinal superior
Síndrome de Cushing
Neoplasias malignas não tratadas
Técnicas:
Bypass gástrico em Y de ROUX
Gastrectomia vertical (Sleeve)
Banda Gástrica ajustável
Cirurgia de Scopinaro ou Switch duodenal
Complicações:
Má absorção de micronutrientes / desnutrição proteicocalorica
Queda de cabelo
Reganho de peso → Não existe consenso sobre o que constitui reganho de peso significativo (> 15% do nadir / + 10 Kg do Nadir) [Média de 30% dos pacientes]
Adaptações metabólicas compensatórias: Redução do gasto energético; Oxidação tecido adiposo e reduçāo leptina; Aumento hormônios orexígenos Ex :grelina → aumento apetite; Fatores hedônicos
Sindromes/complicações gastrintestinais
Dumping/hipoglicemia
Precoce → hiperosmolaridade, clínica de diarreia líquida e hipovolemia relativa
Tardio → hiperinsulinemia incretino-mediada levando à hipoglicema.
Doenças ósseas
Distúrbios psiquiátricos
Excesso de pele
Acompanhamento gestacional
SEMIOLOGIA RENAL
→ Início do funcionamento renal: 10° semana
Sinal de mau-funcionamento renal no feto: Oligodrâmnio
→ Nascimento: entre 600.000 e 1 milhão de néfrons
PIG → menos néfrons do que o normal → maior risco de DRC a partir dos 20 anos
→ Utiliza 20% do débito cardíaco
Perfunde 400mL/100g → mais do que coração, fígado e cerébro.
Utiliza 10% do consumo total de oxigênio corporal
→ Filtração média: 180 litros de sangue produzem 1,5-2L de urina.
→ Uma pessoa normal urina a cada 4-6h e normalmente não urina à noite.
APRESENTAÇÕES CLÍNICAS DE UM NEFROPATA
Alterações urinárias assintomáticas → demandam acompanhamento
Piúria
Hematúria → sem mudança de cor
Proteinúria → sem espuma na urina
Síndrome Nefrítica → igual a Lesão Renal Aguda ou Insuficiência Renal Aguda
Síndrome Nefrótica → insidiosa
Síndrome Renais Tubulares
Maioria se manifesta na infância: túbulo faz equilíbrio ácido-base → doença tubular → criança fica com pH ácido → desgaste ósseo e catabolismo → criança não cresce.
doenças císticas (mais comum em idosos);
poliúria;
glicosúria → glicose é livremente filtrada mas é reabsorvida. Para ser secundária à hiperglicemia a glicemia deve estar >160.
acidose metabólica
Doença Renal Crônica → qualquer alteração renal que dure mais de 3 meses, mesmo com TFG normal
SINTOMATOLOGIA (DADOS SUBJETIVOS)
→ Doenças são muito silencionsas, logo dados são muito confusos ou simplesmente inexistentes.
Alterações da micção → são mais agudas
Polaciúria → aumento de frequência, relacionado à irritação do TU (cálculo, Ca. bexiga)
Poliúria → demanda urina 24h para confirmação
Urgência miccional → relacionado à irritação do TU
Disúria → relacionada à irritação do TU
Nictúria → micção +2 vezes à noite (diferente de noctúria que é a necessidade de se levantar para urinar após deitar para dormir). Relação com tubulopatias mediadas por processos alérgicos, retenção excessiva de líquido e incapacidade de concentração de urina
Incontinência urinária
queixa mais comum em mulheres na menopausa: tônus da musculatura da pelve é dependente do estrogênio → musculatura fica flácida na menopausa (incontinência urinária) → bexiga muda de lugar anatomicamente → presença de urina residual → ITU de repetição
Incontinência urinária paradoxal → relacionada à retenção urinária.
Retenção urinária → Bexiga neurogênica, hiperplasia prostática, estenose de uretra.
Se agudo, há dor suprapúbica.
Alterações do volume
Oligúria [<400ml/dia] → queixa de “urina presa”, mais comum em síndrome nefrítica
Poliúria [>2.500ml/dia] → mais comum em diabetes insipidus (insuficiência na produção ou resposta ao ADH)
Anúria [<100ml/dia] → principal causa é obstrução do trato urinário
Edema
→ Em geral, percebe-se o edema nas regiões periorbitárias (tecido celular subcutâneo frouxo) e nas extremidades inferiores (ação da gravidade).
→ Funcionamento normal: Pressão oncótica é a pressão que mantém o líquido dentro dos vasos pois vence a pressão hidrostática. Porém na porção arteriorlar dos capilares a pressão hidrostática é maior que a oncótica para que ocorra troca (pressão hidrostática glomerular precisa vencer a oncótica) e na porção venosa ocorre inversão e oncótica se torna maior novamente (pode ocorrer “resgate”).
→ Fisiopatolgia do edema
queda da albumina (pode ser por baixa nutrição, baixa produção hepática ou excreção renal elevada) gera queda na pressão oncótica, o que leva à extravasamento (Teoria do underfill). Além disso, extravasamento provoca hipovolemia, que ativa SRAA e consequente leva à retenção de sódio, agravando edema.
doença glomerular → proteinúria seletiva → ativação dos canais de sódio → retençao líquida → aumenta volume extracelular → edema. [Teoria do Overflow]
Alterações da cor da urina
→ Pode ser a primeira queixa, importante perguntar sobre uso de remédios e habitos alimentares
Amarelo-claro/âmbar → normal
Amarelo-escuro → desidratação
Marrom → bilirrubina (colúria), uso de cloroquina ou nitrofurantoína
Laranja → uso de rifampicina
Vermelho/marrom avermelhado → hematúria, hemoglobinúria, necrose tubular aguda (NTA), rabdomiólise, uso de fenitoína, ingesta excessiva de beterraba.
Azul/verde → pseudomonas, uso de amitriptilina
Preto →uso de nitrofurantoína ou levodopa.
Turva/leitosa → infecções, piúria, fungos, cristais de fosfato.
Alterações do odor da urina
Fétido → ITU
Adocicado → cetonúria
Carne podre → proteinúria
Alterações de densidade da urina
→ estão relacionadas ao túbulo
VR: 1015-1025 g/ml → dependente do número de paticular presentes
1.000 → hiperdiluição (diabetes insipidus ou polidipsia psicogênica)
>1030 → glicosúria
>1040 → manitol ou contrastes osmóticos
PARÂMETROS QUÍMICOS
pH → normal da urina é levemente ácido (VR 5,5-7,5).
[alterações de pH da urina podem ser reflexo do pH sanguíneo]
Urina ácida → acidose metabólica; dieta rica em proteínas; depleção de volume (aldosterona)
Urina básica → acidose tubular renal; dieta vegetariana; vômitos; uso de substâncias alcalinas; proteus.
Bilirrubina e Urobilinogênio → VR do urobilinogênio é 0,2-1
Na estase biliar por obstrução ou fármacos, a pesquisa de bilirrubina na urina é positiva
Em hemólise, a bilirrubna indireta (lipossolúvel) aumenta na circulação e pesquisa de bilirribina na urina é negativa
Esterase Leucociária
! Indica lise de leucócitos por PROCESSO INFLAMATÓRIO, NÃO NECESSARIAMENTE INFECÇÃO !
Causas: cólica renal, ITU…
[Falso positivo leucócito/nitrito → contaminação vaginal]
[Falso negativo leucócito/nitrito → hiplerglicemia, albumina, ácido ascórbico, tretaciclina, cefalexina, cefalotina]
Nitrito
→ O nitrato se torna nitrito na presença de algumas bactérias (E. coli, Proteus, Klebsiella). Logo nitrito positivo diz mais a favor de ITU porém nitrito negativo NÃO EXCLUI ITU (algumas bactérias não fazem a conversão nitrato→nitrito: Streptococcus faecalis, Neisseria gonorrhae, Mycobacterium tuberculosis).
Piócitos → podem ter cultura positiva ou não
Cultura positiva → ITU
Cultura negativa:
Processos inflamatórios → nefrite intersticial, nefrolitíase
Bactérias inespecíficas → Tuberculose renal (Koch)
Glicose
Hiperglicemia (<160)
Gravidez
Defeito leve de túbulo proximal
Corpos Cetônicos → degradação lipídica ao invés de carboidratos
diabetes descompensada
dieta restrita
jejum prolongado
Hemoglobina e Mioglobina → hematúria e lesão muscular
Proteinúria
→ Até 150mg/dia é considerado normal:
30-50mg são mucoproteína Tamm-Horsfall → é a matriz de formação de cilindros e a maior constituente dos cilindros hialinos.
Até 30mg são albumina
Restante são globulinas;
200-500mg/24h | +/++++ |
500-1500mg/24h | ++/++++ |
2-5g/24h | +++/++++ |
>7g/24h | ++++/++++ |
→ Determinação quantitativa: Em vez de usar a urina de 24 h, pode-se determinar a quantidade de proteína em relação à creatinina em uma amostra de urina eliminando-se o fator tempo e o grau de diluição urinária.
Normalmente, relação proteinúria/creatinina é <1
Uma razão maior que 3,0 a 3,5 indica excreção proteica superior a 3,0 a 3,5 g/24 h,
Uma razão menor que 0,2 indica menos de 0,2 g em 24 h.
Limitações: subestima proteinúria em individuos musculosos (excretam mais creatinina) e superestima proteinúria em individuos caquéticos (excreta menos creatinina)
! É possivel ter proteinúria normal e albuminúria anormal (>30mg) !
Condições clínicas não renais que podem causar proteinúria:
Proteinúria funcional: proteinúria transitória decorrente de aumento da permeabilidade glomerular por angiotensina II ou noraepinefrina sem causar lesão (ITU, febre, climas extremos, exercício intenso, convuslões).
Proteinúria postural: é detectada durante o dia e desaparece à noite.
Albuminúria
VR: <30mg/24h
Microalbuminúria (ligeiramente aumentado) → 30-300
Macroabuminúria (aumento severo) → >300
SEDIMENTO URINÁRIO/MICROSCOPIA URINÁRIA
Células epiteliais
Aumentam com proteinúria
Se tem inclusão de partículas de gordura são chamados de Corpúsculos Ovais → Patognomônico de síndrome nefrótica e número de corpusculos são proporcionais à proteinúria
[ Fisiopatologia: os processos exacerbados de reabsorção de proteína levam a uma degeneração gordurosa das células epiteliais tubulares, com aparecimento de gotículas de gordura no citoplasma. ]
Leucócitos → podem ser originárioa de qualuqer parte do trato urinário
Indicam inflamação, não necessariamente infecção.
Se cultura negativa → TBC renal, cálculo renal, doençã tubulo intersticial
Hematúria → pode ser originária de qualuqer parte do trato urinário
Hemácia isomórficas: indica lesão não renal.
Hemácias dismórficas: indica lesão renal (acantócitos → doença glomerular).
Hematúria transitória: mais comum em jovens, pode ocorrer após relação sexual ou exercício físico intenso ou por ITU (cistite ou prostatite)
Hematúria persistente: é chamada de alta se glomerular (acantócitos) e baixa se de tratao urinário.
Cilindros
Cilindro hialino → mucoproteínas de Tamm-Horsfall
Cilindro leucocitário → inflamação intersticial
Cilindro hemático → doença glomerular
Cilindro celular/epitelial → lesão tubular
Cilindro granuloso → cilindro celular + células que se desintegraram, Fisiológico ou lesão tubular.
Cilindro céreo → fases finais de DRC e estase urinária
Cilindros gordurosos → síndrome nefrótica
Cristais
→ Presença de cristais de ácido úrico, fosfato e oxalato de cálcio podem não ter significado clínico: cristalização pode ocorrer na amostra de acordo com temperatura ambiente e pH da urina.
Cristais de ácido úrico: se em alta quantiade pode estar relacionado à síndrome de lise tumoral (pós-QT) e pode causar insuficiência renal aguda
Cristais de oxalato de cálcio: presente an maioria das crianças em fa se de crescimento, se elas tem HF de cálculo renal, deve-se investir hipercalciúria. Em adultos, normalmente corre por intoxicação por etilenoglicol.
Cristais de estruvita: litíase associada à ITU por bactéria produtora de urease (Klebsiella e Proteus).
→ Ocorre transformação do fosfato amoníase-magnesiano em estruvita pela urease. A estruvita não quebra com litotropsia, tratamento é cirúrgico.
PADRÕES DE DOENÇAS RENAIS
Hematúria glomerular (codócitos/acantose) + albuminúria = glomerulonefrite proliferativa [Berger, vasculite ANCA, nefrite lúpica]
Albuminúria com ou sem hematúria = Glomerulonefrite não proliferativa [diabetes, amiloidose, nefropatia membranosa, GESF, lesões mínimas]
Celulas granulares e epiteliais = Necrose tubular aguda com lesão renal aguda subjacante
Piúria isolada = infecção ou doença tubuintersticial
AVALIAÇÃO DE FUNÇÃO RENAL
Exame de sangue:
Creatinina → cálculo da taxa de filtração glomerular pela formúla de Cockcroft-Gault
[(140 - idade em anos) x peso corporal em kg] (x0,85 se mulher) / 72 x creatinina sérica mg/dl
TFG → estadiamento da função renal
G1 | >90 |
G2 | 60-89 |
G3 | 45-59 |
G4 | 30-44 |
G5 | 15-29 |
G6 | <15 |
Exame de urina amostra única:
Albumina (albuminúria):
A1 → <30mg
A2 → 30-300
A3 → >300
EXAMES DE IMAGEM
USG e TC → forma, tamanho, obstrução, cálculos, pólipos e massas.
Pielografia Intravenosa → forma, tamanho, obstrução,anatomina caliciforme [necrose papilar e rim esponjoso medular]
Análises com radionuclídeos → obstrução, vazamento de urina, fluxo arterial renal [estenose arterial renal]
Arteriografia renal → fluxo arterial [esternose arterial renal, vaculite, diferenciação massa vascularizada ou sólida]
Cistouretrografia → refluxo vesicoureteral
Pielografia retrógrada ou anterógrada → sítio de obstrução [colocação de stent renal]
RX abdome → forma, tamanho, nefrolitíase (radiopaco), nefrocalcinose
RMN → massa renal, trombose venosa renal
DOENÇA RENAL SECUNDÁRIA
Infecções → HBV, HIV, HCV, Sífilis
Doenças auto-imunes → LES, AR, Sjogren, Doença mista do tecido conjuntivo, Vasculites
Neoplasias → Tumores de orgãos sólidos (proteinúris pode ser a primeira manifestação), Mieloma Múltiplo
Amiloidose
HIPERTENSÃO ARTERAL SISTÊMICA SECUNDÁRIA
→ HAS causada por acometimento do fluxo renal (20% do fluxo global)
Suspeitas: sopro abdominal, HAS resistene, paciente jovem (<30 anos), asismetria de pulsos periféricos ou cardíacos, resposta exagerada à IECA ou BRA, edema agudo de pulmão sem causa aparente.
CÓLICA RENAL
Ondas 20-60 minutos, localização da dor indica local do cálculo
! A dor bedece limites dos orgão genitals externos (nunca vai para as pernas) !
Cálculo na junção uretero-pélvica: dor em flanco + Giordano positivo
Cálculo na porção média do ureter: dor abdominal que irradia para ligamento inguinal e testículos/grandes lábios ipsilateral
Cálculo na junção vesicoureteral: disúria, polaciúria, urgência miccional.
→ Em caso de litíase renal faz-se coleta 24h para descobrir motivo do cálculo
Dosa: cálcio, ácido úrico, oxalato, fósfora, cistina, ácido cítrico (citrato), sódio, magnésio
SOLICITAÇÃO DE COMPLEMENTO
Solicita-se complemento total e fracionado (C1, C2, C3 e C4)
Consumo (hipocomplementonemia)
Glomerulonefrites: pós streptocócica, lúpica, da crioglobulinemia, membranoproliferativa, por endocardite.
Normocomplementonemia
Glomerulonefrite por: vasculite ou anticorpo antimembranoso glomerular basal
Berger (Nefropatia por IgA)
DOENÇAS GLOMERULARES
HEMATÚRIA GLOMERULAR
→ Clínca: urina amarronzada, cor do coca cola. Normalmente não possui coágulos, diferentemente do sangramento de trato urinário
→ Diagnóstico: presença de codócitos e/ou acantócitos em pesquisa de dismorfismo eritrocitário (exame feito com urina amostra única)
A presença de cilindros hemáticos também fala a favor de hematúria glomerular.
→ Principais causas de hematúria assintomática (isolada):
Doença de Berger (nefropatia por IgA tratada com dapaglifozina)
Doença de Alport → Doença do X que causa defeito de colágeno
Doença de membrana fina
PROTEINÚRIA ISOLADA ASSINTOMÁTICA
→ deve ser >150mg/dia e <3g/dia
→ Causas intermitentes:
Exercício físico intenso
Proteínura postural/ortostática → perceptível pela diferença entre uma urina colhida de manhã e outra no final da tarde; mais comum em jovens.
Febre
ICC → descompensada
→ Causas persistentes [evolução pode ser lenta e já demonstrar DRC avançada em estado assintomático
Glomeruloesclerosse segmentar focal (GESF) → pode ser primário ou resultado final de alterações sistêmicas:
HAS → vasoconstrição da artéria arqueada como mecanismo de proteção do glomerulo (HAS não atinge glomerulo diretamente) → isquemia glomerular → menos néfrons funcionantes → hipertensão intraglomerular por sobrecarga → inflamação do endotélio glomerular → esclerose glomerular → GESF
[Angiotensina II promove vasoconstrição arterial, logo o tratamento de escolha para estes casos são IECA]
Nefropatia diabética
SÍNDROME NEFRÍTICA
Clinica → Quadro agudo de:
Hematúria glomerular [obrigatório]
Proteinúria até 3,5g/dia → se ultrapassa esse valor denomina-se Síndrome Nefrítica Nefrótica
Edema → não relacionado à proteinúria pois não tme tempo de perder albumina suficiente para afetar pressão oncótica
HAS → relacionada à retenção volêmica
Oligúria [<400 ml/dia]
! Tratamento sempre envolverá diuréticos e dieta hiposalina !
Etiopatogenia → É uma inflamação que pode ser causada por:
Imunocomplexos: deposição de IC no glomerulo gerando inflamação.
! Quanto mais próxima da corrente sanguínea for a deposição de IC, maior será a quantidade de células de defesa reagindo e, consequentemente, maior será a reação inflamatória !
Glomerulonefrite pós infecciosa → é aguda e auto-limitada
Nefropatia por IgA → é intermitente e crônica
Nefrite lúpica
Anticorpos: doença anti-membrana basal glomerular → é rapidamente progressiva
Pauci-imune: há ausência de depósitos de Ig à imunofluorescência porém acredita-se ter, só não foi comprovado ainda
Vasculites ANCA e outras associada → é rapidamente progressiva
Exames Complementares
EAS: proteinúria não nefrótica + hematúria glomerular + leucocitúria com cultura negativa
Bioquímica: aumento de ureia e creatinina [azotemia]
Eletrólitos: hipercalemia
Gasometria: acidose metabólica
Complemento (C3, C4 e CH50 → total)
SISTEMA COMPLEMENTO BAIXO | SISTEMA COMPLEMENTO NORMAL |
Glomerulonefrite pós-estreptocócica | Vasculites sistêmicas |
Lúpus Eritematoso Sistêmico | Doença de Berger |
Criglobulinemia | Vasculite por IgA |
Glomerulonefrite proliferativa | Doença de Goodpasture → anticorpos anti-membrana basal glomerular |
Endocardite associada a shunt |
Exames complementares direcionados
Suspeita de Hep B. ou C. → dosagem de crioglobulinas séricas
Suspeita de LES → dosagem de FAN, Anti-DNA e Anti-Sm
Suspeita de Vasculite → dosagem de antiproteinase 3 (c ANCA) e antimieloperoxidase (p ANCA)
Suspeita de Doença de Goodpastura → dosagem de Ac Anti-MBG
Suspeita de Endocardite/Abcesso → hemocultura
Principais causas de síndrome nefrítica:
5.1. Glomerulonefrite Pós-Estreptocócica
→ Agente etiológico: Streptococo Beta-hemolítco do Grupo A, C ou G
Cepas nefritogênicas:
Orofaringe: 1, 4, 12 e 25
Pele: 2, 42, 48, 56 e 60
→ Fisiopatologia: depósito de IC no mesângio e subendotélio do glomérulo
→ Idade: pode ocorrer em qualquer idade mas pico é aos 7 anos, o prognóstico é pior em adultos com alta chance de evoluir para DRC
→ Sexo: cepas orofaríngeas predominam no homem e da pele não há diferença.
→ Clinica [ ! Não tem febre pois infecção já aconteceu ! ]:
Frequentemente é subclínico mas pode apresentar dor abdominal, náuseas, vômitos e dor lombar bilateral
Hematúria glomerular é obrigatória
70% tem HAS → é volume dependente e pode se apresentar em edema agudo de pulmão ou encefalopatia hieprtensiva
50% tem disfunção renal.
→ Diagnóstico:
Síndrome nefrítica aguda: 1-2 semanas após faringite ou 2-3 semanas após infecção de pele [demora mais em infecções cutâneos devido à menor vascularização] + Anticorpos: ASLO se faringite [não é confirmatório] ou Anti-DNase se cutâneo + Queda do complemento em até 8 semanas
! Complemento TEM que voltar a aumentar após 8 semanas, se não volta diagnóstico está ERRADO !
→ Diagnósticos diferenciais:
Glomerulonefrite por endocardite bacteriana
Nefrite lúpica
Glomerulonefrite membranoproliferativa
Vasculite por IgA
Nefropatia por IgA
→ Exames específicos:
Histologia: glomérulos aumentados com infiltração de mónictos e células polimorfonucleares
Imunoflurescência: dépositos de C3, IgG e IgM
Microscopia eletrônica: depósitos subendoteliais ao longo da membrana basal glomerulas (humps de camelo)
→ Indicações de biópsia renal:
Idade <2 ou >12 anos
Oligúria >1 semana
Hematúria macroscópica >3 semanas
Queda do complemento por >8 semanas
Proteinúria nefrótica >8 semanas
Proteinúria não-nefrótica >6 meses
Hematúria microscópica > 2 meses
Evidência clínica ou sorologica de doença sistêmica
Glomerulonefrite rapidamento progessiva (GNRP)
→ Tratamento
Dieta hiposódica
Restrição hídrica (<2000ml/dia)
Diurético: Furosemia → utiliza-se diurético de alça pois é o único capaz de eliminar sódio melhor
IECA/BRA tem risco de hipercalemia e consequente piora da função renal
Vasodilatores: bloqueadores de canal de cálcio
Antibióticos: apenas se evidência de processo infeccioso como:
S. epidermidis / S. aureus ativos durante glomerulonefrite
Encocardite
Catater venoso central
Nefrite Shunt
! Imunossupressores não estão indicados !
5.2. Glomerulonefrite Rapidamente Progressiva
→ Conceito: é uma síndrome nefrítica, independentemente da causa, que evolui para insuficiência renal de maneira acelerada e fulminante
→ Possíveis causas:
Glomerulonefrites primárias
Glomerulonefrites infecciosas/pós infecciosa
Doenças glomerulares multissistêmicas
Doenças glomerulares medicamentosas → alopurinol, hidralazina, rifampicina
→ Tratamento: demanda imunossupressão por pulsoterapia (altas doses em pouco tempo) com corticoides e imunossupressor
→ Prognóstico: sobrevida de 80% em 1 ano com apenas 40% retormando função renal
SÍNDROME NEFRÓTICA
Clínica → quadro insidioso com:
Proteinúria >3.5g/dia → paciente pode ter albumina extremamente baixa e não conseguir passar este valor. Logo não é obrigatório
Edema → anasarca
Hipoalbumenimia <3,5g
Hiperlipidemia → tentativa de compensação hepática da hipoalbunemia hiperativa vias lipoproteicas gera aumento de LDL + perda da lipase (hipoproteinemia) gera diminuição do catabolismo do VLDL e aumenta TG.
Lipidúria → corpos graxos ovalados
Etiologia
< 10 anos → 90% por doença de lesões mínimas
10-50 anos → 30% por GESF
>50 anos → 50% por nefropatia membranosa
Avaliação inicial
EAS
Sangue → perfil lipídico, creatinina, ureia, hemograma
Pesquisa da causa:
Glicemia
FAN
Complemento → reduzido pode indicar GNPE, endocardite, abcesso visceral, nefrites pós shunts, LES, crioglubulinemia ou glomerulopatia membranoproliferativa idiopática
Crioglubulinas
Sorologia → Anti-HCV, Anti-HBsAg, Anti-HBc IgM e IgG, Anti-HIV
Anticorpos → ANCA, Anti-MBG
Biópsia renal → indicada apenas quando todas causas secundárias já foram excluídas
Eletroforese de proteínas
Alfa-2 e betaglobulinas aumentadas indicam que hipoalbunemia é crônica
Gamaglobulina normal ou aumentada → síndrome nefrótica secundária à infecções, hepatopatias crônicas ou doença autoimunes
Hipogamaglobulinemia → doença por lesões mínimas, GESF, mieloma múltiplo. [há discussões de que DLM é GESF que irá se desenvolver]
Causas secundárias
Doenças sistêmicas → DM, amiloidose.
vasculites sistêmicas (Wegener, poliangeiíte microscópica)
doenças imunológicas sistêmicas: LES, Henoch-Schonlei, Goodpasture
Infecções → Sífilis, Hep. B, Hep. C, HIV, CMV, Malária, Filaria, Esquistossomose, Strep (raro)
Intoxicação
Drogas → Heroína
Medicamentosa → AINEs, Lítio, Captopril, Clorpromazina, Penicilamina, Rifampicina
Metais Pesados → Ouro, Mercúrio
Neoplasias
Hematológicas estão associadas à doença de lesões mínimas (leucemia, linfoma)
Orgãos sólidos estão relacionados à nefropatia membranosa
Hereditários → Anemia falciforme, síndrme de Alport, síndrome nefrótica familiar
Outros → Nefroesclerose hipertensiva acelerada, pré-eclampsia
Consequência da hipoproteinemia
Albumina → edema
Antitrombina III → estado de hipercoagulabilidade
Globina de ligação de tiroxina → alteração de função tireoideana
Proteína de lifação do colecalciferol → hiperparatireoidismo secundário
Transferrina → anemia hipo micro com resistência à reposição de ferro
Imunoglobulinas → imunossupressão
TROMBOSE DE VEIA RENAL
→ Suspeita-se na presença de varicocele, hematúria macroscópica, mudança no padrão de proteinúria, redução inexplicável do débito urinário, assimetria de tamanho e/ou função renal
→ Causas: glomerulonefrite membranosa (principal), glomerulonefrite membranoproliferativa, amiloidose
DOENÇA POR LESÕES MÍNIMAS
→ Doença de Nil ou nefrose lipoide
→ Maioria é idiopática, estudos sobre ser precursora de GESF
→ Epidemio:
80% dos casos são em crianças, meninos 2:1, com pico aos 4 anos.
20% são em adultos/idosos sem diferença entre sexo com pico aos 65 anos
→ Clínica:
Proteinúria nefrótica
Ausência de cilindros hemáticos ou dismorfismo eritrocitário
Hematúria em 30% doas casos, não altera prognóstico
HAS e 30-40% dos casos
Maioria mantém função renal preservada
→ Associações:
Medicamentosa → AINEs (principal), alfa-interferona, Lítio, ampicilina, rifampicina
Metais pesados → Ouro, Chumbo, Mercúrio
Hipersensibilidade → Alergias, Picadas de inseto, Imunizações
Doenças Malignas → linfomas (Hodgkin e não Hodgkin), tumores sólidos
Infecções → mononucleose, HIV
→ Evolução: segue com remissões e recidivas
Adultos tem pior prognóstico e apenas <5% das crianças cursam com DRC avançada
→ Tratamento:
Corticoidoterapia → se resistente associar ciclofosfamida
IECA ou BRA para tratamento HAS
→ Indicação de biópsia renal:
Hematúria macroscópica
Complemento baixo
<1 ano ou >8 anos
Resistência à corticoide
GLOMERULOESCLEROSE SEGMENTAR FOCAL
→ É a principal causa de síndrome nefrótica em adultos: 25% dos brancos e 50% dos negros.
→ Lesão focal: esclesore e colapso capilar em uma parte dos glomérulos
→ Lesão segmentar: alças capilares dos glomérulos
→ Fatores de risco: HAS, DM, Obesidade, Anemia falciforme, Nefrectomia subtotal, disgenesia renal unilateral, doença cardíaca cianóticas congênita, refluxo vesicoureteral.
[FRs podem causar tanto hipertrofia glomerular quanto perda de néfrons, ambos podem evoluir para GESF]
Outros: lítio, heroína, pamonidrato e infecção por parvovírus B19
GESF Primária
Pico entre 25-35 anos
Homens 2:1
Ocorre lesão dos podócitos por distúrbio dos linfócitos T, imunofluorescência mostra depósitos de IgM e C3
Clinica:
síndrome nefrótica
queda da função renal (25-50% dos casos)
Hematúria em 50% doas casos
Perda definitiva da função renal em 10 anos (25-55% doas casos)
Recorrência em 30% dos casos
Tratamento: prednisona, ciclofosfamida, micofelonato
GESF Secundária à:
Sequela de:
Nefrite lúpica
Vasculite
Berger
Doenças inflamatórias sistêmicas
Sobrecarga por:
Nefropatia por refluxo
nefroesclerose hipertensiva
Nefrectomia parcial >75%
Anemia falciforme
Hiperfluxo por:
Nefropatia diabética
Obesidade mórbida
Anemia falciforme
NEFROPATIA MEMBRANOSA
→ Epidemo: homens brancos >40 anos
→ 75% são primárias
→ Fisiopatologia: espessamento de membrana basal com depósito de IgG e C3 e formação de IC in situ.
→ Causas secundárias:
LES
Hepatite B e C
Sífilis
Malária
Neoplasia de pulmão e TGI
Medicamento: captopril, sais de ouro, penicilamina
→ Clínica
80% tem síndrome nefrótica clássica
70% mantém função renal e PA preservadas
→ Evolução
Doença renal terminal em 10-15 anos
Remissão espontânea
Proteinúria com função renal preservada ou em declínio lento
→ Tratamento
Se proteínuria <4:
PA ideal <130/80 → uso de IECA/BRA se necessário
exame proteinúria e função renal de 3 em 3 meses
Se proteinúria 4-6
IECA/BRA
se em 3-6 meses não houver remissão, iniciar imunossupressão
Se proteinúria >6
IECA/BRA
imunossupressão imediata
DOENÇA RENAL DIABÉTICA
CONCEITO → complicação crônica da Diabetes Mellitus que acomete cerca de 35% dos portadores de DM.
EPIDEMIO → Ao longo dos anos, os desfechos renais na DM estão diminuindo, porém a prevalência de DM está aumentando.
RASTREIO
DM1 → Inicia rastreio renal após 5 anos de doença
DM2 → inicia rastreio renal ao diagnóstico
FATORES DE RISCO
Hiperglicemia sustentada
HAS
Obesidade
Albuminúria
Ativação do SRAA
Alta ingesta proteica
Tabagismo
Genética
Inflamação
Stress oxidativo
DIAGNÓSTICO → É o mesmo que o diagnóstico para DRC, utiliza Taxa de Filtração Glomerular baixa e Relação Albumina/Creatinina alta.
TFG → <60
RAC → >30 [É indicado fazer o primeiro rastreio com amostra de urina isolada e em caso de valores alterados, confirmas com exame de 12 ou 24 horas]
DIAGNÓSTICO DIFERENCIAL
→ História do paciente deve conter:
Passado de controle inadequado da DM
Sinal de lesão de outros orgão-alvo da DM
Ausência de outros sintomas e sinais que indiquem doença de etiologia renal
→ Deve-se excluir doenças sistêmicas como Hep. B, C e HIV
→ Presença de hematúria é forte indicativo de doença renal auto-imune ou imuno-mediada.
CLASSIFICAÇÃO PATOLÓGICA → classificação é histopatológica, logo demanda biópsia.
Classe I (Silencioso) → Aumento da MBG.
Não tem albuminúria
Aumento na PA diastólica discreto por alteração no descenso noturno
Aumento na TFG
Classe II → Expansão mesangial [albuminúria frequente]
IIa (Silenciosa) → Expansão mesangial <25%
Ocorre microalbuminúria apenas em estado hiperglicêmico
IIb (Incipiente: é o início da doença renal diabética evidente)→ Expansão mesangial >25%
Microalbuminúria em níveis crescentes
HAS
Classe III (Evidente)→ Esclerose mensagial: a glomeruloesclerose pode ser difusa (mais comum) ou nodular (Lesão de Kimmestiel-Wilson). Para a classificação de Classe III é necessário mínimo de 1 nódulo de KM.
Declinio progressivo da TFG (ocorre queda acentuada quando comorbidades como a HAS não são tratadas)
Proteinúria >0,5
Classe IV (DRC terminal)→ Esclerose mesangial >50% dos glomérulos + Fibrose intersticial + atrofia tubular
TFG <15%
→ Classes I e II são lesões revesíveis.
→ Sem controle adequado, há média de 5 anos para progredir de classe.
PATOGÊNESE [principal responsável é a hiperglicemia sustentada]
Fatores Hemodinâmicos
Hiperglicemia → liberação de mediadores vasoativos [IGF-I; Óxido Nítrico; VEGF (fatores de crescimento vascular derivado do endotélio) e glucagon] → Vasodilatação de arteríola aferente → Hiperfiltração Glomerular → Hiperperfusão renal → Hipertersão Intracapilar Glomerular + alterações na autorregulação glomerular → Podem gerar dano vascular permanente
Hiperfiltração Glomerular + Angiotensina II → Aumento da reabsorção de sódio tubular [explica benefício de bloqueio de SRAA]
Hiperglicemia → Fatores de crescimento → Hipertrofia celular + aumento na quantidade de matriz extracelular (em primeiro momento) → diminuição de células de matriz + progressiva expansão de seu material (em segundo momento) → Formação de nódulos
Fatores Metabólicos
Hiperglicemia → estímulo de oxidação de glicose no clico do ácido tricarboxílico → gera radicais superóxido no nível mitocondrial → ativa 4 vias:
Via dos polióis → acúmulo de sorbitol
Via das hexosaminas
Via da PCK → induz albuminúria
Produção de AGE (produtos finais da glicosilação avançada) → induzem crescimento celular, inflamação, angiongênese e produção de MBG
As vias dos polióis, hexosaminas e PCK induzem stress oxidativo, o que gera fibrose tecidual e disfunção vascular.
Fatores de Crescimento
Hiperglicemia sinaliza intra-celularmente:
Aumento das moléculos de adesão → ICAM e VCAM
Quimiocinas → CCL2 e CCL5
Citocinas → TNF-alfa, IL-6 e IL-8
Fatores de crescimento → VEFG e PDGF
VEFG → Proliferação vascular + aumento da permeabilidade endotelial = retinopatia e nefropatia diabética
Fatores Pró-inflamatórios e Pró-fibróticos
Quimiocinas → CCL2 e CCL5
Citocinas → TNF-alfa, IL-6 e IL-8
→ Recrutamento de leucócitos causa:
Podocitopenia
Glomeruloesclerose
Atrofia tubular
Fibrose Intertiscial
SCREENING
→ Exames: albuminúria em exame de urina em amostra única + creatinina para cálculo de TFG em amostra sanguínea
→ Temporalidade:
DM1 : 5 anos após o diagnóstico
DM2: imediatamente após diagnóstico
MONITORIZAÇÃO DO TRATAMENTO
Controle Glicêmico → alta dificuldade de manejo da glicêmia em nefropatas diabéticos devido aos fatores de risco para hipoglicemia:
Menor degradação insulínica
Uremia causando anorexia
Menor gliconeogênese renal
→ O monitoramente é feito pela HbA1c e as metas de HbA1c são individualizadas, no geral seguem:
7%: pacientes menores que 50 anos sem comorbidades
8%: pacientes mais velhos ou com comorbidades
→ O controle intensivo diminui:
risco de micro e macroalbuminúria
prevalência de complicações vasculares
[Não há evidências que controle melhore desfechos renais em pacientes com DRC estabelecida nem reverta alterações causadas pela hiperglicemia previamente → Memória Metabólica/Efeito Legado]
TRATAMENTO NÃO-FARMACOLÓGICO
É a base de todo o tratamento de nefropatia diabética e normalmente consiste em:
dietas com restrição de sal
prática de atividades físicas
perda de massa corporal
cessação de tabagismo
TRATAMENTO FARMACOLÓGICO
Hipoglicemiantes → serão escolhidos de acordo com meta glicêmica, efeitos adversos e conveniêncai para o paciente.
Metformina: se TFG <45, dose máxima é de 1g/dia; se TFG <30 é contra-indicada.
Sulfoniureias: alto risco de hipoglicemias
Inibidores de alfaglucosidase: contra-indicado se creatinina <2 ou DRC estágo III ou IV
Glitazonas: a piorglitazona pode ser empragas em qualquer fase da DRC, seu único problema é a retenção hídrica que pode exacerbar uma IC.
A-GLP-1: recomendados com ajuste de dose
IDPP4: recomendados com ajuste de dose
ISGLT2: extremamente recomendados, única questão é a falta de estudos da sua associação com bloqueadores de SRAA, que são ponto-base do tratamento
Insulinas: recomendadas na ausência de resposta a hipoglicemiantes orais
Bloqueadores do Sistema Renina-Angiotensina-Aldosterona (SRAA)
IECA ou Losartana são reocmendados para o traamento da nefropatia diabética em qualuqer estágio
Devem ser usados para a prevenção da nefropatia diabética em pacientes diabéticos hpertensos.
Bloqueadores de aldosterona
A espironolactona pode reduzir proteinúria e retardar a progressão da doença independentemente do bloqueio de SRAA, porém, não existe até o momento recomendação formal para o uso de medicamentos bloqueadores de aldosterona com essa finalidade.
! Risco de hipopotassemia em paciente com TFG mais baixa !
Bloqueadores de canal de cálcio
Alguns estudos demonstraram efeito sobre proteinúria
DOENÇA RENAL CRÔNICA*
_________________________________________________________________________
DOENÇA RENAL POLICÍSTICA AUTOSSÔMICA DOMINANTE (DRPAD)
(Não entedi por que ela está no slide então coloquei aqui em cima)
→ Nefropatia hereditária mais comum, relacionada ao gene PKD1 (mais jovens, aparecimento abrupto) ou PKD2.
→ Sintomas:
HAS
Hematúria
Proteinúria
Disfunção renal
Cólica renal
Nefrolitíase → 20-30% dos casos
→ É uma doença síndromica: possibilidade de cistos hepáticos e/ou pancreáticos
Cistps hepáticos são mais comuns e não levam à insuficiência hepática mas podem fazer efeito de massar, levando à compressão gástrica o que gera empachamento.
→ Principais complicações
Aneurisma cerebrovascular (20%) → morte súbita aos 20-30 anos
Doença valvar cardíaca: prolapso da valva mitral leve e regurgitação aórtica → não têm repercussão hemodinâmica
Divertículos de cólon → aumentam a absroção de oxalato, o que aumenta as chances de cálculo renal
Cistos de vesícula seminal
→ Diagnóstico diferencial: Rim multicístico
Para diagnóstico correto de rim policístico é obrigatório estudo genético ou presença de cistos em outros orgãos + complicações
→ Critérios diagnósticos em exame de imagem:
15-39 anos: 3 ou mais cistos uni ou bilaterais
40-59 anos: 2 ou mais em cada rim → único critério de exclusão e <40 anos e <2 cistos
>60 anos: 4 ou mais cistos em cada rim
__________________________________________________________________
CONCEITO:
Funcional → TFG <60 por mínimo de 3 meses
Quando TFG<60 já existe repercussões clínicas:
Retenção de fósforo
Maior tendência a anemias
Maior tendência a cidose metabólica
Marcadores de dano renal alterado por ao menos 3 meses
Albuminúria >30mg/g
Anormalidades de sedimento urinário → cilindros hemáticos ou leucocitários
Hematúria glomerular → codócitos/acantócitos
Alterações ácido-base
Alterações eletrolíticas
Anormalidades histológicas → esclerose, atrofia, fibrose
Anormalidades estruturais (visualizadas em exame de imagem) → atrofia, rim em ferradura, displasia e agenesia
Transplante renal.
INSIDIOSIDADE
→ A DRC é extremamente insidiosa e a homeostase é mantida até as fases mais avançadas, isso pode ser explicado por:
A capacidade funcional renal é vastamente superior ao mínimo necessário para alguém viver. Seres humanos conseguem viver com 10% ou menos da função renal normal.
Néfrons tem alta capacidade de se adpatar à injúria renal crônica, multiplicando o ritmo de trabalho em várias vezes
FATORES DE RISCO
Doenças cardíacas → HAS e cardiopatias em geral
Doenças metabólicas → DM e obesidade
Fatores exógenos → toxinas, poluição, mudança climática
Fatores sociais → pobreza, pouco letramento em saúde, moradia em local remoto
Infecções → HIV, TBC
Doenças auto-imunes
Neoplasias
Anormalidade congênitas
PIG
→ Grupos de risco: hipertensos, diabéticos, cardiopatas, familiares nefropatadas crônicos e obesos. Demandam rastreio:
dosagem de creatinina → cálculo da TFG
dosagem de albuminúria em amostra única de urina
FISIOPATOLOGIA
Desequilíbrio Hidroeletrolítico
Hipervolemia → HAS gera retenção de água e sódio pelo SRAA (risco de intoxicação hídrica)
Hiponatremia → mesmo com a ativação crônica do SRAA, a perda da capacidade renal de excreção de água livre leva a diluição do sódio plasmático
Sintomatolgia: cefaleia, confusão mental, desorientação, náuseas.
Hipercalemia → Única excreção de potássio é feita pelos rins. No geral, néfrons conseguem manter níveis normais em aumentos séricos de potássio em até 30%. Porém se passado disso, consequências sérias como arritmias podem ocorrer.
Sintomatologia: dormência, fraqueza muscular, vômitos e bradicardia
Conduta:
Gluconato de sódio + solução injetável de glicose 5% → cálcio antagoniza efeitos do potássio na membrana e a sgi 5% é compátivel para solubilização [dura 3 min]
Insulina + glicose → inuslina carrea potássio intracelularmente e glicose evita hipoglicemia
Diuréticos (furosemida)
Resinas de troca?
Diálise
Hiperfosfatemia → A absorção de fosfato não depende muito da Vit. D, logo não é muito afetada pela DRC, enquanto sua excreção é quase proporcional à TFG, que pode estar muito diminuida na DRC, levando à retenção de fosfato
Processo fisiológico ao aumento do fósforo: aumento de PTH → bloqueio de canais de fósforo nos túbulos proximais → diminuição da reabsorção → fosfatúria [porém na DRC não é suficiente para manter níveis séricos normais]
Sintomatologia: prurido e calcificação metástatica
Favorece complicações como calcificações extraósseas e doenças cardiovascular
Hipocalcemia: rins sintetizam calcitriol (froma ativa de Vit. D), e a diminuição leva a diminuição drástica de absorção de cálcio no intestino. Este quado leva a um hiperparatireodismo secundário como tentativa compensação, conseguindo manter níveis de cálcio pouco abaixo do normal ou até normais em alguns casos, porém custando reservatório ósseo.
Queda de cálcio → aumento crônico do PTH → desmineralização óssea
Sintomatologia: cãibras, formigamentos e cólica abdominal
Hipermagnesemia (>4,5): a diminuição gradual da TFG leva a diinuição da excreção de magnésio e consequente acúmulo sérico.
Sintomatologia: ausência de reflexo tendinosos (Mg inibe liberação de Ach nas terminações nervosas e reduz excitabilidade de membranas), frequeza muscular e respiração lenta
Acidose Metabólica
→ Incapacidade renal de excretar ácidos + distúrbio de produção renal de bicarbonato e reabsorção dele = acidose metabólica
Consequências:
Aumento da reabsoção óssea (cálcio e fosfato são alcalinos) → osteopenia/osteoporose
Aumento do catabolismo proteico [acidose → ativação da via ubiquitina-proteassoma → liberação de aminoácido musculares como a glutamina → utilização para formar bicabornato] → diminuição da massa muscular
Insuficiência cardíaca [acidose → distúrbio na entrada e liberação de cálcio intracelular no miocárdio → dificuldade de contração cardíaca → inotrópio negativo → ICC]
Aumenta resistência a insulina [acidose gera disfunções na via da insulina e GLUT-4] → intolerância a carboidratos
Aumenta resistência ao GH [diminui expressão dos receptores] → distúrbios do crescimento
→ Tratamento é a reposição do bicarbonato a partir de cálculo do déficit.
Distúrbios da Mineralização Óssea
Terminologia: DMO-DRC → Distúrbios Minerais e Ósseos da DRC
Fisiopatologia: TFG <60 → diminui produção de calcitriol e excreção de fósforo → aumento do PTH:
Fosfatúrico
Aumento da reabsorção óssea
Formação renal de calcitriol → aumento da abrsorção intestinal de cálcio
Aumento da reabsorção de cálcio em néfrons distais
As osteodistrofias renais (termo restrito para achdos histológicos) pode ser dividas em dois grupos: Doenças de Alto Remodelamento Ósseo (Alto Turnover) e Baixo Remodelamento Ósseo (Baixo Turnover)
[Remodelação = Absorção (osteoclastos → dura de 30-40 dias) + Formação (osteoblastos → dura aproximadamente 150 dias)]
Anemia
Alterações da Homeostasia
Alterações Endócrino-metabólicas
CLÍNICA
Estágio 1 e 2 → desordem de sono, edema de MMII ou face
Estágio 3 → Ansiedade, depressão, piora do edema, constipação
Estágio 4 → Sobrecarga de volume, prurido, câimbras, disfunção cognitivas, náuseas e vômitos, pernas inquietas
Estágio 5 → Síndrome urêmica
→ Síndrome urêmica: não possui sintomatologia específica mas necessariamente deve ter TFG baixa (VR é <15 com exceções)
Ureia é depressora de SNC logo podem haver sintomas psiquiátricos
Sintomas cardiovasculares
Sintomas TGI
Anorexia
Disfunção sexual
Sangramentos
Aumento do prurido
DIAGNÓSTICO
USG
TRATAMENTO
→ O tratamento da DRC consiste em reduzir o declínio da TFG, como?
Tratando a doença de base, seja ela metabólica ou auto-imune
Reduzindo o s níveis de hipertensão glomerulas
Reduzindo os níveis das moléculas pór inflamatórias (citocinas e quimiocinas)
→ Terapia de substiuição renal: hemodialiase, dialiase peritoneal e transfusão renal
diálise:
indicada para diabéticos com TFG <14
indicada para não diabeticos com TFG <12
indicada para qualquer DRC com quadro de: edema agudo de pulmão, hiperpotassemia e acidose metabólica que não possam ser controlados de outra maneira
No geral, a dialise peritoneal será indicada para pacientes que não possuem acesso vascualr adequado
Transplante renal: uma TFG <20 já indica o transplante
Não cura causa base! Poderão haver recorrências:
DM2: 100%
Berger: 20-50%
GESF: 30-40%
GNM: 5-20%
LES:2-10%
INTRODUÇÃO À REUMATOLOGIA
DOR
→ Duração:
Aguda: <4 semanas
Subaguda:4-6 semanas
Crônica: >6 semanas
→ Evolução-
Aditiva: soma
Migratória: resolve-se e migra para outro local
→ Peridiocidade
Remitente: períodos de exacerbação e período de dor leve
Intermitente: crises com períodos sem dor
Constante
→ Padrão/Ritmo
Inflamatório:
piora em repouso → associada a despertar noturno
dor noturna e na manhã
alivia com movimentação
rigidez marinal >30 minutos
Ex: LES, AR, EA
Mecânico:
doi ao movimento
pior no final da tarde
alivia com repouso
rigidez <30 minutos
EX: OA
FIBROMIALGIA
DEFINIÇÃO
→ É uma síndrome de dor musculo-esquelética crônica, difusa e não-inflamatória.
EPIDEMIOLOGIA
→ É a principal causa de dor musculo-esquelética generalizada.
50-60 anos
Mulheres 3:1
Comum associação à outras doenças reumatológicas
PATOGÊNESE
A dor da fibromialgia é do tipo nociplástica, ou seja, ocorre um aumento da sensibilidade do sistema nervoso e os receptores nociceptivos são estimulados de dor desproporcional ao estímulo ou sem causa evidente.
Esta dor possui duas bases patogênicas:
Resposta anormal e inadequada do SNC à estimulo perifértico → hiperexcitabilidade neuronal → amplificação da dor
Reestruturação do sistema nociceptivo → perpetuação da dor, mesmo sem estímulo periférico
ETIOLOGIA
[É uma condição multifatorial]
→ Mecanismo periférico:
Estímulos periféricos dolorosos inciam ou aumentam o processo nociplástico → alterações dos nervos periféricos (disfunção das pequenas fibras)
→ Mecanismos centrais:
Aumento da substância P no LCR
Redução dos receptores opiodes
Atividade doparminérgica reduzida durante o estímulo doloroso
Aumento de NT excitatórios (glutamato)
Redução dos níveis da GABA
Fenômeno wind-up [forma de plasticidade de curta duração que ocorre no corno dorsal da medula como aumento do número de potenciais de ação iniciados por um estímulo]
→ Disfunções do SNA
Hiper ou hipoativação do SNA
Redução dos níveis de catecolaminas
Níveis séricos de cortisol aumentados
→ Genética:
Parentes de 1° grau tem 8,5x mais chance.
Existem genes e polimorfismo que envolvem o metabolismo e transporte relacionado à neuromodulação da dor
→ Infecções virais podem desencadear o processo da doença:
Hepatite C
HIV
Covid-19
Parvovírus B19
→ Sensibilização cognitivo-emocional: ativação em áreas do cerébros relacionadas à dor.
Embotamos da respotas ao stress
Níveis reduzidos de estratégias de entrentamento
Catastrofização
→ Possível correlação imune:detecção de subtâncias e citocinas pró-inflamatórias
CLÍNICA
Sintomas cardinais:
Dor musculoesqueléticas generalizada (multissítio)
Pode ser em queimação, peso ou parestesia
Altralgia de duração <30 minutos
Alodinia (redução do limiar doloroso → não demanda estímulo)
Hiperalgesia (respotas aumentada a estímulos dolorosos)
Sensação de inchaço
Fadiga mental e física
Sensação de cansaço sem justificativa, pode ter diferentes graus.
Distúrbios do sono
Insônia
Despertares frequentes
Sono não-reparador
Outros sintomas:
Psiquiátricos:
Ansiedade
Depressão
PTSD
TAB
Ideação suicida
Sindrome das pernas inquietas
Disfunção cognitiva [Fibrofog”]:
Hipotenacidade
Perda de memória
Hipovigilância
Hipersensibilidades
À extímulso externos
Fotossensibilidade
À sons
À lugares cheios
Síndromes dolorosas regionais:
Dispepsia
Síndrome do intestino irritável
Dismenorreia
Vulvodínea → dor crônica na vulva
Dor pélvoca/vaginal
Cefaleia
Dor na ATM
Distúrbios autonômicos:
Xeroftalmia
Fenômenos de Raynauld
Hipotensão ortostática
Variação na FC
Perda auditiva
Outros: urgência miccional e turvação visual
EXAME FÍSICO
No geral, paciente é BEG sem alterações em exame articular com sensibilidade leve/moderada em locais de tecido mole na maioria da s vezes, principalmente nos tender points
Tender Points → Occipito, Trapézio, Cervical Inferior, Super-espinhoso, 2a costela, Epicôndio lateral, Glúteo, Trocanter e Joelho
CRITÉRIOS DIAGNÓSTICOS:
Dor generalizada há pelo menos 3 meses sem explicação: avalia 9 regiões e índice de dor
Gravidades dos sintomas (0-3) na última semana: fadiga, sintomas cognitivos, sono nao-reparador
Últimos 6 meses: presença de dor ou cãibra na parte infeiror do abdome, depressão, cefaleai
→ Dor generalizada: dor me pelo menos 4 das 5:
Região superior esquerda
Região superior direita
Região inferior esquerda
Região inferior direita
Região axial
DIAGNÓSTICO → >/= 6 locais de dor + distúrbios do sono moderados a grave ou fadiga + sintomas há pelo menos 3 meses
! A presença de outro distúrbio de dor ou sintomas relacionados não exclui o diagnostico !
INVESTIGAÇÃO
→ Não existe um exame que confirme o diagnóstico porém são importantes para descartar diagnósticos diferenciais
Laboratorial:
Hemograma
PCR
VHS
TSH
T4 livre
HBsAg
HIV
Auto-anticorpos apenas se alta suspeita de doença auto-imune
Imagem → São desnecessárioa pelo alto risco de falsos positivos, resultado em tratamento desnecessário
TRATAMENTO
→ Tem como base 4 pilares:
Educação do paciente: conhecimento sobre a doença e autogestão
Atividade Física: 30 minutos no mínimo de aeróbico 3x por semana
Pode ter piora no inpicio porém atividade tem alto nível de evidência
Psicoterapia: TCC e estrategias de enfrentamento
Farmacologia: decorre de decisão comportilhada e fatores como comorbidades, efeitos adversos, acesso/custo e sintomas predominante devem ser levados em conta
ADT (IRSN) → Amitriptilina ou nortriptilina
Atuam na dor, sono, fadiga e depressão
Tme bom custo-benefício porém tolerância é baixa.
Relaxantes musculares de ação central → ciclobenzapina
É um tipo de agente triciclíco que atua na dor e no sono
Duais (IRSN) → Duloxetina ou venlafaxina
Atuam na dor, fadiga e alterações do humor
Anticonvulsivantes Moduladores do Canal de cálcio Alfa-2-ligantes → Pregabalina e gabapentina
atuam na alodinia, hiperalgesia, sono e fadiga
IRSR → Fluoxetina
Atua na dor e humor
[ ! AINEs e Corticoides são ineficazes ! ]
→ Em caso de refrateriedade (mínimo de 1-3 meses de tratamento) deve-se rever a desão e então mudar a conduta:
Mudar mecanismo de ação
Associar: IDT + ISRS ou Dual + Anticonvulsivante
Clínica da dor
Tramadol (opióide)
Tratamentos alterantivos → acunpuntura, ioga
Neuromodulação cerebral
Cannabis
PROGNÓSTICO
O prognóstico é afetado por fatores psicossociais e demográficos, a maioria dos paciente vem de maneir normal porém em alguns a dor e a fadiga pode persistir e afetar a capcidade de trabalho
Fatores de risco para pior prognóstico: mulheres, depressão, desemprego, obesidade e baixo nível econômico.
OSTEOARTRITE
CONCEITO
→ Osteoartrite = osteoartrose = artrose
→ É uma doença degenerativa e inflamatória das articulações sinoviais, principalmente por insuficiência cartilaginosa e comprometimento do osso subcondral. Porém, acomete todas as estruturas da articulação.
EPIDEMIOLOGIA
É a doença reumatológica mais comum e uma causa importante de incapacidade e morbiadde.
A incidência aumenta com a idade e a prevalência aumenta conforme envelhecimento populacional e aumento de taxas de obesidade
É mais comum em mulheres e local mais comum de acometer são os joelhos
FATORES DE RISCO
Fatores locais → fatores que influenciam como a carga é distribuida pela articulação
Joelho valgo ou varo
formato e congruência do quadril
lesão ou cirurgia prévia em aritculação
fraqueza muscular
desigualdade no comprimento das pernas
Fatores sistêmicos
Idade avançada
sexo feminino
susceptibilidade geneética
Doenças sistêmicas: obesidade, artropatia inflamatória, doença de paget, doença de wilson, osteonecrose, hemocromatose, artropatia por cristais, hemofilia, hiperparatireoidismo, amiliodose
FISIOPATOLOGIA
Os fatores de risco levam a ativação de vias:
vias das citocinas pró-inflamatórias
via da sinalização WNT-Beta Caternina
Via da sinalização de fatores de crescimento de fibroblastos
Via do óxido nítrico
A ativação das vias leva a:
ativação e hipertrofia dos condrócitos com desregulação anabólica e catabólica
Produção da proteasses
Inflamação sinovial
Apoptose de condrócitos
Os possívels resultados são:
Ruptura da cartilgaem rticular
Hiperplasia sinovial
Degeneração meniscal
Osteófitos em margem mensal
Fibrose capsular
Esclerose e remodelação óssea subcondral
CLÍNICA
→ Início é insidioso (anos a décadas) e pode ser mono, oligo ou poliarticular.
→ A dor tem ritmo mecânico e protocinético
→ Riidez costuma durar <30 minutos e é desencadeada por inatividade longa
→ ! ausência de sintomas sistêmicos !
→ Locais típicos: 1a CMC [base do polegar], IFDS, IFPS, joelhos, pés, coluna lombar e ccervical, coxofmorais.
Osteoartrite generalizada → acometimento de mão, pés, joelhos com ou sem quadril
→ Locais atípicos: Punhos, MCF, omboas, tornozelos e cotovelos
! Nestes casos, pensar em causas sistêmicas !
→ O exames físico no geral irá procurar por: aumento do volume articular, defornimidade, artrite, desvios, desalinhamento, redução, de amplitude, crepitações e instabilidade
Gonartrose [Joelhos]
Procurar por desalinhamento em varo (desalinhamento femorotibial medial) ou em valgo (compartimento femorotibial lateral)
Coxoartrose (quadril)
Procurar por dor em região da virilha ou emparte anterior do quadril que pode irradiar para região medial ou anterior da coxa, dor aumenta em rotação ou flexão do quadril
A perda mais precoce é da rotação interna e evolução leva a posição em flexão, rotação externa e leve adução com marcha antálgica
Mãos
Rizartrose → é a osteoartrite na 1a CMC
Clínica é de crepitações frequentes e perda imporante de funcionalidade
Osteófito de IFD → nódulos de Heberden
Osteófito de IFPs → Nódulos de Bouchard
Em <10% dos casos a osteoartrite da mão é erosiva, normla é nodal
Pés
Local típico é a 1a MTF e a deformidade típica é hálux valgo (joanete)
Evolução leva a háluz rígido
Coluna
Tipos: uncoartrose, atrose facetária /interpofisária, discoartrose
Locais:
Carvica: C5-C6 e C7-C8
Lombar: L4-L5 e L5-S1
! osteófitos podem levar a compressão radicular !
EXAMES COMPLEMENTARES
Laboratorial: exclusão de diagnpstico diferencia ou investigação causa secundária
RX: Pode ter dissociação clínico-radiológica.
Achados: redução do espaço articular, esclerose óssea subcondral, osteófitos, cistos
Análise do liquido sinovial: Líquido sinovial não inflamatório (<2000
células)
Cuidado: cristais de pirosfosfato de cálcio são frequentemente encontrados em articulações com OA
TRATAMENTO
→ O objetivo do tratamento é otimizar a função, aliviar a dor e modificar o processo de dano articular
! Não existe medicação modificadora do curso da doença !
Não-farmacológicos: educação do paciente, atividade física, equipamento auxiliar, TCC, perda de peso, acunpuntura?, frio local em caso exacerbação e calor local tem benificio a curto prazo para mão e joelho
Farmacológicos
AINEs oral ou tópico
Corticoide intra-articular para joelho e quadril [indicado máximo 3-4 vezes/ano]
Duloxetina (IRSN) para generalizada
Capsaicina tópica para joelho → gera dessensibilização das fibras nervosas da pele
Suplementos nutricionais não são recomendados, exceto condroitina para mãos
analgésicos simples tem pouco beneficio
Acido hialuronico é controverso
Opiódes são pouco eficazes mas se precisar usar, optar pelo tramadol
PROGNÓSTICO
Aumentos da mortalidade por todas causa, principalmene DCV
Piora funcional progressiva
FR: intesidade da dor, IMC>25, sintomas bilaterais e depressão
GOTA
CONCEITO
→ Artrite inflamatória e destrutiva resultante da deposição de cristais de monourato de sódio.
[hiperssaturação de ácido úrico sanguíneo → cristais de monourato de sódio]
EPIDEMIOLOGIA
Prevalência: 2-7% da população, mais comum em negros e aborígenes
30-60 anos
Homens → estrogênio promove excreção de ácido úrico e “protege” mulheres, incidência aumenta no sexo feminino após menopausa.
HF positiva em 40% dos casos
FATORES DE RISCO → aumentam uricemia
Não modificáveis:
Homo sapiens → não possui uricase
Sexo
Idade
Etnia
Genética
Modificáveis:
Síndrome metabólica → 70% dos pacientes
DRC
DCV
SAOS [Síndrome da Apneia Obstrutiva do Sono]
Dieta → carne vermelha, álcool, frutos do mar, bebida açucaradas, frutas ricas em frutose
Medicamentos → AAS (>60mg), cliclosporina, diuréticos, ácido nicotínicos, betabloqueadores, BRA não losartana, IECA, Levodopa, QT citotóxica, tacrolimus
Doenças inflamatórias/proliferativas → pré-eclâpsia, psoríase, cetoacidose diabética, acidose lática, estado hipoxemico, DRC, distúrbios hemolíticos, Síndrome de Batter e Gitelman, Neoplasias (principalemnte hematológica/linfroproliferativa pois tem maior lise tumoral)
FATORES DE PROTEÇÃO [Promovem uricosúria]
→ Modificáveis:
Dieta: café, laticínios, cereja, óleos saudáveis, suplement de vitamina C
Medicamentos: Losartana, bloqueadores do canal de cálcio, fenofibrato, atorvastatina, sevelamer
ETIOPATOGENIA
Em homeostase, o ácido úrico sanguíneo origina-se 80% do metabolismo endógeno das purinas e o restante da alimentação. 1/3 é eliminado pelo intestino e 2/3 são eliminados pelos rins.
A gota ocorre por um estado de hiperuricemia crônico (>7,5) levando à formação de cristais.
Fatores que facilitam a formação de cristais: pH baixo, baixa temperatura, desidratação, presença de restos de colágeno, cartilagem, condroitina e hialuronato normalemnte liberados por osteoartrite ou trauma.
[Em 90% dos casos o aumento ocorre pode hipoexcreção (correlação direta com DRC) e 10% por hiperprodução.]
CLASSIFICAÇÃO
Primária/idiopática: hipeuricemia sem causa identificada (maioria)
Secundária: hiperuricemia pode ser explicada
ESTÁGIOS
Hiperuricemia Assintomática [ác. úrico >6,8]
Clínica assintomática mas exames de imagem evidenciam microtofos sinoviais e de tecido mole.
O risco de desenvolver gota aumenta proporcionalmente ao nível urêmico sendo um risco de 50% quando ác. úrico >10
NÃO HÁ NECESSIDADE DE SER TRATADA! (reforçar MEV)
Gota Aguda
Ocorre pela liberação aguda de mediadores inflamatórioa sendo as principais células os macrofágos e neutrófilo e as principais citocinas a IL-1B e IL-18
Clínica: dor intensa e inchaço que pode ser acompanhados de calor, eritema, redução de funcionalidade e movimento, frebe/calafrios e fadiga.
Costuma iniciar como mono ou oligoarticular migratória e evoluir para poliarticular em crises subsequentes [mulheres tendem a já iniciar poliarticulares]
Pico: 12-48h
Articulações acometidas:
1° metatarsofalangiana (podagra) → principal
Tarso
Joelho
Punhos
Dedos
→ Pode ter bursite olecraniana e tenossinovite/tendinite em aquiles.
As crises tendem a acontecer à noite:
Queda da temperatura corporal
Queda dos níveis de cortisol
Desidratação relativa
Acidose respiratória leve
Fatores desencadeantes de crises:
Libação alcóolica/alimentar
desidratação
trauma articular
sepse
medicamentos hiper ou hipouriceminates
As crises em estágio inicial desaparecem em 3-14 dias sem tratamento graça às NETSs (armadilhas extracelulares dos neutrófilos) e mediadores anti-inflamatórios
A subsequência de crises pode aumentar:
A gravidades delas
A frequencia cias
A duração delas
[Após 1 ano, 60% das pessoas tem nova crise e após 3 anos, 80%]
Propedêutica em caso de crise: Artrocentese → fundamental para descartar diagnósticos diferenciais, principalmente a artrite séptica
Período Intercrítico
É um intervalo de ausência completa de sintomas, normalmente dura de 6 a 2 ano mas pode chegar até 10 anos.
Tende a diminuir com o tempo
Hiperuricemia ainda está presente e a formação e a deposição de cristais pode continuar, assimc om uma inflamação subclínica contínua
Gota Avançada
A gota grave de longa duração sem tratamento pode levar a formação de tofos, aproximadamente após 10 anos de crises agudas contínuas, a à poliartrie crônica.
Tofos → cleção organizada de cristais de monourato de sódio rodeado por células inflamtória e tecido conjuntivo com aparência de nódulo subcutâeno drenantes ou semelhantes a giz.
Locais: articulações e dedos, bursa do olécrano, tendão de aquiles, pavilhão auricular, olhos e válvulas cardíacas
EXAMES COMPLEMENTARES
Exames Laboratoriais
Ácido úrico → 4 semanas após crise
Uricosúria
Função Renal (creatinina e ureia)
Função Hepática (albumina e bilirrubinas)
Provas inflamatórias (PCR, VHS)
Pesquisa de DM e DLP (GJ, HbA1c, CT e frações e TG)
Análise de líquido sinovial [padrão ouro]
visualização dos cristais → análise microscópiccom luz polarizada permite ver cristais com forte birrefringência negativa, em formato de agulha
celularidade (<2000) + viscosidade + turvação [pode ser semelhante à artrite séptica, são sinais de inflamação]
Exames de imagem
Rx simples → possível ver presença de tofo e erosão em saca bocado
USG articular → possívei ver depósito de cristais e sinal do duplo contorno
TC → Tc de dupla energia (DECT) evidendia tofos bem
RMN
TRATAMENTO
Tratamento Não-Farmacológico:
Mudanças de estilo de vida → diminui 10-20% dos níveis de ácido úrico
Perda de peso
Atividade físicad
Diminuição da ingesta de álcool e bebidas açucaradas
Diminuição da ingesta do EXCESSO de carne vermelha, miúdos, embutidos e frutos do mar
Tratamento da crise aguda → primeira linha: AINEs, corticoide e colchina.
(Não tem diferença de eficácia, escolha depende de carcaterísticas do paciente)
AINEs → naproxeno, ibuprofeno, diclofenaco, meloxicam, celecoxibe ou indometacina em dose plena por ao menos 3 dias: idealmente, início será nas primeiras 48h para melhor efetividade e após resolutação completa descontinuar em 2-3 dias.
Cuidados: HAS mal controlada, IR, IC, história ou risco de doença ulcerosa péptica.
Glicocorticoides → normalmente se utiliza predniso VO mas pode-se usar corticoide IM, metiprednisona EV ou intração intra-artciular.
Cuidados: HAS mal controlada, IC, DM ou suspeita de infecção
Colchicina → anti-inflamatório que inibe movimentação de leucócitos e fagocitose
No geral, mantém uso como profilaxia mas tem efeito terapêutico. Deve ser usado nas primeira 12-36h
Cuidados: toxicidade, DRC, idosos, disfunção hepática e interações medicamentosas
Outras opções em casos de refratariedade ou CI aos tratamentos de primeira linha (pouco disponíveis):
Anti-IL-1 → Anakira, Canaquinumabe
Corticotropina
Analgésico transdérmico alcalinizante
Profilaxia → o ínicio do tratamento hipouricemiante altera os níveis de ácido úrico rapidamente, o que pode desencadear um crise. Logo é importante que haja uma profilaxia prévia ao tratamento.
Colchina
Tratamento hipouricemiante
Indicações → 2 ou + crises/ano; DRC GIII; Urolitíase; Tofos; crises agudas graves difíceis; sinal de dano articular em Rx; 1 crise + ácido úrico >9
Primeira Linha → Alopurinol
Inibe xantina oxidase
Pode se rinicado junto a tratamento de crise e não deve ser suspenso em caso de uma.
Dose depende do ácido úrico alvo: <6 ou <5 se há presença de tofo.
Hipersensibilidade (SSJ) está relacionada ao alelo HLA-B*5801
Efeitos adversos: alterações de TGI, citopenias, Rash, hepatotoxicidade
Segunda Linha → Fenobuxostate; Uricosúricos
Indicados em caso de CI, toxicidade ou falha do alopurinol em máxima dose tolerada.
Uricosúricos → Probenecida; Benzbromarona (disponível no brasil)
inibem URAT 1
CI → TFG<30 ou história pregressa de litíase urinária
Terceira Linha → Pegloticase
É um imunobiológico que induz a conversão de ácido úrico em alantoína (mesmo processo da uricase)
Indicado em caso de atrite gotosa grave cronicamente incapacitante refrária à tratamento de primeira e segunda linha
Tratamentos complementares:
HAS → preferir losartana e/ou anlodipino e suspender hidroclorotiazida se possível.
Hipertrigliceridemia → preferir fenofibrato
LÚPUS ERITEMATOSO SISTÊMICO
EPIDEMIOLOGIA
Mais comum em muheres em idade fértil (relação com estrogeno)
CLÍNICA
40-80% tem manifestçoes sistêmicas: febre, fadiga, perda de peso e mialgia
70-80% tem manifestações cutaneo-mucosas
Manifestações cutaneo-mucosas não especificas: fenomeno de raynaud, vasculite uritcariforme [mais prolongada que a normal (>24H) e quando some deixa hipo ou hiperpigmentação], úlceras mucosas indolores (25%), alopecia não cicatricial (50%)
93% tem artite ou artralgia
5-25% tem fibromialgia
50-70% tem nefrite lúpica → pode acometer qualquer parte do rim mas mais comum é glomerular
proteinúria
cilindros celulares
hematúria glomerular
nefrite intersticial é menos comum
→ A classficação é pela biópsia renal, classes proliferativas são as mais graves [4 é a mais comum e 5 e a que mais se asoscia à síndrome nefrótica]
Das manifestações pulmonares a serosite é a mais importante
Das manifestações cardíacas o derrame pericárdico é o mais comum e a pericardite vem em seguida.
Em caso de endocardite, ela é asséptica (endocardite de Libman-Sacks)
Das manifestações psiquícas acomentimento é mais central
Psicose lúpica esta relacionada ao anti-P-ribossomal
Manifestações hematlógicas podem ser anemia por doença crônica ou ferropenica
Manifestações de TGI e oftalmológicas são menos comuns
LÚPUS CUTÂNEO-MUCOSO
→ É possível ter o lúpus cutâneo isolado, pode ser:agudo, subagudo ou crônico
Lúpus Cutâneo Agudo
Tem muito relação com a fotossensibilidade
Pode ser localizado (Asa de borboleta/rash malar) ou generalizado
Não deixa cicatriz
90% de asssociação com LES
Lúpus cutâneo subagudo
Pode despigmentar e gerar teleangiectasia
Tem forte assciação com Anti-Ro/SSA
Pode ter lesões anulares ou papuloescamas ( psoriaseforme)
Lúpus cutâneo crônico
Deixa cicatrizes
Tipos: discoide (muito eritematoso), profundus, perniose, liquenoide
EXAMES COMPLEMENTARES
Sangue: Hemograma, VHS,Anti-DNAds, complemento (C3, C4, CH50) [marcadores de atividade da doença] PCR, Creatinina, Albumina, Eletroforese de proteínas, Dosagem de Imunoglobulinas: IgG, IgM e IgA, lipidograma, glicose jejum, HbA1c
Reticulócitos, LDH, bilirrubinas, haptoglobina, coombs direto [pesquisa de anemia hemolítica auto-imune]
Urina: Urina rotina, Relação proteína/creatinina, Proteinúria 24h, Clearence de creatinina
Imagem: RX-T, TC, RNM SNC, ECOTT e ECG
Biópsia renal e pele se necessário
AUTO-ANTICORPOS
FAN
1:80 e acima são mais significartivo
Padrão Nuclear pontilhado fino denso é prevalente em hígidos
Padrão Nuclear homogêneo é específico de LES
Anti-DNA
É mais específico para LES
Serva para marcador de atividade e nefrite
FAN nuclear homogêneo
Anti-Sm
É mais específico para LES
FAN nuclear pontilhado grosso
Anti-histona
Relacoinado a LES induzido por drogas
Anti-Ro
Relacionado ao Les Neonatal [Anti-Ro atravessa a placentea pdoe causam LES neonatal, o principal sintoma é BAV]
Anti-fosfolípides
Tem relação com trombose e morbidade gestacional
São eles: anti-cardiolipina, anticoagulanete lúpico, anti-beta2-glicoproteína1
TRATAMENTO
Hidroxicloroquina
Todo paciente exceto se CI deve usar !
Avaliação oftalmológica inicial e anualmente após 5 anos
Não-farmacológica
Educação do paciente
Controle pressórico
Vacinação
Fotoproteção
AAS → se presença de anticorpos anti-fosfolípides
SÍNDROME DE SJOGREN
CONCEITO
Doença autoimune crônica caracterizada por infiltração linfocitária das glândulas salivare,s lacrimais e outras glândulas exógenas.
→ Pode ser primária ou associada a outras doenças reumáticas
EPIDEMIOLOGIA
Prevalência: 0,05-0,7%
Sexo: 10-13 mulheres para 1 homem
Idade: diagnóstico costuma ocorrer entre 40 e 60 anos
ETIOPATOGENIA
Fatores Genéticos:
Polimorfismos de nucleotídeo único [HLA-DQ e HLA-DR → DR2 e DR3] em:
Fator des transcrição do fator 5 regulatórioa de interferon → direciona a produção de IFN-alfa e citocina prói-inflamatórias
Ativador e transdutor de sinal da transcrição 4 (stat 4) → estimula a produção de IFN-gama e IL-17
Fatores Ambientais:
Infecções virais (CMV, EBV, HTLV) → aumento de interferon → indução de autoantígenos
Diminuição de hormônios sexuais (estrogênio e di-hidroepiandrosterona)
FISIOPATOLOGIA:
Genética + Gatilho Ambiental → ativação anormal do epitélio glândular [epitelite auto-imune] → recruta sistema imune → infiltração linfocítica das glândulas exócrinas → células epiteliais passam a produzir e expressar auto-antígenos → infiltrado inflamatório se intensifica e predomina linfócitos T, B e células dendríticas → Linfócitos B tornam-se hiperreativos → produção de autoanticorpos [Anti-RO/SSA e Anti-LA/SSB] → destruição progressiva das glândulas exócrinas pela inflamação e infiltração crônica.
SINTOMATOLOGIA
Sintomas Secos:
Xerostomia (boca seca), que pode ser percebida através de: halitose, perda dentária, cárie, candidíase, disfagia.
Xeroftalmia (olho seco), que pode ser percebido através de: prurido, hiperemia, ceratoconjuntivite, irritação.
Xerodermia (pele seca), que pode ser percebida através de: prurido, lesões
Xerotraqueia, que pode ser percebica como uma tosse seca
Secura vaginal que pode ser percebida através de: dispareunia, atrofia vaginal, candidíase
Sintomas constitucionais:
Fadiga
Sintomas Musculoesqueléticos:
Poli (artrite/artralgia) inflamatória não deformante
Miosite
Mialgia
Sintomas Glandulares
Aumento de glândulas lacrimais e salivares → um exemplo comum é a parotidite
Sintomas Dermatológicos:
Fenômeno de Raynauld
Eritema anular
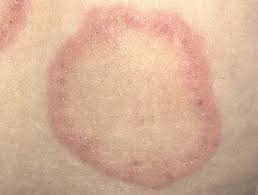
Queilite angular
Eritema nodoso
Livedo reticularis → mancha violácea em forma de rede
Líquen plano
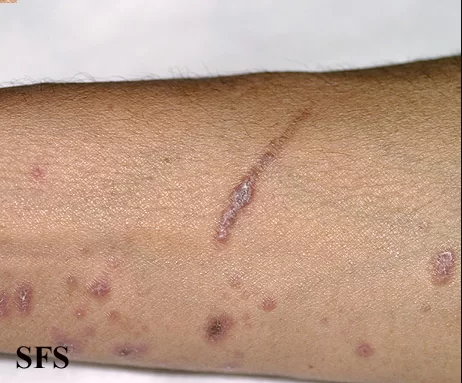
Vasculite cutânea.
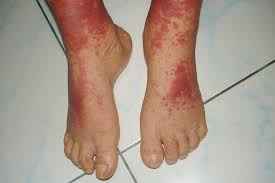
Sintomas renais:
Nefrite intersticial → comum acidose tubular renal.
Glomerulonefrite
Sintomas neurológicos centrais
Desmielinização
Neurite óptica
Mielite transversa
Disfunção cognitiva
Vasculite
Sintomas neurológicos periféricos:
Polineuropatia axonal sensorial e sensitivo-motora,
Neuropatia de pequenas fibras
Neuropatia sensorial atáxica (ganglionopatia)
Mononeurite múltipla
Neuropatia trigeminal
Neuropatia autonômica
Polineuropatia inflamatória desmielinizante crônica (PIDC)
Radiculoneuropatia desmielinizante
Sintomas cardiovasculares:
Pericardite
Distúrbios de condução
Aumento do risco cardiovascular
Sintomas Pulmonares
Doença pulmonar intersticial → PRINCIPAL [cerca de 10-20%, principalmente a doença pulmonar intersticial não-específica]
Doença pulmonar cística
Hipertensão pulmonar
Derrame pleural
Sintomas Gastrointestinais
Disfagia
Esofagite de refluxo
Dispepsia
Pancreatite
Gastrite atrófica
Gastroparesia
Colangite biliar primária
Hepatite autoimune
ALTERAÇÕES LABORATORIAIS
Sintomas Hematológicos
Citopenias (anemia de doença crônica ou hemolítica) → Leucopenia, neutropenia, linfopenia, plaquetopenia;
Linfocitose e monocitose
Hipergamaglobulinemia policlonal
Gamopatias monoclonais
Presença de crioglobulinas
Linfonodomegalias
Llinfoma
Aumento de provas inflamatórias (PCR, VHS)
Consumo de complemento
Autoanticorpos positivos → os autoanticorpos podem estar positivos anos antes das manifestações clínicas. Os principais são Anti-RO e Anti-LA [1/3 tem ambos, 1/3 não tem nenhum], normalmente cursam com FAN padrão fino pontilhado porém FAN pode estar negativo (mais comum quando Anti-RO52 +).
Anti-RO
Anti-RO52 → tem maior risco de doença pulmonar intersticial e pode ter FAN negativo
Anti-RO60
Anti-LA → positivo isoladamente apenas em 2,3% dos casos
Fator Reumatóide → positivo em 40-70% dos casos
Anti-Centrômero → positivo em 5-10% dos casos. Relacionado a maior risco de colangite biliar primária
PROPEDÊUTICA
Biópsia de glândula salivar menor (padrão-ouro)
Resultado esperado em Sjogren: sialoadenite linfocítica focal com presença de um ou mais foco em 4mm quadrados de tecido glandular
Foco = 50 ou mais linfócitos agrupados → A presença de 4 ou mias focos ou de centros germinativos indica risco de linfoma
Testes Objetivos de Olho Seco
Teste de Schimmer → mede-se a quantidade de lágrimas produzidas com fita estéril
Resultado sugestivo de olho seco: <5mm/5min em pelo menos 1 olho
Coloração da SUperfície Ocular → utiliza-se Fluoresceína para córnea lisamina verde para conjuntiva
Utiliza-se dois scores:
Ocular Staining Score ≥ 5 (0 a 12)
Van Bijsterveld Score ≥ 4 (0 a 9)
Exames complementares para avaliação de xerostomia:
Sialografia → exame invasivo que utiliza contraste
Cintilografia → Hipocaptação ou hipoexcreção do radiofármaco. Baixa sensibilidade, alta especificidade
USG de glândulas salivares → Áreas hipo ou anecoicas. Correlaciona-se fortemente com a presença do anti-Ro.
Sialometria (medida de fluxo salivar) → <0,1ml/min durante 5-15 minutos
CRITÉRIOS DIAGNÓSTICOS
→ Critérios de Inclusão (basta um dos tópicos):
Xeroftalmia ou Xerostomia com mínimo de 2 destes 5:
Olho seco, diária e persistentemente ao menos 3 meses?
Sensação de areia nos olhos?
Uso de lágrima artificial mais de 3x/dia?
Sensação de boca seca diariamente há > 3 meses?
Frequentemente ingere líquidos para facilitar a deglutição, principalmente sólidos?
Na presença de envolvimento extraglandular característico definido pelo ESSDAI
→ Critérios de Exclusão (basta 01)
História pregressa de tratamento com radiação de cabeça e pescoço;
Infecção ativa por hepatite C;
Síndrome de imunodeficiência humana adquirida (SIDA);
Sarcoidose;
Amiloidose;
Doença enxerto versus hospedeiro;
Doença relacionada a IgG4
→ Critérios Classificatórios: mínimo de 4 pontos
Biópsia de glândula salivar com sialoadenite linfocítica focal com pontuação de foco >/=1 | 3 |
Anti-RO positivo | 3 |
OSS (Ocular Staining Score) >/=5 em pelo menos 1 olho | 1 |
Schirmer </=5mm/5 minutos em pelo menos 1 olho | 1 |
Fluxo salivar não estimulado </= 0,1ml/min | 1 |
TRATAMENTO
Medidas Gerais:
Hábitos de vida saudáveis
Vacinação em dia
Monitorização do risco cardiovascular
Xeroftalmia:
Tratamento não-farmacológico:
Colírios sem conservantes
Gel ocular;
Barreiras físicas (óculos que conservem a umidade);
Modificação do ambiente (ex:umidificadores);
Compressa morna
Tratamento Farmacológico:
Imunomodulador tópico como ciclosporina ou tacrolimus.
Colírio com vitamina A.
Corticoide tópico.
Soro autólogo (pouco disponível)
Agonistas muscarínicos (pilocarpina, cevimelina)
Suplementação oral com ômega 3 (óleo de linhaça)?
Imunossupressor sistêmico?
Tratamento Cirúrgico: oclusão de ponto lacrimal
Xerostomia
Tratamento não-farmacológico:
Higiene oral (pasta com flúor)
Estimulantes gustatórios (balas ácidas, pastilhas e chicletes sem açúcar e/ou com xilitol)
Saliva artificial
Clorexidina (enxaguante bucal)
Limpeza dentária a cada 3 meses
Gotas de limão em copo d'água
Fatias de frutas secas como pêssego.
Tomar goles regulares de água (ou chupar gelo)
Evitar bebidas ácidas (coca, café, energético)
Adicionar omega-3 de cadeia longa líquido à água.
Manutenção de vias nasais abertas para evitar respiração pela boca.
Tratamento Farmacológico:
Agonistas muscarínicos (pilocarpina e cevimelina): paciente
ainda precisa de saliva residual.
Mucolítico (acetilcisteína)
Imunossupressor sistêmico?
Opções de imunossupressores e imunomoduladores:
Hidroxicloroquina
Metotrexato
Leflunomida
Ciclosporina
Azatioprina
Micofenolato de Mofetil
Ciclofosfamida
Rituximabe
Belimumabe
Glicocorticoide
Imunoglobulina
Colchicina
SEGUIMENTO
→ Os seguintes exames devem ser solicitados com periodicidade:
Eletroforese de proteínas
Crioglobulinas,
C3, C4,
Gasometria Venosa
EAS
→ Monitorização da atividade da doença se faz através do índica ESSDAI.
PROGNÓSTICO
Maior mortalidade está associada a:
Vasculite
Hipocomplementenemia (principalmente C4)
Crioglubulinemia
Risco de Linfoma Não Hodgkin → a síndrome em si já aumenta o risco de 5-9 vezes comparado a outras pessoas
Normalmente ocorre 6-7 anos após o diagnóstico, local mais comum é na parótida e tipo mais comum é MALT
Achados suspeitos de LNH
Aumento persistente das parótidas e consistência endurecida
Vasculite cutânea
Púrpura palpável
Neuropata periférica
Perda de peso
Esplenomegalia
Linfadenopatia
Desenvolvimento de glomerulonefrite
Febre
Proteína Monoclonal IgM Kappa
Neutropenia
Linfopenia
Fatores de risco para LNH
Crioglobulinas circulantes
Hipocomplementenemia (principalmente C4)
Escore focal >3-4
Presença de centro germinativo
Fator Reumatóide em alto títulos
Desaparecimento do Fator Reumatóide
ARTRITE REUMATÓIDE
DEFINIÇÃO
Doença sistêmica, inflamatória, autoimune, crônica e progressiva, sendo o sítio primário a membrana sinovial das articulações.
→ A artrite reumatóide tem como impacto: a perda da capacidade funcional e laboral; Diminuição da qualidade de vida; Significativo impacto social e econômico
EPIDEMIOLOGIA
Prevalência 0,4-1% e incidência de 0,2-0,4 a cada 1000
Mulheres 3:1 homem
Inicio 30-50 anos
FATORES DE RISCO
Genética:
Taxa de concordância para gêmeos idênticos: 12 a 15%.
Parentes de 1o grau tem risco 3x maior.
HLA-DRB1: epítopo compartilhado.
Genes: PTPN22, STAT4, TRAF1-C5, PADI4
Ambiental:
Tabagismo
Doença periodontal (Porphyromonas gingivalis)
Microbioma intestinal (Prevotella)
Infecções virais
Exposição à sílica
TEPT
FISIOPATOLOGIA
Citrulização e carbamilação de proteínas por indução/expressão de proteínas (peptidil arginina deiminase) → criação de neoantígenos na membrana sinovial → quebra da tolrância imunológica → Reconhecimento pela células T → Ativação de linfócitos B com produção de autoanticorpos + secreção de citocinas (TNF, IL-1, IL-6, IL-7) com recrutamento e ativação de macrófagos, fibroblastos, osteoclastos e acrondrócitos e angiogênese → Forma Pannus
Pannus: tecido composto por células inflamatórioas em membrana sinovial que invade cartiglageme osso, destruindo-os.
→ A sinovite crônica é mantida pelas interações entre as células nativas da articulação e células inflamatórias recrutadas para a articulação, com o subsequente estabelecimento de redes de citocina que perpetuam a inflamação
CLÍNICA
→ O padrão mais comum é de instalação insidiosa (semanas-meses)
Sintomas constitucionais: Astenia, Fadiga, Mal-estar, Febre baixa e Perda de peso
Sintomas Periarticulares: Artralgia de ritmo inflamatório; Poliartrite aditiva e simétrica de pequenas e grandes articulações; Acometimento mais periférico; Poupa as IFD;
Rigidez matinal > 30min e Tenossinovites
Sintomas extra-articulares: Ocorrem em 40% dos pacientes ao longo da doença, sendo a maioria homens.
Fatores preditores para essas manifestações: Tabagismo, presença de autoanticporpos, HLA-DRB1*04
Pele: Nódulos reumatiodes são a MEA mais frequente, podem estar em qualquer lugar do corpo. Característica indolor, firme e imóvel são preditores de gravidade da doença
Outros: Síndrome de Sweet; Pioderma gangrenoso; Dermatite neutrofílica e Úlceras cutâneas
Hematológicas: Anemia de doença crônica; Trombocitose; Linfadenopatia; Linfoma não Hodgkin
Síndrome de Felty [AR + esplenomegalia + neutropenia, está relacionado ao HLA-DRB1*0401 e Associação com leucemia linfocitária granular]
Respiratória:
Pleurite → Relação com FR em altos títulos e nódulos
subcutâneos.
Derrame pleural → Assintomático em até 70%. Exsudato. Geralmente pequeno e unilateral
Doença pulmonar intersticial → Manifestação pulmonar mais comum. Principal: PIU
Síndrome de Caplan: AR + pneumoconiose: múltiplos nódulos pulmonares
Obstrução de via aérea → Artrite da cricoaritenoide
Cardiovascular
Pericardite está presente em mais de 50%, mas maioria assintomática
Aterosclerose: A doença cardiovascular é a principal causa de morte nos pacientes com AR
Vasculite Reumatoide [Inflamação de vasos de pequeno e médio calibre→ MEA mais grave com 40% óbito em 5 anos; Associada a AR grave, ativa, soropositiva e com anos de evolução
Manifestações: vasculite cutânea com úlceras, infartos ungueais, gangrena digital, púrpura palpável, neuropatia dos vasa nervorum, aortite, acometimento ocular, vasculite renal e mesentérica.
Olhos
Ceratoconjutivite seca é mais comum (15-40% dos pacientes). Xeroftalmia. A AR e a doença de Sjogren frequentemente se sobrepoe. Risco de síndrome da ceratomalácia (grave).
Sistema Nervoso Periférico
neuropatias compressivas (STC), neuropatias periféricas (sensorial, sensitivo-motora, autonômicas)
Sistema Nervoso Central
Mielopatia Cervical → É uma urgência neurociúrgica.
Por subluxação atlanto-axial (C1-C2) secundária à tenossinovite do ligamento transverso de C1.
Fatores de risco: sexo feminino, presença de autoanticorpos, uso de corticoide em longo prazo, doença de longa data, doença erosiva, níveis elevados de PCR e VHS.
SINAIS E EXAME FÍSICO
Aumento de volume nas articulações MCF e iFP.
Subluxação das MCF com desvio ulnar dos dedos e polegar em Z
Subluxação do punho; tenossinovite extenssores com ruptura de tendão extensor
Dedo em botoeira: flexão das IFP e extensão das IFD
Dedo em pescoço de cisne: flexão das IFD e extensão das IFP
→ Exame das IFD: palpação em 4 pontos
→ Exame da MCF: palpação dorsal e volar
COMORBIDADES
→ O risco de desenvolver muitas destas comorbidades aumenta como consequência da inflamação contínua ou das suas sequelas:
Doenças cardiovasculares e seus fatores contribuintes (HAS, DLP)
Tromboembolismo venoso
Malignidades
Doenças metabólicas (obesidade, sarcopenia)
Osteoporose
Doenças mentais (depressão, ansiedade)
Fibromialgia
Infecções
EXAMES COMPLEMENTARES
Hemograma: Anemia de doença crônica, trombocitose
VHS e PCR: Avaliação de atividade de doença e resposta terapêutica
Auto-anticorpos: Fator reumatoide e Anti-peptídeo citrulinado cíclico
Ambos tem relação com prognóstico.; Podem estar presentes anos antes da doença clínica e Ambos podem ser NEGATIVOS (AR soronegativa).
Fator Reumatoide: anticorpo que tem como alvo a porção Fc da IgG. Presente em 70-80%.
Presente em: idosos, após vacinação ou transfusão, se familiar de 1o grau com AR. Pouco específico (+ em outras dças crônicas - LES, Sjogren, hepatopatia crônica, sarcoidose, fibrose pulmonar, mononucleose, hepatite B, TB, hanseníase, sífilis, endocardite, leishmaniose, paracoco, esquistossomose, malária)
Anti-CCP: ANTI-CCP: sensibilidade semelhante porém mais ESPECÍFICO (95-98%) que o FR.
Raio X: método mais utilizado na avaliação do dano. Pouco útil na fase inicial.
Achados iniciais: aumento de partes moles e osteopenia justa-articular. → Com a progressão da doença: subluxações e anquilose.
Lesões mais características: erosões ósseas e redução dos espaços articulares.
USG: Sensibilidade superior ao RX na detecção do dano. Útil para detecção precoce e na dúvida entre dano e atividade.
Power Doppler é útil no monitoramento da doença (hiperfluxo indica a presença de sinovite ativa).
RNM: Método mais sensível para detecção de alterações iniciais. Identifica edema ósseo, sinovite e erosões.
CRITÉRIOS CLASSIFICATÓRIOS
→ Pontuação minima de 6, analisa:
Artrite de articulações grandes ou pequenas e quantas
VSH e/ou PCR aumentados
Fator reumatoide e/ou anti-CCP positivo
Duração por menos ou mais de 6 semanas
TRATAMENTO
Não-Farmacológico: Intervenções psicossociais; Educação do paciente; Atividade física e reabilitação; Medidas de proteção articular e Vacinação
Farmacológico
→ Há janela de oprtunidade de 6-12 meses após inicio dos sintomas para o tratamento efetivamente mudar o curso da doença a longo prazo
DMARDs (drogas modificadores do curso da doença sintética) → Metotrexato é a 1a escolha, outros: leflunomida, sulfassalazina e anti-maláricos
Corticoides podem ser usados em curtos períodos no início da doença e em perídos de exacerbação/recidiva
Outras possibilidades: DMARD biológicos e DMARD alvo especificas
SEGUIMENTO
→ Objetivo do tratamento sempre será atingir remissão ou baixa atividade da doença em 3-6 meses de tratamento.
A atividade da doença é verificada por progressão radiográfica, impacto funcional, exame físico e índices compostos de atividade da doença
Índicede atividade de doença 28 (DAS28)
Índice simplificado de atividade de doença (SDAI)
Índice clínicode atividade de doença (CDAI)
Nem FAN nem anti-CCP ser alteram com o tratamento, logo sua função prognóstica é apenas no diagnóstico inicial.
PROGNÓSTICO
→ Fatores agraventes do prognóstico: Tabagismo; Início de doença em idade mais precoce; Baixo nível socioeconômico; Manifestações extra-articulares; FR em títulos elevados; Positividade do anti-CCP; Provas inflamatórias elevadas (persistente); Grande número de articulações edemaciadas; Índices elevados de atividade de doença; Erosões precoces; Presença do epítopo compartilhado
ESPONDILITE ANQUILOSANTE
CONCEITO
Espondilo = vértebras
Anquilosante = fusão
Espondilite anquilosante = espondiloartrite axial
É um acometimento inflamatório das ênteses [local em que tendões, ligamentos, cápsular, artricualres e fáscias se ligam no ossopondiloartrite axial
EPIDEMIOLOGIA
→ A prevalência depende da frequência de HLA-B27 em uma população
Mais comum em caucasianos e homens
Risco de 10% se parente de 1° grau e 20% de HLA-B27 presente
Sintomas costumam inicar 20-30 anos
ETIOPATOGENIA
Genético → HLA-B27
hipóteses relacioadas a patogenese → peptídeo atritogênico, mimetismo molecular, cadeia pesada livre, proteína desdobrada
HLA-B27 + tem risco 50-70% maior de desenvolver mas 0,5% dos positivos realmente desenvolvem a doença
presença do gene está relacionado ao prognóstico e existem subtipos que não estão relacioados à EA.
Ambiental
Estresse mecânico em estruturas alvo (coluna e calcanhar)
Imunológico
Disfunção microbiota intestinal
Infecções
FISIOPATOLOGIA
articulações sacro-ilíacas + artciulações facetárias → sinovite ou osteíte → erosão → neovascularização e osteoproliferações → anquilosa + sindesmófitos
Sindesmófito → é uma proliferações óssea que ocorre no ânulo fibroso (êntese vertebral → componente externo do disco intervetebral que insere-se nos corpos vertebrais adjacentes)
SINAIS E SINTOMAS
Sintomas articulares:
Dor lombar inflamatória → critérios (mínimo 4 de 5)
Idade < 40 anos
Início insidioso
Melhora com exercício
Sem melhora com o repouso
Dor à noite (com melhora ao se levantar)
Dor em glúteos (unilateral, bilateral ou alternante)
Rigidez marinal por mais de 30 minutos
Artrite periférica → tipicamente é oligoarticular em MMII
Entesite (32%)
Local característico: Inserção do tendão de Aquiles.
Dactalite (6%) → Dedo em salsicha
A dactalite é a combinação de Polientesite + tenossinovite + artrite
Síntomas extra-articulares
Oculares → Uveíte anterior aguda é a manifestação extra-articular mais comum (25-35%)
Clínica: Unilateral ou alternante. Dor, hiperemia, lacrimejamento, fotofobia [Recorre em 50%.]
Cutâneas → Psoríase (10%)
TGI → Doenças inflamatória intestinal [Chron ou retocolite ulcerativa] 4-6%
ACV → Distúrbios de condução, aortite com ou sem insuficiência aortica
AR → Fibrose pulmonar apical, distubio ventilatorio restritivo
Renais → Nefropatia por IgA, amiloidose renal
TÉCNICAS DE EXAME FÍSICO
Manobra de Patrick Fabere → Flexão, Abdução e rotação externa de um MI, se há dor glútea contralateral é sinal de sacroiliíte
Manobra de Gaenslen → flexão máxima de um lado e extensão contralateral como teste bilateral da scroilíaca
Teste de Schoeber
paciente em pé e descalço
Linha entre espinha ilíacas postero-superiores
Linha 10cm acima da primeira linha
Flexão da coluna ao máximo sem dobrar os joelhos
Esperado em pessoa hígida é aumento de minimo 5cm entre as linhas
Teste da expansibilidade torácica → medida do 4° EI-anterior em inpiração e expiração máxima, em pessoa hígida espera-se aumento de 2cm no minimo.
EXAMES COMPLEMENTARES
Hemograma → anemia de doença crônica, leucocitose e trombocitose
VHS e PCR → aumentados em 50-60% dis casos.
São preditores de progressão radiográfica e servem para acompnhamento da resposta ao tratamento
Teste antigênito → HLA-B27 por PCR (reação em cadeia da polimerase)
Fosfatase alcalina óssea específica → fica aumentada
IgA → Aumenta
Raio X de articulações sacroilíacas → indicado em caso de dúvida diagnóstica ou para diferencias atividade x dano da doença:
Grau 0 → sem alterações
Grau 1 → alterações suspeitas
Grau 2 → áreas localizadas de erosão ou esclerose + espaço articular normal [alterações mínimas]
Grau 3 → Anormalidade moderdas
Grau 4 → Anquilose total
ALTERAÇÕES AGUDAS | ALTERAÇÕES CRÔNICAS |
Edema medular ósseo | Erosões |
Sinovite | Anquilose |
Capsulite | Metaplasia gordurosa |
Entesite | Esclerose |
Visível em: T2-FAT-SAT, STIR | Visível em: T1 |
RNM de Coluna Vertebral
Locais de lesões inflamatorias
Canto vertebral
Placa terminal vertebral
Lateral torácica (articulações costovertebrais)
Articulações facetárias
Elementos posteriores (ligamentos espinhais articulação costotransversa)
CRITÉRIO CLASSIFICATÓRIO
[2009]
→ Aplicar em pacientes com dor lombar > 3 meses e início antes dos 45 anos.
Sacroiliíte em imagem com 1 ou mais característica de espondilite anquilosante
HLA-B27 positivo com 2 ou mais característica de espondilite anquilosante
→ Características de espondilite anquilosante: Dor axial inflamatória, artrite, entesite de calcâneo, uveíte, dactilite, psoríase, doença inflamatória intestinal, boa resposta à AINE, história familiar de espondiloartrite (presença em parentes de primeiro ou segundo grau de EA ou uveíte anterior aguda), PCR elevada, HLAB27
AVALIAÇÃO E SEGUIMENTO
→ Índices de atividades: BASSDAI (Doença atividade >4 pontos); ASDAS (mudança de 1,1 é melhora e >2 é melhora importante, se total <1,3 = doença inativa); MASES (avalia 13 locais, pontuação de 0-13), BASMI
→ Avaliação radiografica: mSASSS [avalia partes anteriores das vértebras cervicais e lombares em perfil]
TRATAMENTO
Não farmacológico: educação, atividade física (alongamento, aeróbico e musculação), interrupção de tabagismo e alimentação com fibras e ácidos graxos de cadeia curta
Farmacológico:
Primeira linhas: AINEs
Manter contínuo enquanto doença ativa. Pode manter sob demanda posteriormente.
Mudar tratamento se refratário a 2 AINEs por 4 semanas cada.
2o linha para quadro axial: DMARD biológicas (Anti-TNF ou Anti-IL-17) e medicamentos alvo-específicos
2a linha para quadro periférico: DMARDs (sulfassalazina e e metotrexato)
! Não tem eficácia para quadro axial !
3a linha para quadro periférico: DMARD biológicas (Anti-TNF ou Anti-IL-17) e medicamentos alvo-específicos
→ O bloqueio de citocinas depende da área com manifestações
TNF: Pele, articulaçoes periféricas e axiais, intestino e olho
Il-17: Pele, articulaçoes periféricas e axiais, intestino e dor generalizada
PROGNÓSTICO
Baixa chance de remissão espontânea
Alta chance de recorrência após suspensão do tratamento
Se doença leve, maioria mantém capcidade funcional e ocupacional
Complicações extramusculares com risco de vida são raras
AVC ISQUÊMICO
EPIDEMIOLOGIA
2a maior causa de morte no mundo
Principal causa de sequelas neurológicas no Brasil
1 a cada 4 pessoas no mundo terão um AVC, sem incluir fatores agudos
87% dos AVCs são isquêmicos, enquanto 13% são hemorrágicos
PROBLEMA
internações prolongadas
reabilitações prolongadas
complicações secundárias com internações
FATORES DE RISCO
NÃO MODIFICÁVEIS
Idade
Gênero → M>F exceto extremos de idade [pode estar relacionado comm comportamento]
Etnia → afrodescendentes > latino > asiático > europeu
Genética
MODIFICÁVEIS
HAS → Principal!
Cardiopatias → FA (escala de CHADS-VASc → se for mulhere 2 já é indicado anticoagulação, se homem, apenas 1), IAM, Doença Valvar Mitral
DM
Tabagismo
Hiperlipidemia
Obesidade
Sedentarismo → indicada atividade física de moderada a intensa pelo menos 40 minutos 3 a 4 vezes por semana
Excesso de álcool
Contraceptivo Oral → quando não usar: mulheres com exaeuca com aura não controlada, tabagista, historia de aborto espontâneo
Apneia do sono
→ Portadores de estenose de carotida assintomatica ja devem receber AAS e estatina
CLASSIFICAÇÃO DE TOAST - causas de AVCI
Aterosclerose de grandes artérias (trombose/embolia)
Placa ateromatosa → estenose do vaso → oclusão [se não houver embolia ou trombose, oclusão é assintomática pois há tempo para formação colateral]
Rompimento da placa → trombose aguda → AVC [MLEHOR REMÉDIO PARA PREVENÇÃO É ANTIAGREGANTE PLAQUETÁRIO]
Rompimento de placa → embolização de peuqneas porções de placa → pequenos AVCIs de repetição em área de irrigação de artéria específica
Oclusão de peqeunas artérias (lacunares) [microangiopatia]
Artérias perfurantes (derivam da cerebrla media, não tem camada média) → sofrem com HAS, dislipidemia, tabagismo
Maior parte da substância branca é area não eloquente (assintomatico), paciente vai tendo micro AVCs mas não faz nada → desenvolve Microangiopata obstrutitva crônica [sintoma é demência]
PREVENÇÃO É ANTIANGREGANTE PLAQUETÁRIO
Cardioembólico
Principal causa é FA (estase na auricula esquerda) [PRINCIPAL FORMA DE PREVENÇÃO É ANTI-COAGULANTE]
Trombo → oclui grandes vasos (AVCs catastróficos)
Outras causas identificáveis
Dissecção arteria
Maior causa de AVC em jovens (<50 anos)
Causas:
Trauma
Atividade sexual
Tosse e espirro
Yoga
Exercício físico intenso
Montanha russa
Dança, natação, tênica, voleibol, basquetebol
Quiropraxia
Condições predisponenetes: displasia fibromuscular (parede do vaso fica irregular, costuma ter alteração simpática) síndrome de ehlers-danlos e Marfan
Prevenção de AVC na dissecção é AAS
Vasculites
Podem ser sistêmicas ou Primárias do SNC:
Arterite de Takayasu
Arterite de células gigantes
Poliarterite nodosa
Doença de Kawasaki
Angeíte primária de SNC (PACNS)
Secundárias: Colagenosas (LES), Infecções (meningites virais ou bcaterianas, HIV, sífils, TBC)
Inflamação → contração → estenose do vaso pr acumulo temporário de sangue
Estados Pró-Trombóticos
Causas:
Síndrome do anticorpo anti-fosfolípide (LES e Covid-19 geram essa sindrome transitória]
Mutação do Fator V de Leiden (pirnicpal)
Mutação de MTHFR
Deficiencia de proteina C ou S
Deficiencia de antitrombina
Mutação de protrombina
Elevação da liporoteina
Pode ser secundaria: síndrome paraneoplásica
Doenças Metabólicas e Genéticas
Raros
Doença de Fabry (polineuropatia, cardioparia, alterações cutaneas)
CADASIL (cefaleia, déficit cognitvo progressivo)
Medicamentoso ou Drogas Ilícitas
Substâncias simpaticomiméticas → hipertensão, vasoespasmo e/ou vasculite:
Anfetaminas
Cocaína
Heroína e outros opiódes
Maconha
Canabióide sintético (spice)
GHB (boa noite cinderela)
Criptogênico (30% dos casos)
Provavelmente um grande percentual é cardio-embólico → FA é paroxístca, pode chegar no PA sem estar arrítmico
! FA faz o átrio esquerdo crescer → é possível pesquisar isso !
Prevenção é com AAS de acordo com protocolo, porém prevenção real seria com anti-coagulante
ATAQUE ISQUÊMICO TRANSITÓRIO
Não gera lesão, nem sequela, mas mostra um risco de AVC (alta morbimortalidade)
Principal causa é doença aterosclerótica
→ CLASSIFICAÇÃO DE ABCD2 (risco vascular)
<4 baixo risco e acima é alto risco
se <4 → iniciar AAS e estatina
se >4 → iniciar dupla anti-agregação (AAS + Clopidogrel)
Pode ser cardioembólico, porém é mais raro (trombo pequeno, é dissolvido endogenamente)
AVC AGUDO
Triagem → F.A.S.T.
B - Balance (ataxia
E - Eyes
F - face (desvio da comissura labio
A - arm (perda de força
S - speech (afasia)
T - Chamar ajuda
Suspeita clínica
! ALTERAÇÕES SÃO SÚBITAS !
Confusão mental, dor de cabeça forte sem causa conhecida, afasia, perda de força, perda focal súbita
Escala de NIH (0-42)
Ferramenta mais efeitiva para avaliar gravidade.
<4: quadro leve, a não ser que sintomas sejam de fala
<6: AVC moderado
>10: AVC grave
>20: AVC muito grave
Fiosopatologia
CORE: area que morre na hora → aumenta a cada minuto.
Penumbra: recebem sangue das colaterais. Área depende da qualidade das colaterais
Trombolíticos
1995: Alteplase rt-PA → trombolítoco de meia-vida curta, tempo de tratamento é de 1h em bomba de infusão.
Wake-up stroke: paciente acorda com o déficit
O tempo que o AVC demora para aprecer no Flair, é em torno de 4:30h, no de difusão é em torno de 30 minutos
Mismatch: sintoma clinico pior que se ve na imagem, significa que imagem irá piorar.
2023: Tenecteplase → estudo demonstrou que são igualmente eficazes e ela tem meia-vida é muito maior
Trombectomia em grandes vasos até 24h → estudo da penumbra.
Opções de tratamento de reperfusão
trombólise venosa em alteplase ou tenecteplase em até 4,5h
trombolise venosa com alte ou tenecteplase em paciente que acordam com deficit mas tem mismatch difusão/FLAIR
Em paciente trombolisados mas com oclusão de grande vaso, avaliar trombectomia
Trombectomia em oclusões proximais em até 24h quando há mismatch perfusão/clínica
Unidade de AVC
Stroke-team cuidando em unidade exclusiva de AVC
Recuperação maior e mais precoce
AVC HEMORRÁGICO
PADRÕES
Hipertensivo → mais comum
Vasos lacunares são muito sensíveis e se rompem por HAS. Apesar de pequenos, são vasos arteriais, então tem ghrande fluxo e pressão.
São sempre profundos, pois é onde artérias perfurantes irrigam (ventriculo, nucleos da base)
Paciente normalmente chega no PA com pressão extremanete alta → é errado tentar abaixá-la muito rápido pois pode ocorrer hipoperfusão cerebral (PA alta esta vencendo PIC alta) → transitoriamente, paciente pode ser tratado com manitol (diurético osmótico, que puxa o liquido intersticial, sem risco de hipoperfusão), não resolve o problema.
! Max 15% da PA pode cair por hora !
Tratamento é cirúrgico → drenagem e alguns casos expecíficos, trombolíticos
Má formação arteriovenosa (MAV)
angiogênese anormal a nível capilar
pode ocorrer em qualuqer região do cérebro
pode atingir tamanhos significativos
MAV pode sangrar de forma recorrente, se cortical pode causar crise epiléptica, pode crescer (síndrome expansiva)
Sinais: déficti focal subito, déficits transitórios recorrentes, deficit gradual e progressivo
Angiopatia Amiloide
Complexo amiloide se deposita em vasos cerebrais, tornando-os frageis e friáveis → alto risco de sangramento
É a principal causa de hematoma lobar → sangramento de tronco arterial
Possibilidde de ver angiopatia amiloide antes de sangramento → RMN Eco: ve hemossiderina preta → tem oxigenio → varios microsangramentos
Não há nivel de evidência sobre prevenção.
Não ha evidencia sobre relação com demencia de alzheimer (são coplexo beta-amiloides diferentes)
Paciente nunca deve ser anticoagulado
Hemorragia Subaracnóidea
Alta taxa de sequelas neurológicas e mortalidade, mas alta chance de tartamento sem sequelas se precoce.
Maior causa é aneurisma sacular → bifurcação de artéria é repetidamente injuariada por pressão sanguinea, forma aneurimas sacular (apenas 10% sangram)
Maior parte das bifurcações importantes é no polígono de Willis → explica alta mortalidade
Se individuos está vivo, é por que sangramento parou → formação de trombo e mudança de plasitcidade cerearteral em tantiva de parar sangramento. Se sangramento continua é morte súbota
Diagnóstico:
Cefaleia súbita e intensa (normalmente diferente de qualquer outra que ja teve)
Perda de consciência, convulsão, náusea/vômitos
Rigidez de nuca pode demorar várias horas para se instalar → è o sintomas clínico clássico, porém ausencia não descarta hemorragia.
HSA em até 25% de pacientes atendidos com cafélias súbita e intensa
sinais de gravidade de cefaleia (sempre fazer exame de imagem): dor de cabeça sem precedentes + qualquer déficit focal (exame neurológico tronco central) + qualquer sintoma clínico que remeta a imunodeficiencia ou contexto infeccioos (HIV, corticoide, febre, prostração, neoplasia)
Exames de imagem:
TC de crânio → altisisma sensibilidade para avliar sangramenro NAS PRIMEIRAS 24 HORAS (hemossiderina é degrada)
RMN → mais sensível que TC
ANGIO-RMN e ANGIO-TC (SÃO IGUAIS PARA ESTUDO) → principais para depois de 24h. Muita sensibilidade, consegue descartar
Punção lombar → líquor fica xantocrômico (sinal de sangramento)
Problemas: nao se vê o aneurisma, conduta é impossibilitada. Logo paciente vai ter que fazer exame de imagem de qualque jeito. Queda na pressão intracranian pode mover trombo que esta estancando sangramento
Arteriografia → tamanho de aneurisma, É a mais sensível
Desfecho
Destino → morte súbita ou estado muito grave.
Diagnóstico → deve ser feito nas primeiras 48h
Tratamento cirúrgico → cateterismo com coil, stent, ou cirurgia aberta
Suporte intensivo → prevenção do vasoespasmo, identificação do vasoespasmo , tratamento do vasoespasmo [aumento da pressão arterial + dilatação do vaso com nimodipina + doppler transcraniano → velocidade aumentada de fluxo indica vasoespasmo, faz todos os dias]
vasoespasmo é a principal complicação que sobrevive sangramento, sangue em contanto com parede externa do vaso leva a espasmo
<3mm → risco extremanete baixo de compplicação
3-5mm → risco pode aumentar de acordo com local e características do aneurisma ( pode já fazer intervanção)_
5-7mm → aneurisma com risco significativo a não ser que haja CI absoluta
7-10mm → risco altíssimo
>10mm → aneurismas gigantes, pode ter efeito tumoral
Triagem (RMN) → familiar de 1° grau que teve aneurisma
MENINGITE E MENINGOENCEFALITE
DIFERENÇA → meningoencefalite tem acometimento focal → rebaixamento do sensório
Nível de consciência
Estado e consciência
A meningite bacteriana sempre vai evoluir para meningoencefalite
SINTOMAS
Cefaleia com febre não é sinônimo de sinusite
Cuidado com Rx de seios da face → marioais vai ter velamento, não exclui outras infecções sisteminas ou meningites
Tríade clássica da meningite bacteriana:
Rigidez de nuca, febre e alteração do sensório
Ausência dos três descarta meningite BACTERIANA
Sinal de Kernig e Burdzinski tem baixa sensibilidade
Petéquias → meningococcemia [coropo todos, só no rosto podem ser apenas esforço de vômito)
DIAGNÓSTICO
Tres exames mais importantes:
Lab → hemograma [leucocitose x leucopenia], hemocultura [positiva em 50-90% dos casos], função renal [creatinina, ureia, Na, K, Ca, Cl, Mg, Gasometria venosa e urina: EAS e relação albumina/creatinina em amostra única]
! também pedir glicose para interpretação do líquor !
TC de crânio sem contrates → pode visualizar abcesso associado ou hemorragia sub-aracnoidea
Líquor: aumento de celularidade [VR 5 celulas por mm3] (polimortfo → bac ou linfomorfo → viral); consumo de glicose [ VR até 2/3 da glicemia]; valor de proteína; gram e cultura; Látex (procura antígenos especificos bacterianos mas tem alto percentual de falso positivos)
pulurento = bacteriana
aumento de linfocitos + consumo de glicose → TB e carcinomatose meningea
TRATAMENTO
O atraso em mais de 3h no diagnóstico é FR independente de mortalidade, sendo maior que infecção por um microorganismo resistente a penicilina
Iniciar ATB logo após punção lombar
Corticoide → se susperita de meningite pneumocócica usar antes ou junto com dexa
Não usar coirtiocide se houver iniciado ATB ou imunosuprimidos ou desnutridos
Interromper o corticoide se ainfecção não por pneumococo
Se corticoide + vancomicina, associar rifampicina
RN → bacteria do perineo maternos, tratamento empíroco é ampicilina + cefotaxina
3-5 meses → transmissão via oral (inluenza e meningococo)
A partir dos 5 anos, principal causa é pneumococo (não transmite por via oral) → se não tem punção lombear, dar ceftriaxona + aciclovir
Prevenção contato para agente de saúde → ciprofloxacino dose única
MENINGITE ASSÉPTICA
Causa mais comum é enterovírus → meninigite muito comum, benigno, não se trata.
Pode ter causas não infeccionas → autoimune, drogas, paraneoplasico
MENINGOENCEFALITE VIRAL
Causa mais comum é o herpes virus 1
Tratamento : aciclosvir; se imunodeprimidos: anfotericina B
Muito grave: tropismo por lobo temporal
CEFALEIA AGUDA
INCIDÊNCIA
18% da população; 3:1 mulheres (está relacinado ao estrógeno)
52% possuem crises esporádicas de cefaleia tensional (mulheres = homens)
Principais causas de cefaleia secundária → sinusite, meningite, lesão expansiva, hemorragia intracraniana
Principais causas de cefaliea primária → enxaqueca, tensional e cefaleia em salvas
SNOOP
Pode ser resumido em 3 (que são imperativos solicitar TC
Dor sem precedentes
Alteração de exame neurológico
Presença de sinais sistêmicos ou história de imunossupressão (corticoide, oncológico, HIv +)
S → Sistêmica
N → Neurológico
O → Onset
O → Outros (especificidades)
P → Padrão
MANEJO CLÍNICLO NO PA
Objetivos
fluxograma
CEFALEIAS PRIMÁRIAS
Maior causa é cefaleia tensional mas maior causa no PA é enxaqueca
Enxaqueca
Patologia da enxaqueca → Disautonomia cerebral: liberação exagerada de NT (nora).
uso de beta-block: bloqueio de NA
uso de tricíclicos e duasi: controle de liberaçãode NA
uso de topiramata ou valporato de sódia: modulaçõa de atividade elétrica
[Tentativa de modulação da atividade cerebral]
A aura deve vir obirgatoriamente antes ou junto da dor (dura de 5 minuto a 1h]
Critérios diagnósticos:
Pelo menos 5 crises
Duração 4-72 horas
Pelo menos 2 desses: dor unilateral, pulsátil moderada ou intensa, piora com atividade física
Pleo menos 1 dos seguintes [DD com cefaleia tensional]: náusea e/ou vômitos, fotofobia e fonofobia, não atribuída a outra causa.
→ PROVA: Um paciente que tenha neuasea ou vomito/ fotofobia e fonofobia NÃO É CEFALEIA TENSIONAL
Sintomas premonitórios (pode ser várias horas antes da dor): Irritabilidade, raciocionio e memorização mais lentos, desânimo, avidez por alguns tipos de alimentos
Aura
Típicos: escotomas luminosos ou negativos
Perda de distorção de um dos hamicampos visuais ou partes deles
Às vezes associam-se a parestesia unilateral e/os disfasia
Vertigem → enxaqueca vestibular
Tratamento
Dimenidarato (antiemético)
Dipirona
Cetoprofeno
Sumatriptana subcutâneo → triptanos agem com bloqueio agudo da CDRT (calcitonina) gera vasoconstrição quase totalmente seletiva cerebral
→ Associação se enxqueca crônica ou se cetoprofeno não funcionou: Dexametasona (se não melhorar: clorpromazina IM e SF 0,9%)
→ Não prescrever opióde para a crise de enxaqueca!!
Complicações
Estado migranoso: duração maior que 72h com intervaldos de no maximo 12h. É uma dor que melhora ma svolta
Infarto migranoso: aura persistente por mais fede 60 minutos e exqueca que não melhora, em RMN há AVC
Mingrânea crônica
Cefaleia Tensional
Desregulação adrenérgica: grau de tensão relacionado à dor
Sono ruim → relaxamento musuclar ocorre apenas a aprtie da fase 3 do sono
Má postura → presença de pontos de gatilhos
Pelos menos 2 dos seguintes: dor bilateral, pressão ou aperto não pulsátio, dor leve a moderada, não piora com atividade física
Não pode ter: nasusea ou vomitos, fotofobia e/ou fonofobia
Mínimo de 10 crises, duração de 30 minutos a 7 dias
Tratamento: Paracetamol, dipirona, ibuprofenos, diclofenaco
Associação com cafeína a eficácia analgésico
Cefaleia Em Salvas (Cluster Headaches)
Mais presente em homens
Dor desautonômica levando a processos inflamatórios e vasodilatação
c→ vasodilatação muito intensa, tem sintomas parecidos com sinusite
Mlehor remédia: O2
Vem em blocos
Pelo menos 1 : injeção conjutival ipsilateral e/ou lacrimejamento; Congestão nasal e/ou rinorreia; Edema palpebral ipsilateral; Miose e/ou ptose ipsilateral
FLUXOGRAMA
Dor aguda, emergente, sem febre → TC com contraste
Dor aguda emergente com febre
com sinais de alerta (qualquer alteração no tronco neurologico) → Meningite
sem sinaisd e alerta → sistemico
Dor crônica progressiva sem febre → HIC idiopática (prestar atenão em mulheres que tiveram alteração d epeso subito)
Dor crônica progessiva com febre
Dor crônica não progressiva → abuso de analgesio
REVISÃO
12 QUESTÕES
principal causa de avc isquemico em <50 anos [independente de FR]: dissecção arterial
pct que acorda com deficit neurpologico sem certeza exata de que horas o avc foi → com a RMN se tem restrição à difusão mas sem alteração do flair pode-se afirma que tem <4,5 horas = pode-se avaliar trombolise
cefaleia subita intensa sem precedentes há 72 h → procedimento mais adequeado para avaliar aneurisms roto é ANGIOTC arterial cerebral
prinicpal causa de avc hemorragico intraparenquimatose → HAS
cefaleia tensional x enxaqueca → fotofoibia, fonofobia, nausea ou vomito NÃO É TENSIONAL
cefaleis em salvas tratamento de crise → OXIGÊNIO (12L/min, MASCARA FCIAL Fo2 100% POR 20 MINUTOS)
Meningite x encefalite → encefalite tem alteração do exame neurologico
Meningite bacteriana líquor → neutrofilos com consumo de glicose e aumento de proteina
crises focais perceptivas x disperceptivas → alteração de memoria ou consciência é disperceptiva (diferente de ausencia)
delirium → base orgânica (alcool e BZD podem causar delirium tanto pelo uso quanto pela abstinência)
comprometimento cognitivo leve amnesico e não amensico → amnesico mais relacionado ao nivel de alzheimer
anticorpos monoclonais trazem risco de edema ou sangramernto cerebral (removem complexo beta-amiloides → alterações de imagem relacionada a amiloide)
profilaxia secundaria apos avc isquemico agudo → lacunar é dupla-antiagregação por 21 dias (AAS e clopidogrel), depois muda para só AAS; avc grande é só AAS. Ambos tem estatina
EPILEPSIA
CONCEITO
→ Pré-disposição de ter crises epilepticas, pode ser genética ou adquiricas e pode ser parte de uma doença ou puramente primário
! sem controle, aumenta em 3 vezes o risco de morte → alto risco de acidentes !
ETIOPATOGENIA
→ diminuição do limiar convulsivo (pode ser primário ou secundário → alcool) ou foco com estimulo eletrico maior que proteção natural consegue segurar (traumas, cicatrizes)
→ Pode ser focal ou generalizada (generalizada = acomete os dois hemisferio, nao necessariamente o cerebro todo.
PROVA
TIPOS DE CRISE FOCAL
Perceptiva (crise parcial simples) → nivel de consciencia, estado de conscienciae memória são preservados
Disperceptiva (crise parcial complexa) → perda de nivel de consciencia ou estado de consciencia ou memoria
CLASSIFICAÇÃO
Início Focal
Motor
Não-motor
Início Generalizado
Motoras
Tonico-clonicas
Outras clonicas
Não motoras
Ausências
Início desconhecido
CRISES GENERALIZADAS
Crise Tônico-Clônicas Generalizadas
Perda de consciência
2 fases:
Fase tônicas → com enrijecimento de todos os músculos
Fase clônica → com movimentos repetitivos
Salivação excessiva, moderdura de língua e bochecha, possibilidade de liberação de esfíncteres, estado pós-comicial (sempre|)
Crise de Ausência (pequeno mal)
Perda total da consicencia sem perda de tônus por fração curta de tempo mas pode ocorrer dezenas de vezes ao dia
Olhar vago
Muito cuidados com diagnóstico diferencial de TDAH
Crise Mioclônica
Contrações musculares bruscas de 1-2 segundos
Podem ser únicas ou repetidas
Pode ter eprda de consicnecia durante a crise, mas normlamente não ´percebida pela rapidez da sua duração
Importância: crise mioclônica juvenil
Crise atônica
assemelha-se a um desmaio → não existe diferença fora do EEG
Crises secundárias:
abusos de SPA
metabólica
Infecciosa
Lesão cerebral
Vascular
Trauma
Tumoral
Inflamatória
CONDUTA PÓS 1a CRISE
Exame de imagem → TC ou RMN
Verificar motivo da crise
Manterm observação por minimo 6h
CRISE EPILEPTICA EM PACIENTE EPILEPTICO
Verifica motivo da crise
falha na tomada da medicação
Intercorrência clinica
Interação medicamentosa
Posologia inadequada
Outros fatores pedisponentes
Condutas de prevenção até a avaliação especializada
ESTADO DE MAL EPILÉPTICO (Status Epilepticus)
Crise convulsiva que dura mais de 5-10 minutos OU crises convulsivas reentrantes sem recuparação dea csnciencia entre eles
Porcesso de retroalimentação do curto circuito
A partir de 30 minutos, cerebro começa a queimar
Complicações: rabdomiolise, IRA< acidose laticas, morte neuronal, pneumonte aguda
Protocolo:
estabilização (ABCDE)
Glicemia
Criança: midazolam nasal, retal ou pelo IOT
Diazepam venoso até 40 mg (1omg de bolus) → Fenitoina (Levetiracetam se disponível) → se não funcionaou → Midazolam (dose anticonvulsivanete é 4x maior que a sedativa) ou fenobarbital venoso (paciente precisará ser entubado)
COMPROMENTIMENTO COGNITIVO
DECLÍNIO COGNITIVO NATURAL DA IDADE
Bascaimente existe declinio MAs não há perda de conhecimento ou hbialidade que a pessoa já tem e não ha perda da capacidade de aprender coisas novas
DELIRIUM
alteração do estado e nivel de conscientia por base orgânia
I WATCH DEATH
! Pode coexistir com demeência → deliconoi cognitivo que piora subitamnete não faz parte da historia natural da doenaç !
DIAGNOSTICO DIFERENCIAL
Causas secundárioas → deficiencia de B12, B1 (wernicke-korsakof), disfunção tireoideana, neurosífilis, etilismo crônico, uso cronico de BZD.
CONCEITO DEMÊNCIA
Compromentomento de pelos menos 1 domínio cognitivo que é progressivo, disfuncional sem outras causas possíveis
aprrendizado e memoria
linguagem
função executiva
atenção complexa
perceptomotor
cognição social
COMPROMETIMENTO COGNITIVO LEVE (CCL)
Preservação das atividades diárias
Pode ocorrer por depressão, desuso
CCL Amnésica → risco maior para Alzheimer
CCL não-amnésica → risco maior para outras demências
Tratament é psicoestimulo : atividiade fisica, atividade intelectual, vida social, higiene do sono
Alertas
→ Lecanemabe: funciona para CCL ou DA leve com comprovação de beta-amiloide em PET ou líquor