BIOC15 Final
Table of Contents
Mutations
Chromosomal rearrangements
Sequencing
Ploidy and Molecular Genetics
Gene Regulation and Adaptation
Mutations
Early evidence
DNA is attached to backbone of phosphate group. Polymer length of bases together.
The phosphate group makes it a nucleotide, without it it’s just a nucleoside. It has what differentiates it as a purine or pyrimidine, but we don’t care to make the difference unless it’s a nucleotide.
Hydrogen bonds: two parallel lines, base pairing, what can bind to what?
Rosalind Franklin found the opposite directions I believe.
Mutations happen anywhere in the genome and this causes a forward mutation. If the same mutation reverts back to the original (same mutation at same place) this is a reverse mutation. 5000 opportunities to induce the phenotype.
Types
Substitutions
If you have an original base changing
Transition: purine for purine, pyrimidine for pyrimidine
Transversion: purine for pyrimidine, pyrimidine for purine
SNPs (single nucleotide polymorphisms): point mutations. Can be classified based on effect of phenotype. Codon table and codon code show several ways of coding for the same amino acid. Depending on what codon is there, mutation might or might not effect changes.
Synonymous mutation: amino acid doesn’t get changes
Non-synonymous mutation: will affect protein function - new codon encodes a different amino acid.
Nonsense mutation: protein doesn’t get made. The new codon produced is a stop codon. Premature termination.
Deletions and Insertions
Can be a single base pair or many thousands of base pair.
Single (point mutation) or several nucleotides/large fragments.
Can lead to deletions of genes or frameshift mutations. Sometimes genome doesn’t code for any genes, they don’t often really affect genes.
Frameshift mutations: Everything is shifted downstream. It still tries to get read, but everything has been shifted. In-frame you lose maybe three codons, and frameshift you lose an uneven number or random 3 that weren’t previously grouped as a codon.
Mechanisms
Errors in DNA replication
Complex comes in and pushes DNA strands apart, synthesize new complementary strands. Working in opposite directions. Genetic machinery linking can only work on the 3’ end.
Template DNA needs a primer. dNTPs could be a,c,t,g. Gets added in.
Steps:
Initiation: Cell doesn’t have any DNA primers. Along the DNA double helix, when the region is actively replicating, the two strands are separating. Single strand binding proteins keep things apart. Protein unwinds the double helix.
Elongation: Continuous synthesis from 3’ to 5’/ Lagging on 5’ to 3’. Polymerase going in small chunks, will link back up.
Errors can occur due to DNA polymerase not being 100% efficient. Mutations come from DNA replication. 1 in a million chance it just makes a mistake. Sometimes it can tell and repair, other times it cannot tell.
Errors can occur due to unstable genomic regions (trinucleotide repeats). Polymerase has trouble reading it and falls through, slips and new synthesized strand falls. Next round of DNA replication, decreases in number of copies. They may cause insertion or deletions, expanding or contracting the repeat regions. Many genes link this.
Trinucleotide repeats: Expansion of repeats in a few specific genes has been associated with specific neurological disorders. Fragile X syndrome (Fmr1 gene). Huntington’s disease (HTT gene). X-linked spinal and bulbar muscular atrophy (SMN1). Myotonic dystrophy (DMPK gene). Spinocerebellar ataxia type 1 (ataxin-1 gene). Dentatorubral-pallidoluysian atrophy (atrophin-1 gene).
Concept is similar. Wild-type allele with the lowest number, it varies based on how many alleles it has from a wild-type allele.
Other natural causes
Depurination: hydrolysis of a purine (A or G). Happens about 1000x per hour in a cell. A random base is introduced after depurination. A mutation occurs three-quarters of the time. Cells will try and repair purine but will not necessarily know where. Might put in A or C by accident. ¼ of time no mutation, ¾ of time a mutation occurs.
Deamination: Removal of amino group from cytosine or adenine. Problem with uracil, if a cytosine is changed to uracil, the polymerase will think uracil is a thymine. It will introduce an A instead of a G like it should. Not read the right way. Will read G as G, doesn’t result in mutation then.
C → U, G→ T and A later on.
Tautomers: identical chemical formulas, but different arrangements of atoms. Bases convert between different structures. Can result in different base pairings that introduce mutations. C opposite G that was missing. Mutation is introduced. Slightly altered bases. Can’t recognize as the right base.
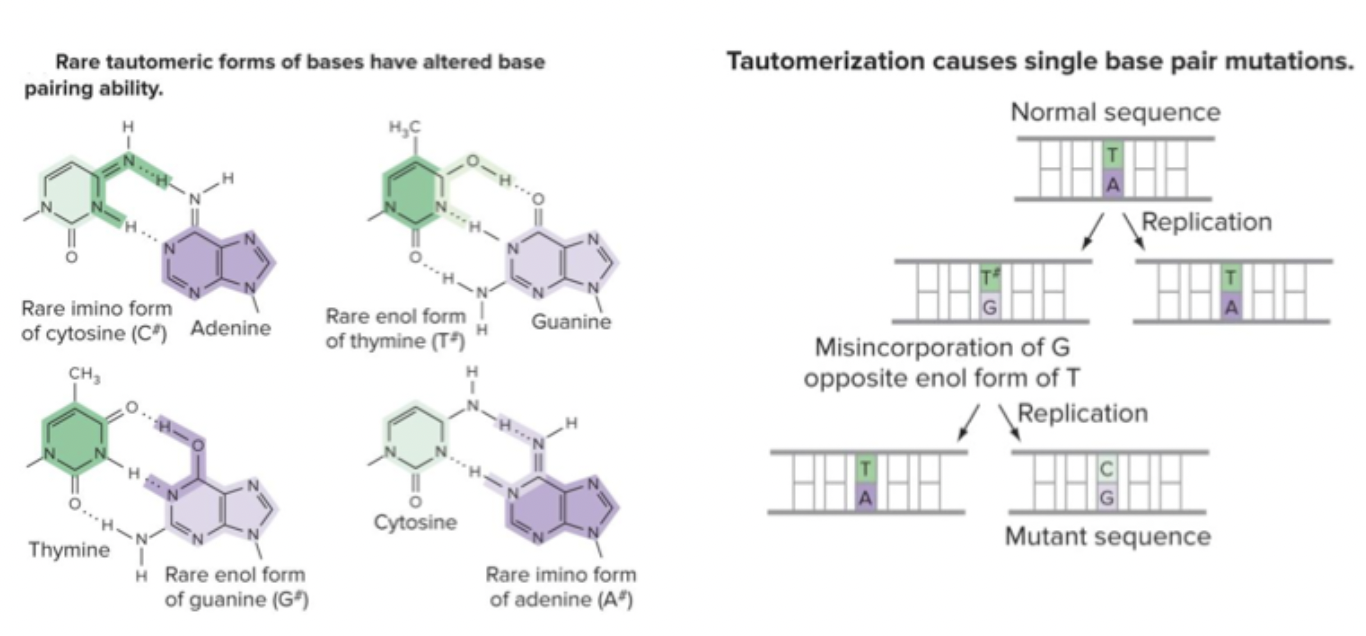
Mutagens
External factors can induce mutations:
X-ray break the back bone of DNA (double-stranded breaks, can’t reintroduce). Why dentists put blanket on you during x-ray. Researchers often die from cancer, not that damaging anymore.
UV light is why it is important to maintain ozone layer. Three types of UV-light, C most powerful, doesn’t come through. Power - the wavelength. Induces the thymine dimer - polymerase gets to it and stalls. May stop and fall off. Mutational age hits. Linked to skin cancer.
Oxidation (oxidative radiation): nuclear radiation is a type, multi-generational reports link nuclear plants and atomic bomb disasters to cancer. Oxidation forms nucleotides, does replication, pairs the wrong nucleotides. GO (oxidative guanine) get’s randomly paired to A instead of to C or A to T. T now exists instead of C.
Chemicals: base analogs, chemical structure almost identical to normal base but pair with different bases. Many in the environment → 5 bromouracil. Not super prevalent chemical, caused many issues in labs. Base analog in the cell - paired with wrong base. 5BU is a tautomer resembling C.
Hydroxylating agents and an -OH group, altering base structure. Chemicals in latex and nylon production. C gets hydroxylated and can’t pair with G, but pairs with A.
Alkylating agents - add ethyl or methyl groups. EMA used in labs, plant systems. Try and select new mutants. How we get a lot of GMO plants.
Deaminating agents: remove amine -NH2 groups. Nitric acid used in synthetic dyes. Changes C to be read as U. Uracil pairs like T instead of with G or whatever.
Intercalators: insert between bases causing insertion and deletions. Polymerase has issue getting passed. Disinfectants. Even though it is considered safe now, people still wear gloves.
Why are mutations relatively rare? → We have repair mechanisms. DNA is always under attack, but also always working to fix itself. Severe consequences to all life otherwise.
Repair
DNA polymerase has a proofreading function
Cuts out (exonuclease activity) and then introduces the right base. Gives polymerase a second chance to add the correct base. Polymerase quality is based on fidelity. Engineered to be high fidelity , 100x higher. Specialized enzymes, damaged to enzymes.
Specialized enzymes can reverse damage to nucleotides
Through absorption of light, removes thymers. Alkyltransferases repair guanine modifications. Photolyase repairs thymine dimers.
Base excision repair
Big protein context fixes all this. Mistakes can be recognized. Mismatch pairing is recognized, glycolases remove the base that’s wrong. Nuclease that gets cut. Open gap. Synthesis by DNA polymerase, needs a big gap to do it.
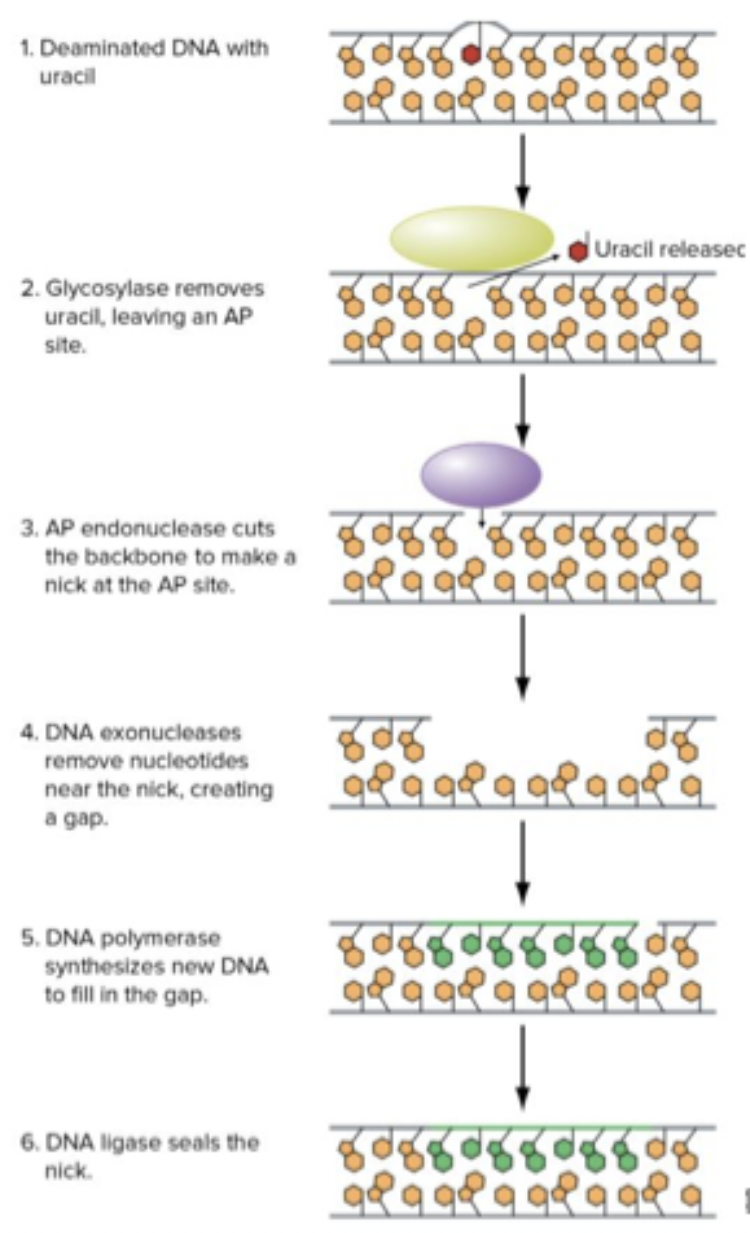
Nucleotide excision repair
Response to UV exposure. Fixing thymine dimer. Nips strand around bases that were fused together, DNA ligase closes the gap.
Exposure → thymine dimer form → UvrB and C endonuclease chop strand with dimer → damaged fragment chopped → DNA polymerase fills gap → DNA ligase seals gap
There may be mispairing or proper nucleotides. In bacteria people have seen methylate mismatch. New strands not methylated yet. Mechanism of strand recognition still not known in eukaryotes.
Double stranded break repair (Homologous recombination/NHEJ)
Homologous recombination: Whole DNA molecule is broken. If DSB occurs during time of sister chromatid in the cell. DSB repaired uses sister chromatid as framework. Not the same as recombination itself.
Nonhomologous end-joining (NHEJ): NHEJ is most common way of fixing, where there is not a way of homologous recombination - cleans up ends
Last resort (error prone)
‘sloppy polymerase’ (bacteria): If DNA is really damaged, when polymerase stalls and can’t continue they basically incorporate random nucleotides. As soon as they get into region of DNA.
Microhomology-mediated end-joining (MNEJ): When 2 strands of DNA are broken apart, a group of enzymes starts restricting till it gets to an end where it can start resection with each other. Cut back on either side till there is a compatible region (annealing, flap removal, fill-in synthesis, ligation)
When things are not corrected, they become permanent. DNA damage (natural process) or DNA base sequence error (natural DNA replication) → replication → mutation.
Somatic tissue: until the death of the individual, the mutation will exist
Germ-line: throughout generations unless eliminated by natural selection or reversal. Could become a wild-type allele.
Mutation rates
Plasmids are important for transgenic work. Bacteria are infected with viruses. Bacteriophages tend to kill bacteria by hooking and implanting a viral genome in cell.
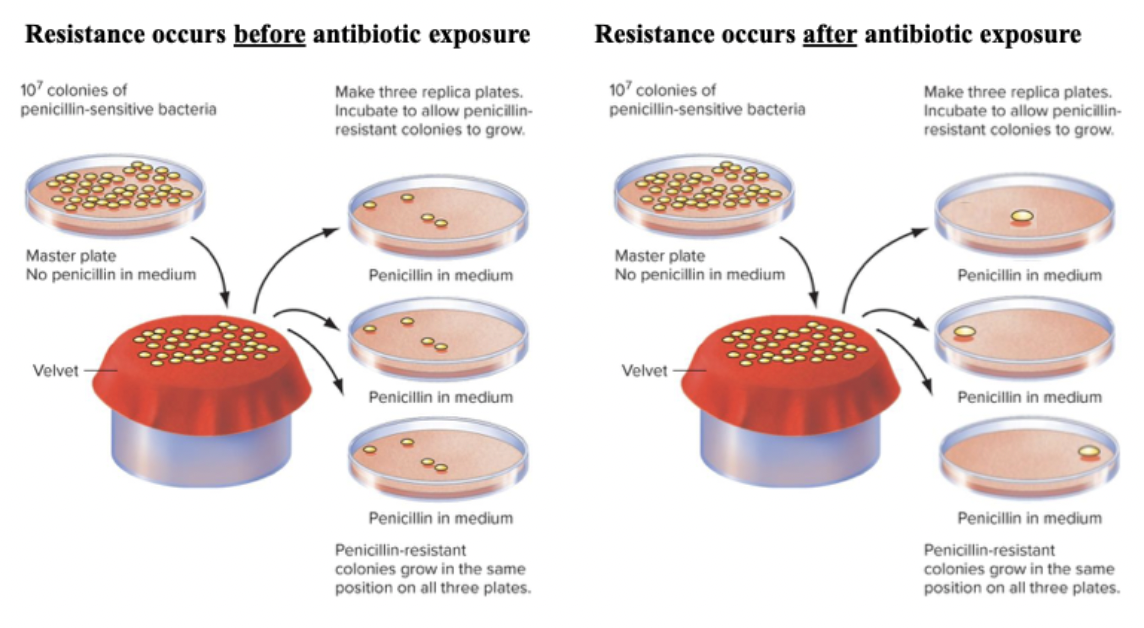
Luria-Delbruck fluctuation test (1943):
Examined origin of bacterial resistance to phage infection.
Majority of cells die, but a few cells can grow and divide.
Two hypotheses for the origin of bactericide resistance.
They grew bacteria in a culture, added bactericide, spread on dish and watched colonies grow. If the resistance is physiological it will form, if mutation is responsive, the population will randomly have the mutation after exposure. If it’s random, mutation will arise early and some colonies may have none.
Results: some petri dishes have more or none resistant. Mutations are random, not responsive.
Lederberg’s replica plating:
You have a master plate with a series of bacterial colonies.
Forward mouse colour mutations: The average rate of one mutation in 2-12 million gametes is variable across genes. Can’t mutate same mouse colour at same rate.
Reverse mouse colour mutations: average rate of one revese mutation in 0-2.5 million gametes. there is a rare reverent.
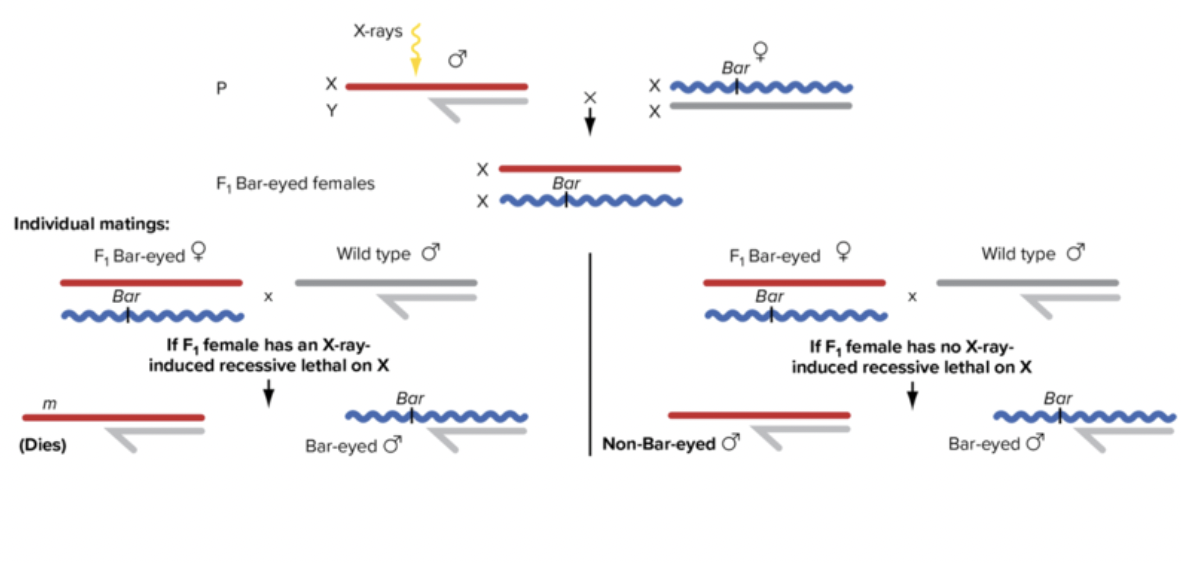
Muller’s lethal tagging test
Evidence that mutagens actually cause mutations
Followed the X-chromosomes
Some new mutations are bound to be lethal. Bar marker. Female progeny have a bar marker. Radiating chromosomes with new mutations. Might not be lethal. Couldn’t uncover non-lethal mutation. You get more with mutagens than without.\
Ames test:
Assesses the rate of reverse mutations in bacteria with and without a mutagen.
His- can’t grow without histidine, they would start growing. Add whatever mutagen and check how many bacteria colonies you get with a functional histidine. Very mutagenic has a lot of bacteria. None only gets some bacteria.
Chromosomal Rearrangements
The three main categories:
Deletions and insertions: a big part of chromosomes being deleted or inserted.
Inversions: happen on one chromosome
Reciprocal translocation: happens on two chromosomes.
Sometimes crossing over happens by mistake, causing chromosomal modifications. Double stranded breaks followed by non-homologous end joining. Illegitimate crossing over.
Deletions:
During repair process, you loss some stuff from the space, might effect many genes
Deletions result in homozygosity → lethal or harmful, depending on size
Deletions may be heterozygotes → have mutant phenotype due to dosage effects, increase risk of phenotype due to recessive mutant alleles. Basically increase risk of recessive allele taking over.
Greig syndrom: intellectual disability
Deletions can help locate genes. Complementation test,
Examine phenotype of heterozygote for recessive allele and deletion → mutant gene must lie in deleted region, if phenotype is wild-type, the mutant gene must lie outside.
We don’t know where a gene is necessarily. Progressively cross all the lines in a gene. As soon as deletion line hits the new region, you’ll see the mutation. Genes in region tell us where a mutation is → deletion mapping.
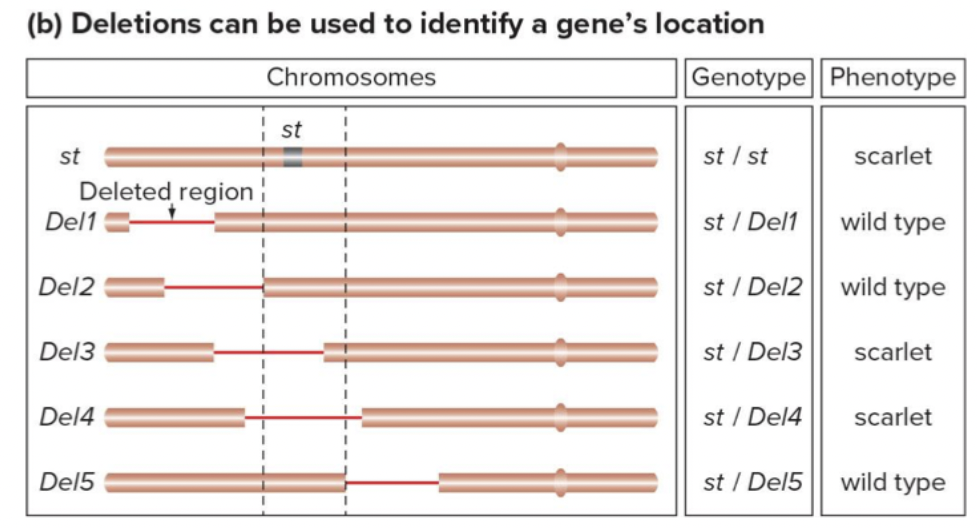
Only work on finding recessive mutations. Coordinate of X chromosome. All red bars are deletion lines.
Deletions help study gene functions. Studying RNA helicase dimers. They need to form dimer to exercise their function in the cell. What is needed to form dimer. When you delete something, what reaction do you get.
Deletion affect gene mapping by recombination frequency: Deletion loop that cannot alight with homolog, results in underestimating difference.
Recombination between homologs can occur only at regions of similarity. No recombination can occur within a deletion loop. Genetic map distances in deletion heterozygotes will not be accurate.
Duplications:
One region of genome is duplicated. Double stranded breaks on both homologous chromosomes. Genes transferred between chromosomes.
Tandem duplications → occur on same side of the chromosomes leg/right next to each other (can be same order, reverse order).
Most common (84% of all rearrangements)
Nontandem (dispersed duplications) → occurs on same chromosome but on different sides/not right next to each other. (can be same order, reverse order)
Phenotypic and genetic effects of duplication: As harmful as having too few chromosomes (triplosensitivity). Whole chromosomes duplication may happen (trisomy).
Microduplications: affect a single gene. all patients had duplication effecting the same gene. Triple sensitivity of the gene.
MECP2 - duplication syndrome. intellectual disability.
Potocki-Lupski syndrome (deletion of same region → Smith-Magenis syndrome). Developmental delay, behavioural issue, metabolic disease.
Where breaks occur in the first place matters. In between two breakpoints - falls exactly on or off the break point. If genes are on breakpoint - some genes are not stitched back together to make whole genes. Inverted duplication may end up with intact version of genes

Unequal crossing over between duplicated regions on homologous chromosomes can result in increased and decreased copy number. Similar to trinucleotide repeats, but at a larger scale. Basically homologous chromosomes don’t line up properly and you have unevenly sized arms.
May increase or decrease copy number. Over time, copy number accumulation ends up in the region. Duplication results in bar eye. Tends to be an unstably genomic region. Double bar. Pair together. Depending on what paits. Reduction of copy numbers in next generation.
Inversions:
Gene flips.
Pericentric inversion: centromere is within the inverted segment
Paracentric inversion: centromere is not within the inverted segment
Inversion can disrupt genes. y+ gene was on a side, got interrupted and flipped.
Paracentric inversions can act as crossover suppressors. In inversion heterozygotes, no viable offspring are produced that carry chromosomes resulting from recombination in inverted region. If you follow line of recombination, you will get two chromosomes where recombination happened. 2 centromere. 1 lacks centromere. Acentric fragment is lost.
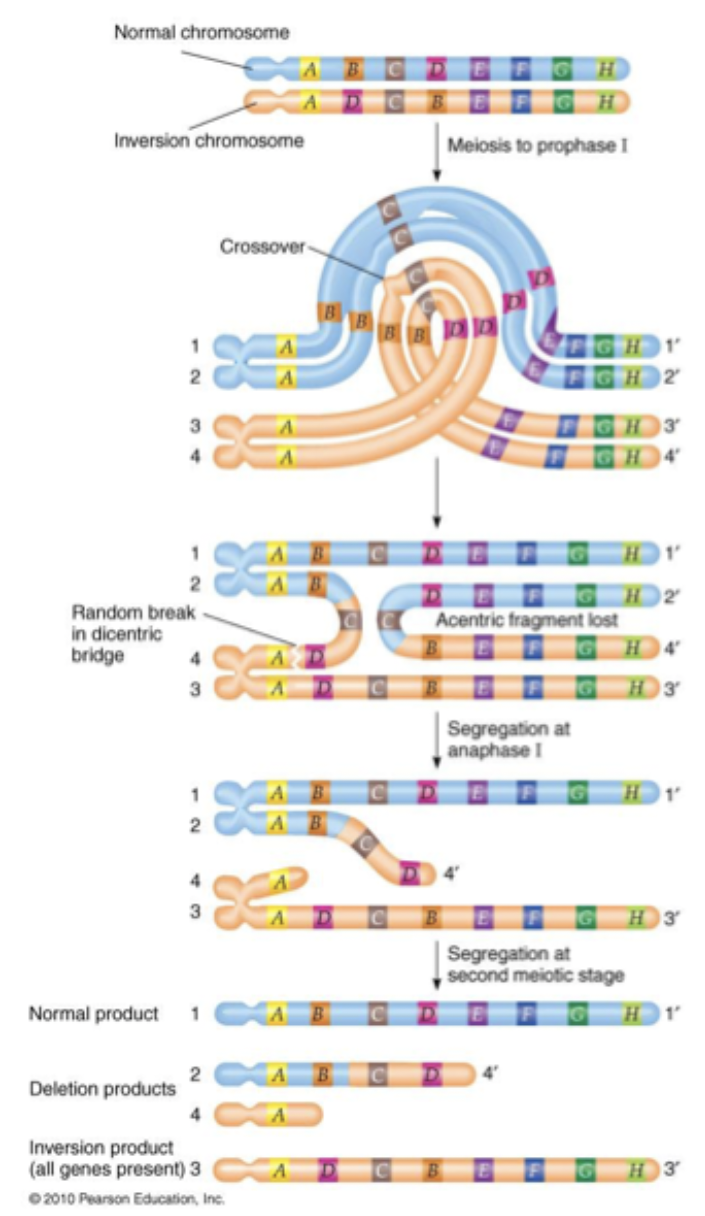
Inversions reduce fertility
Pericentric: Often becomes unviable. Each recombinant chromatid has a centromere, but each will be genetically unbalance.
Paracentric: Is unviable. One recombinant chromatid lacks a centromere and the other has two. Only produce half gametes.
Inversion can be genetic tools
Balancer chromosomes in Drosophila. Have multiple, overlapping inversions. Have a dominant, visible, recessive lethal marker. No viable recombinant progeny will be produced because of crossover. All viable progeny have the normal chromosome unchanged by recombination. Hetero is the way to go in this case of curly wings.
Translocations - two different non-homologous chromosomes. Requires double strand breaks on both. Still have fully complemented genes, just carried through elsewhere. The effect depends on where break occurs.
If the breaks occur within genes, those genes are disrupted. Cancer (somatic tissue), XX male syndrome, Segregation occurs (meiosis). Cell cycle issue - bunch of different lymphomas because of translocation
Reduce fertility - mission some genes. Usually non-viable
Pseudo-linkage: what happens may not be linked on same chromosome because of translocation between chromosomes.
Robertsonian: When you have breaks in 2 acrocentric chromosomes. The chromosome had a very small or big arm.
break near centromere 13, 14, 15, 21, 22 (very small arms)
The small chromosome may be lost from the organism
Down syndrome - 4% of cases of this translocation. Possible recombination of 14/21.
Aberrant crossing-over can cause all four types of chromosomal rearrangements
Somatic cells not doing meiosis when it should be happening. Homologous regions on same chromosome. Interviewing product in the loop.
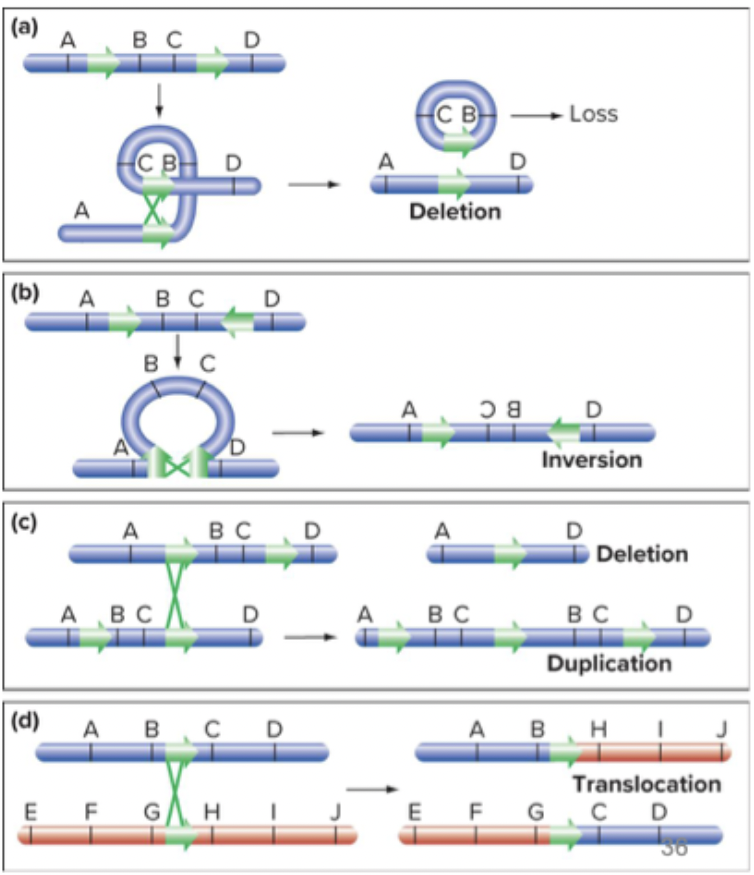
Transposable elements
Aberrant crossing over can be caused by naturally similar genomic region or by transposable elements. → segments of DNA that can move from place to place within a genome.
TEs exist in all organisms and can be present in hundreds of thousands of copies per genome.
Repetitive regions
Discovered by Barbara McClintock - Mottling of kernels caused by TE - the TE elements are very mobile and jump around a lot, spreading.
Retrotransposons: Long term null repeat regions. Within stuff, DNA transposons work differently.
LTR retrotransposons, non-LTR, DNA transposons.
How they jump?
Copy and Paste (ERV and LTR, Non-LTR retrotransposons:
DNA carries transposons inside of a sequence, the enzymes and everything it needs to make mRNA for its business. That transcribed into DNA. Integrated in new location. Copy is reintegrated somewhere else.
Target primer reverse transcription. Transcribed directly in. Result is the same. Small homology region.
Cut and paste (DNA transposons)
Don’t get copied. Homologous region don’t get increased. Excision process is not always very accurate. As you get excised, integrated elsewhere, this may disrupt gene it is integrated in.
SINEs and LINEs in human genome are the most common and are often defective. Deletion of promoter. Carry such a large portion - not active. Fully functional or not. They’re missing parts from other. Can’t jump without any help.
Autonomous TEs: non-deleted TEs that can transpose on their own
Nonautonomous TEs: defective TEs that require the activity of non deleted copies of the TE for movement.
TE insertion can result in an altered phenotype: insert within coding region, insert near gene affecting expression, associated allele may be unstable.
TE can trigger spontaneous chromosomal rearrangement. (unequal crossing over between TEs)
Gene relocation due to transposition. Instead of two sections jumping, everything is jumping. May take a gene with them.
Spontaneous mutation in the white eye gene arising from TE insertions. A lot of TEs have been associated with mutations in drosophila. Slightly different eye colour - different exons and promoter regions.
Alternative splicing of transposase gene limits TE movement: one splice form produces transposase. Other produces a repressor. Repressor competes with transposase for binding to the inverted repeats.
piRNAs regulate TE transcripts → very active, cells limit activity, but they still jump around.
Source of variation:
genes at or near rearrangement breakpoints may have new expression or get fused with another gene
Transposable elements can alter patterns of expression, inactivate a gene, promote illegitimate crossing over.
Duplicated genes may acquire new functions (gene families - genes slightly different function, very related).
Two main themes:
Karyotypes generally remain constant within a species (most genetic imbalances result in a selective disadvantage)
Related species usually have different karyotypes. Closely-related different by only a few arrangements. Correlation between karyotypic rearrangements and speciation.
Chromosome rearrangement can lead to speciation. One population separated into 2 → grew to be unable to produce viable offspring
Somatic recombination
Can be a mistake linked to cancer (illegitimate recombination), in the immune system creating new antibodies, or increasing functional diversity in complex nervous systems (neurodegenerative disease)
V(D)J recombination in the adaptive immune system. 3 of top 10 infectious diseases. Big health threat.
Infectious agents are diverse. How we distinguish us vs. them:
Initial recognition of a pathogen challenge is likely to be through Toll-like receptors. TLRs can recognize molecules on pathogens.
The adaptive immune system is composed of cells that create an antigen-specific memory through antibodies.
In response to an antigen - B cells are triggered to make antibodies (immunoglobulins).
through generating diversity, VDJ in form of IgH. Two families of light chains. Each chain has an N-terminal. Responsible for antigen binding. After antigen binds, elicits following response. Also a C-terminal. N recognize, C mediates.
Igs are heterodimers

Heavy chain locus, longest one. Exons and introns. Light chain.
These different regions in the genome, can be divided into D and J region. What happens in the germline is that you get random recombination. Different amount of exons. Our bodies manage adaptive immunity without.
Recombination between variable (V), diversity (D), and joining (J) regions generates over 15 million possible combinations.
Imprecise recombination and mutation increase the variability into billions of possible combinations.
V(D)J recombination initiation → RAG-1 and -2. Generally expressed DNA repair protein then carry out the joining reaction.
Illegitimate recombination happens in human tumours. Happens more often. Lymphomas originate through translocation. Make fusion proteins. Fusion proteins is constitutively activated. Chronic myeloid patients. Misfunction by machinery does somatic recombination. May have no effect until different translocation.
Sometimes additional mutations exacerbate the effect of a translocation (fibroblast growth factor - cell division regulator).
We share 88% of our genome with fruit flies. Somatic recombination in the nervous system may have an important role. Amyloid plaques - seen in alzheimer patients. Accumulating outside cell, block neuronal function. Variants arise through somatic recombination within the nervous system.
Alzheimer patients have a big variability gene → Non-functional present in all.
Most out-turn of gene products. Many different exons with many different introns. Drosophila feels important. Way of maintaining size, adding functional diversity. APP exercised similar to splicing, through genomic recombination. Recombining, if you make it, might result in different version of APP. expereince change neurons permanently. Linked to activity. what makes cells fundamentally different.
DNA Sequencing
Polymerase - polymers made out of monomers, need a free hydroxyl group. Need triphosphates (dNTPs).
Everything is extended basically through primer extension. Raise temperature until DNA denatures, as temperature lowers, the primers anneal with template. Primer then gets extended using polymerase.
Sanger sequencing: Chain termination method. You have strand and gene of interest, did the denaturation and primer is attached. Add polymerase (dNTPs) and then ddNTPs (missing OH group). Oxy-bridge cannot form and chain terminates. the ddNTPs are fluorescently tagged. We only know where the terminal base is. Nested fragments. Smaller fragments run faster. Gell is designed to tell 1 BP apart. Get 5’ to 3’ sequence. 3’ ddNTPs. Smaller fragments close to the 5’ section of the gel. A computer can detect the lights. Each land displays the sequence of one DNA sample + primer. The gels are scanned and transmitted into computer - detects the lights.
Output: sequence complementary to the template (read 5’ to 3’). Chromatogram, text readout of sequence ATGC.
We run trace (chromatogram) on multiple wells. Stacking multiple times, slightly different results. Slightly variable reads. Blue bars are high coverage. At no point do we not know the sequence. Gels should look very neat.
Small fragments may be hard to distinguish - accuracy of gels lessens.
Epstein Barr: didn’t know anything about the genome before sequencing it. Use to inform how we put sequences together. HGP was very expensvie - 3 billion dollars used Sanger sequencing. First time we could look at genome in and out.
1980s it blew up, and is still used today
Benefits:
Highest fidelity of sequencing method
Reads can be up to 900 bp long
For many smaller applications, it’s cost effective and practical
Issues:
Sequencing is slow for genome / expensive
requires initial mapping and scaffolding - then more sequencing to fill gaps.
Bad when less data available, and you need to fill gaps.
Genome sequencing - realistic process is much easier. We want to compare different species, we can’t use Sanger for these scales.
Shotgun sequencing - used to help sequence the human genome. Principles are that it breaks genome into overlapping fragments. Sequence lots of fragments randomly, assemble sequences base on overlap to form contigs.
Contigs: contiguous sequence - output of sequencing is reads. You don’t go from a read to a genome. You overlap and assemble this and try to close the gap. Produce a whole genome.
Issues with repeat regions - low selective powers - lose some repeats.
Paired-end sequencing: sequences two DNA reads separated by an insert of a known size. Reads + inserts = fragments
Next-generation Sequencing
Whole genome broken up and sequenced at once. High-throughput - generates a very high volume of reads. Based on sequencing by synthesis. Engineered polymerases - addition of dNTPs.
Illumina sequencing:
Sample preparation. High quality check - for purity concentration of DNA to work (using photo spectrophotometer). Adaptors with speciific sequence added to each end. Flowcell adaptor growing like grass on a lawn. First converted to a single strand
Cluster generation. Fragment of interest wasted away. Forms a bridge, connecting with its opposite. They match up all complimentarity.
Sequencing. Sequencing is repeated for the reverse of the fragment. Tagged dNTPs. Identity of last base pair added is noted until the read is done. Sequence denatures - see it on the computer. Now flip.
You end up with detected, paired up reads. Early version. Want to sequence specific example. Reads far away. Paired reads, known inserts.
Benefits of next-gen sequencing
High volume of reads
Much lower per-bp cost
Less time- and labour-intensive
Issues with next-gen sequencing
Higher error rates. High FNA concentration and purity needed.
Errors with assembling longer repeat regions (shorter regions)
Third-generation Sequencing
PacBio sequencing - owned and run by pacific bio resources.
Nonpore large scale by themselves.
The read lengths are super long and very important.
SMRT Sequencing (PacBio): very long fragment or could be whole prokaryotic genome. Must be fairly high quality. Adaptors are added on → create a circular molecule. Single molecule being sequenced at the end of the day. After denaturation, it’s synthesized all at once repeatedly to get many reads. Output is fragmented.
Channels - ends have a DNA polymerase that is anchored to bottom of channel. Fluorescently tagged dNTPs. Detective only able of seeing fluorescent. DNA polymerase goes through single molecule. Get an output. Physical stategy.
Reads are about 5 kb in length, up to 25 kb.
High fidelity (>99.9% accuracy)
Fewer reads than Illumina sequencing. Easier for assembly. Get less reads overall.
Nanopore sequencing (ONT): passing DNA through a membrane pore - hard to actualize and do practically. Artificial membrane. All tiny pores. Enzyme captures DNA from 5’ end, unzips it - forces single strand through. Maintained across the channel.
Batches of based take different amount of time to pass. Amount og time is an addition point of data.
Benefits:
Can sequence native DNA or RNA
Generates ultra-long rads (10-300 kb is routine, up to 4Mb)
Issues:
Higher error (improving)
Needs careful DNA extraction and library prep
More expensive per-bp compared to short-read sequencing
Using sequence data
PHRED scores. Quality of each base pair. Done according to logarithmic score. Log(E) probability of being error. High quality score - lower chance of error.
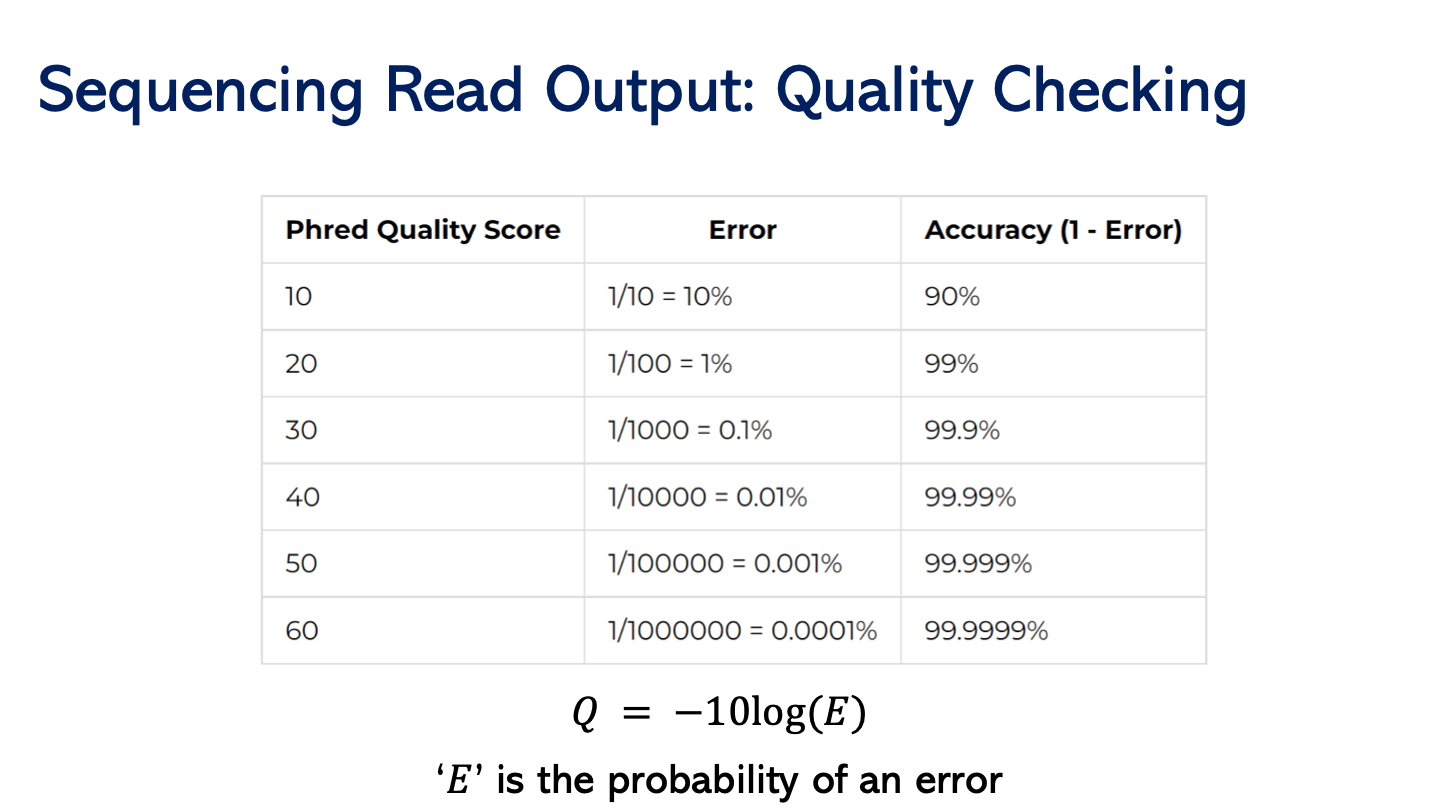
Low quality scores cannot be trusted. Replace base pairs of low quality. It can tell us if it’s likely G.
Assembling reads (reference assembly): you can assemble data with or without reference. Known genome has previously been sequenced - read output. Align reads by consensus. Consensus sequences. Joined to be continuous, assembled into whole genome. Overlapped reads are joined to form a consensus sequence.
de novo Assembly: assembly from fresh, without previous data. Algorithmic approaches. Ways of arranging big data. More likely outcomes. Know reference. You compare reads to one another. Using complex algorithms. Create into contigs. Get scaffolds. You don’t need reference but it takes longer and it is less reliable. Assemble reads using complex algorithms generating de Bruijn graphs to create contigs.
Annotation: identifying genes and other genomic elements. Compare to gene and genome sequences uploaded to public repositories
Comparative analysis: comparing genes and genomes from different taxa. Classifying homologous genes.
Orthologs: gene variations created by speciation events
Paralogs: gene variation created by gene duplication events (often form gene families)
Comparing genes in the presence and absence in different species.
Comparing RNA: Find what genes are present, but mainly what genes are expressed. RNA sequence data can be used to infer differential expression of genes or gene ontology.
DNA sequencing tach has allowed us to explore the living world around us much more thoroughly, has led to the advancement and creation of fields of science, the advent of personal genotyping, the advent of the personalized medicine movement.
Ploidy and Molecular Genetics
Ploidy
Euploid: whole set of chromosomes, could be 1n, 2n, 3n, just depends on the species
Aneuploid: mission one or more chromosomes from a set, or you have extra chromosomes
Loss or gain of one or more chromosomes
Euploidy = 2n
Nullisomy = 2n - 2 (lost homologous chromosomes)
Monosomy = 2n - 1 (lost one chromosome)
Trisomy = 2n + 1 (gained a 3rd)
Some organisms are haploid - many eusocial species are haplo-diploid species → bees are female diploid and male diplo-haploid,
→ plants, fungi, and algae can switch between haploid and diploid stage (alternation of generations). Switching between, not like gametes or grown organisms are different, but fully grown organisms are different. Grow into haploid plants.
Some organisms are polyploid - variation very rare in mammals, more common in reptiles. One rat has too many damn chromosomes people can’t tell if its tetraploid or not.
Variation may occur due to duplication. May be lethal or be speciation who knows. Rapid period of evolution. Two rounds of whole genome duplication in the vertebrate lineage leading to humans (but polyploidy is not viable in humans).

n is always half. When 6x = 42, n = 21.
Creation of ploidy
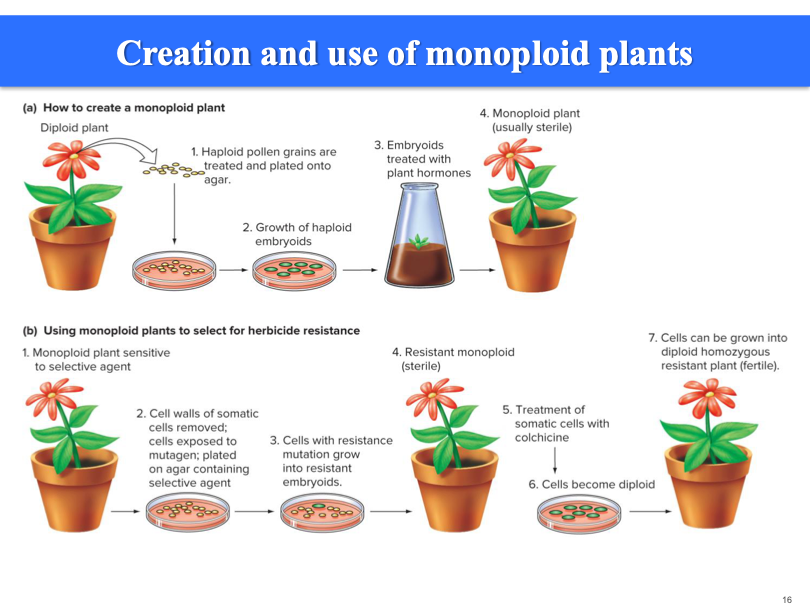
Trying to create a plant with resistance to a herbicide. Plants are usually sterile, but can be used to help pick and put together. Colchicine: spots plants from dividing again. Make the cell diploid again.
Formation of triploid organisms: while a diploid results in a union, triploid plants result from the union on monoploid and diploid gametes. Diploid gametes may arise from 4x parent or diploid with meiosis error.
Meiosis in a triploid organism is weird. Almost always unbalanced. Fertilization with gametes from triploid organisms does not usually produce viable offspring. Meiosis in a triploid organism that produces balanced gametes. If x is small, occasionally a meiosis will produce balanced gametes by chance.
Generation of tetraploid cells: tetraploid cells occur during mitosis in a diploid when chromosomes fail to separate into two daughter cells. If tetraploidy occurs in gamete precursors, diploid gamete are produced.
Union of two diploid gametes produces a tetraploid organism. Not a big deal cuz they pair in an even way. Pairing of chromosomes as bivalents generates balanced gametes. 4 copies of each group of homologous pair two by two to form two bivalents. 2x gametes.
We usually see mendelian genetics in tetraploid anyways - 1:2:1
In agriculture, 1/3 of all known plant species are polyploid. Increased size and vigor. Many polyploid plants have been selected for agricultural cultivation.
Autopolyploid: all chromosome sets are derived from the same species
Allopolyploid: hybrids in which chromosomes sets come from distinct, but related species
Amphidiploid: has two diploid genomes, each from different parental species. Two fully different parents. Hybrid of wheat and rye.
In the rice genome, many blocks of homologous gene sequences are found on two different chromosomes. Two copies of genes are not identical.
Circus plot: edges has different chromosomes - lines are connecting regions of homology - same on different chromosomes. say s most likely have duplicated chromosomes. Through translocation - separated into different things. Used to study and identify in evolution studies.
Molecular biology and recombinant DNA technology
Hybridization techniques
hybridization: pairing of complementary sequences.
Hybridization techniques are based on homology. Based on designing probes. Either DNA or RNA. Probes are complementary to specific genes. Manufactured by attaching dyes. Need to know DNA sequence targeting ahead of time - can’t manufacture something complementary otherwise.
In Situ hybridization: multicolour banding produced by using FISH probes specific for regions of chromosomes. you can see large deletions. Detect chromosome rearrangement with this under a microscope.
Short hybridization probes can distinguish single-base mismatches. Hybridization of short (<40 bases) oligonucleotides to sample (target) DNAs (Allele-specific hybridization)
Mismatch - hybrid will not be stable at high temperatures
Probes are usually used in microarrays for genotyping. Allele-specific oligonucleotides (ASOs) are attached to a solid support. Specific SNP that increases diabetes for example. Probes one with SNP and one without.
Genomic DNA used to probe the ASO chip (microarray).
Preparation of genomic DNA - fragmented, adapter attaches, amplifies by PCR and denatures to make single stranded, fluorescent dye coupled to end of single-stranded DNA. Extract DNA or RNA and put it on the chip.
Anneal with the chip it’s matched to. A lot of loci can be genotyped for just $100. Identify where SNPs are with the fluorescent signals.
Microarrays are used for 23andMe kits. 640K SNPs used. Ancestry uses more.
Admixture: estimating ancestry from SNPs → Looking at disease alleles, most likely to come from one population, so you’re likely from there. Can be difficult to tell though using percent likelihood if they’re too close.
Inferring health data from SNPs (BRCA1/BRCA2): Gene known to increase risk of breast cancer, doesn’t test all variants though. Keeps checks on mitosis. Without checks → many mutations could develop. Shows increased risk, but not much else.
Risk =/= disease; big limitations on inferring. Genome-wide association pretty much every genomic population study. Those with A have and those with C don’t - may be because of them coming to a more related population just by chance.
Ethical consideration of who owns data and who can use it. Should they be allowed to withhold health information for paid subscriptions.
Cloning
Replicating a fragment of DNA so that there is enough to analyze. Plasmids remove the gene. Marker and anti-resistance gene, allows us to see the air in some way (selectable marker - something that allows us to detect what is there).
Cloning general principles:
Isolate DNA of interest
insert into vector (plasmid, bacterial artificial chromosome, yeast artificial chromosomes)
Insert vector into cells (use non-pathogenic E. coli)
Select and grow transformed cells.
Plasmids have landing sites. Origin of replication, marker gene, and landing site for cloned fragment.
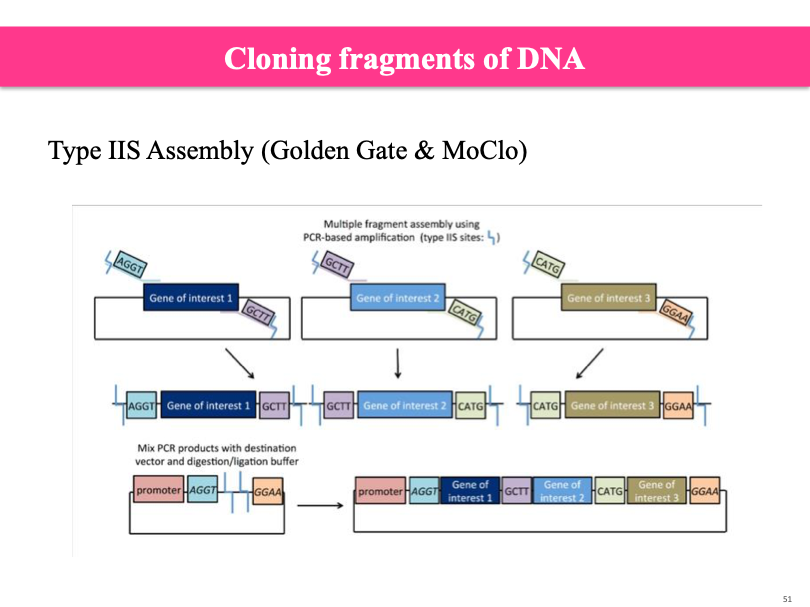
Cloning fragments of DNA:
Cloning used to be hard and done with restriction enzymes. Added sequence of restriction site/enzyme. Overlap of two restriction sites on either end. Need make cells and amplify them.
Gateway cloning - relies on base-pairs and LR clonase. With primers, you now add. Donor vector gets flipped. Flipped into vector.
TOPO cloning: Comes with a backbone that is part of a kit - cut it up. Backbone keeps the rest of it together i guess. You cut out the PCR product.
Gibson assembly cloning:
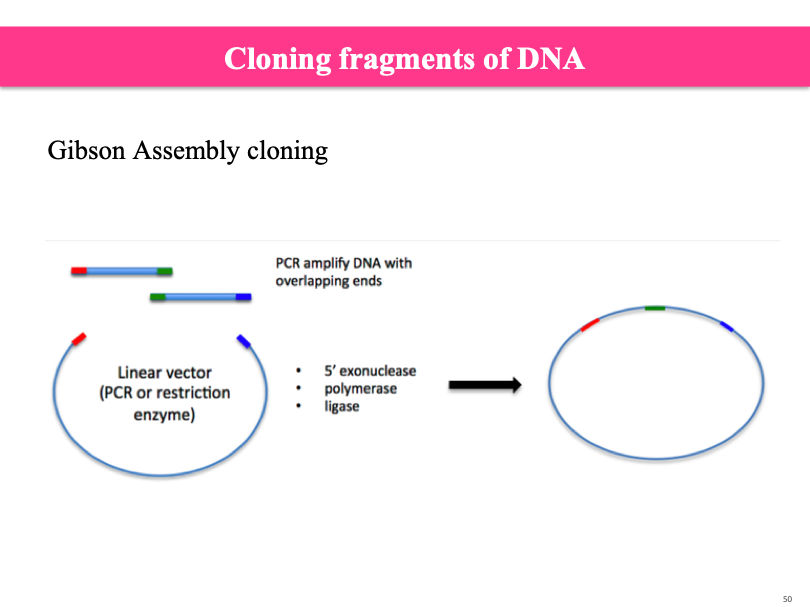
Cloning for whole genome sequence. You need a lot of DNA ot do it (like with Sanger sequencing)
E.coli - only 0.1% of cells will be transformed with plasmids.

Like McDonald’s monopoly. Why sequencing becomes so fundamental. Cloning is still widely used to make transgenic organisms.
Genomic libraries made sequencing of many animals possible. Genomic libraries became mostly obsolete with next gen sequencing. Cloning still widely used to make transgenic organisms.
Transgenic organisms
Contains a transgene from another individual of the same species or a different species.
Clone transgene → introduce transgene into the genome of the germ cells → select for transgenic individuals
You can integrate foreign DNA in an organism by injecting plasmids without genomic integration. Works for yeast and prokaryotes. Eukaryotes need integration into the genome.
You can also inject transgene into DNA followed by random genome integration.
Fertilized mouse embryo before the cells fuse. Put injected embryo into female again. When mated, offspring should have it. Very inefficient.
You can also have P-elements mediate genome integration of plasmids. Excise and integrate into DNA. Separate two components of the P-elements. If you separated two regions and put gene of interest in between, transposase gene will insert into genome. Transposase gene not being here anymore.
You can also have viral vector or agro-bacterium mediate transgenesis. Help a plasmid that has genes needed to integrate, then have what you want to introduce, flanked by region of homology. Naturally infect cells.
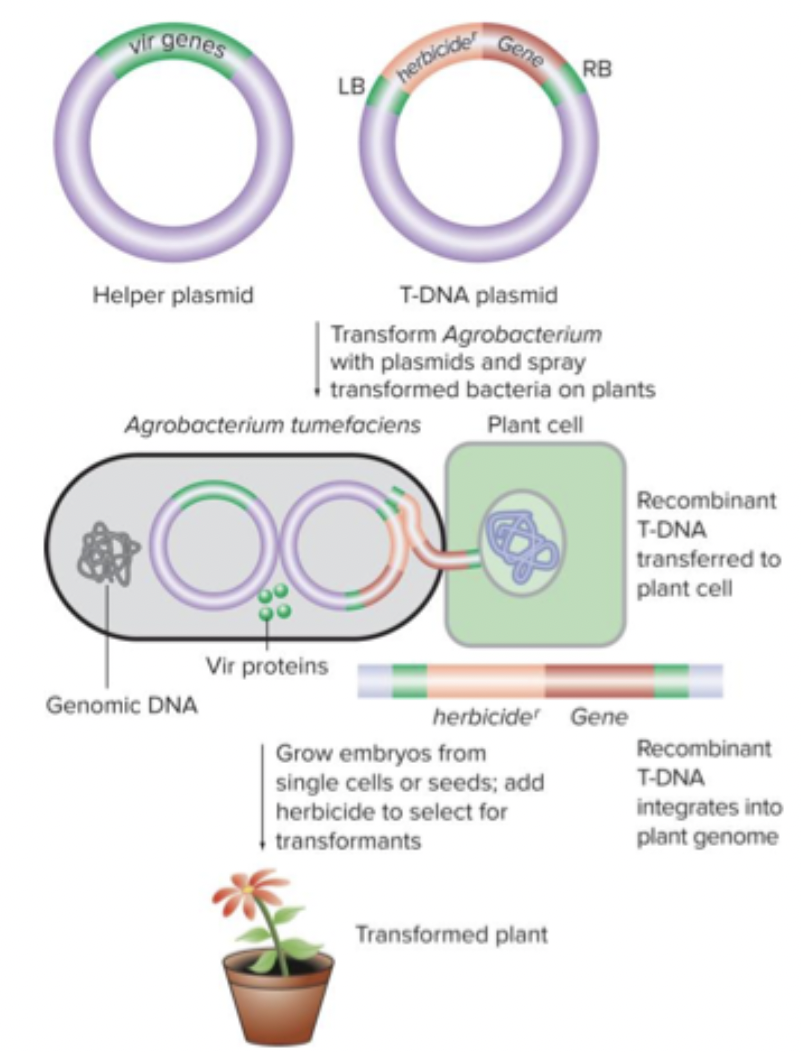
Targeted mutagenesis: Add regions of homology to whatever you’re cloning, target different region of genome you’re trying to target. Gene targeting with homologous recombination. CRISPR (predominant).
Spacers in the CRISPR locus of bacteria are fragments of bacteriophage genomes from previous infections. Viral genome is recognized by specific sgRNAs generated from these CRISPR sequences; targeted for destruction.
Cut it up (double strand break), then introduce Cas9. Donor sequence - has homologous region, broken DNA, will introduce during repair process.
Transgenic organisms can help with connecting genes and phenotypes. A mutation that deletes two genes results in malformed Drosophila eyes. Which two genes are involved? Restore the genes - two genes, one in each, and decide which gene is affecting eye shape.
Conditional knockouts: cross-over caused by protein to overcome lethality. Knockout (kills mouse after a generation because it could only delay lethality or something idk).
Transgene → report genecan investigate gene expression, reporter constructs used to identify enhancers, introduced into eukaryotic organisms as transgenes.
Transgenic mammalian cells in liquid culture is used to make pharmaceutical proteins.
Pharmacy can use transgenic animals and plants to produce human protein drugs.
BioSteel - goats produce golden orb weaver spider silk protein in their milk. 20 x as strong as steel, very stretchy and highly resistant to low and high temperatures.
Somatic nuclear transfer used to create reproductive clones. Diploid nucleus of a somatic cell from one individual inserted into an egg cell whole nucleus has been removed. Made Dolly and CC (demons/abominations).
GMO crops are widely used. Round up, corn Bt protein. GM salmon in 2015.
Concerns:
a few large agricultural companies have all the power. Farming communities may be disrupted. Potential environmental consequences, such as transfer of transgenes to wild organisms.
Phenotypes don’t always recapitulate well. FGFR3 gene in mouse with same mutation as humans. just makes mouse really small and weak limbed.
Gene therapy to cure disease:
Disease caused by loss-of-function → add wild-type copy of gene
Disease caused by gain-of-function → therapeutic gene must inactivate disease gene or protein product
Disease with complex origin → target gene for a genetic pathway involved.
In vivo gene therapy: therapy delivered to somatic cells in the body (Ex. injected into retinal cells or inhaled into lungs)
Ex vivo therapy: cells removed from body, treated, then put back in (Ex. bone marrow cells).
We’re able to treat SCID-X1 (severe combined immune deficiency) (ex vivo modification of bone marrow cells), congenital blindness (recombinant AAV vector injected into retinal cells), repair sickle cell allele with CRISPR/Cas9 (red blood cell precursor removed or express fetal hemoglobin in patient to prevent mutant cells sickling).
CRISPR baby controversy. CCR5 gene deleted (a gene that allows HIV to enter the cell) - Unexpected adverse side effects. Gene editing is dangerous without complete understand of all the effects/interactions of a gene.
Gene Regulation and Adaptation
A primer into gene regulation
Gene: a region of DNA that encodes a biologically functional molecule
Gene has been getting a broader and broader definition recently. Genes make molecules out of RNA and also protein. Does a massive amount of things as RNA.
miRNA: block mRNA from becoming proteins
Genes have specific features oftentimes. Transcribed but not translated. Regulatory region. Cannot be mutually exclusive - introns control what’s there. Coding region part of gene that makes useful molecule. Regulatory region part of the gene that controls how much of the protein is made. Genes can also encode for functional nucleic acids, called non-coding genes.
Stuff interacts with your DNA leading it to be something. Hox gene cluster - humans have hox gene cluster. Genes have duplicated. Fundamentally the same group gene.
A gene is “on” if RNA-polymerase is walking on it, making RNA from the DNA. A gene is “off” if RNA-polymerase. Fundamentally genes are on or off.
How to be on or off:
DNA accessibility
DNA needs to be sufficiently unwound. Of course your DNA is tightly packaged. If your gene is going to be transcribed, DNA needs to be unwound.
DNA wrapped around nucleosomes, become wrapped to chromosome. Piece of chromosomes needs to loosen. Heterochromatic (tight state).
RNA Pol-II physically can’t access gene (gene inactive) vs. physically can access gene (gene active)
DNA methylation is simple. Covalently bonds to base pair, more difficult for protein to bind (not always the case), but most often.
Methylation: A small molecule that binds to DNA, making it (usually) harder for other molecules to bind to DNA
Transcription factors
A protein that lives in the nucleus, can bind to DNA, and can attract or block RNA polymerase II. Interact with DNA, recruiting RNA polymerase.
A gene may want multiple TF binding sites to interact with the environment. For gene to be regulated, they need to do some stuff. Protein will say okay. Transcription factor insulin response - will bind in response. You need regionalization and responding to insulin. With all three - the gene will still turn off. Repressor is a little bit dominant I guess.
These components of gene expression let us make specific cells in a dish. We’re able to use these principles to make specific human cell types in the lab. These heart cells are our natural pacemaker.
Complex traits and the “Omnigenic” model
Complex trait is not necessarily complicated in whatever, but in the numbers. Lots of genes associated, environment is also a big issue. Genetics and environment play a dual role. If you only take genetic factors alone, you can still only predict 10-40%. G x E Interplay - Aa affected by nutrition.
The genes and environments factors in a complex trait communicate through gene regulation. Pre-transcriptional - what is binding, methylation status, are transcription factors themselves showing anything. Post-transcriptional - what is being shown, introns or exons involved. mRNA should be stable. Genes are regulated based on “variable on”
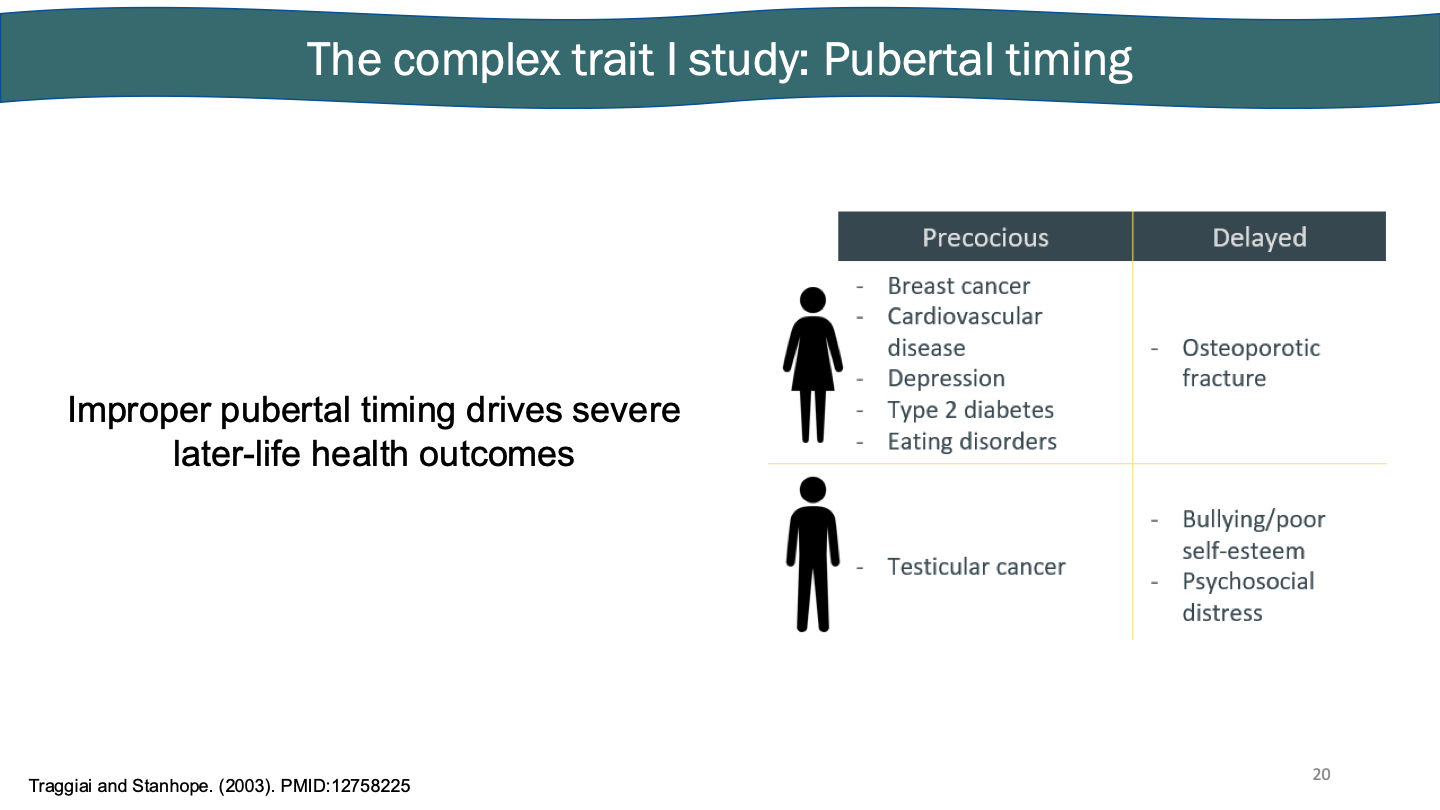
The complex trait I study: puberty and the hypothalamus
Powerful way of looking how genes interact
Puberty is considered the start of the hypothalamus
Negative regulation of estrogen, pituitary gland responds.
Feedback loop of hormones showing stuff.
Hypothalamus → GnRH → anterior pituitary → LH, FSH → Gonads → sex hormones
Pubertal timing: highly veritable (50-70%). Conserved signaling and regulation. Hundreds of associated genetic factors. Hugely environmentally sensitive. Puberty serves as a model to study gene regulation and plasticity in a non-disease context.
Improper pubertal timing drives severe later-life health outcomes
Omnigenic model
Lots of cell types, we’re trying to get it to all happen. Transcription factors - directly interacts, indirectly important. Peripheral - likely interacting with 100s of genes. Really important regulatory gene on core gene. Disease studies can show what is important.
Many cell-types (neuron, glia, etc.) work together to produce a complex trait. Many genes work together to make sure each cell is working properly. Some genes are way more important:
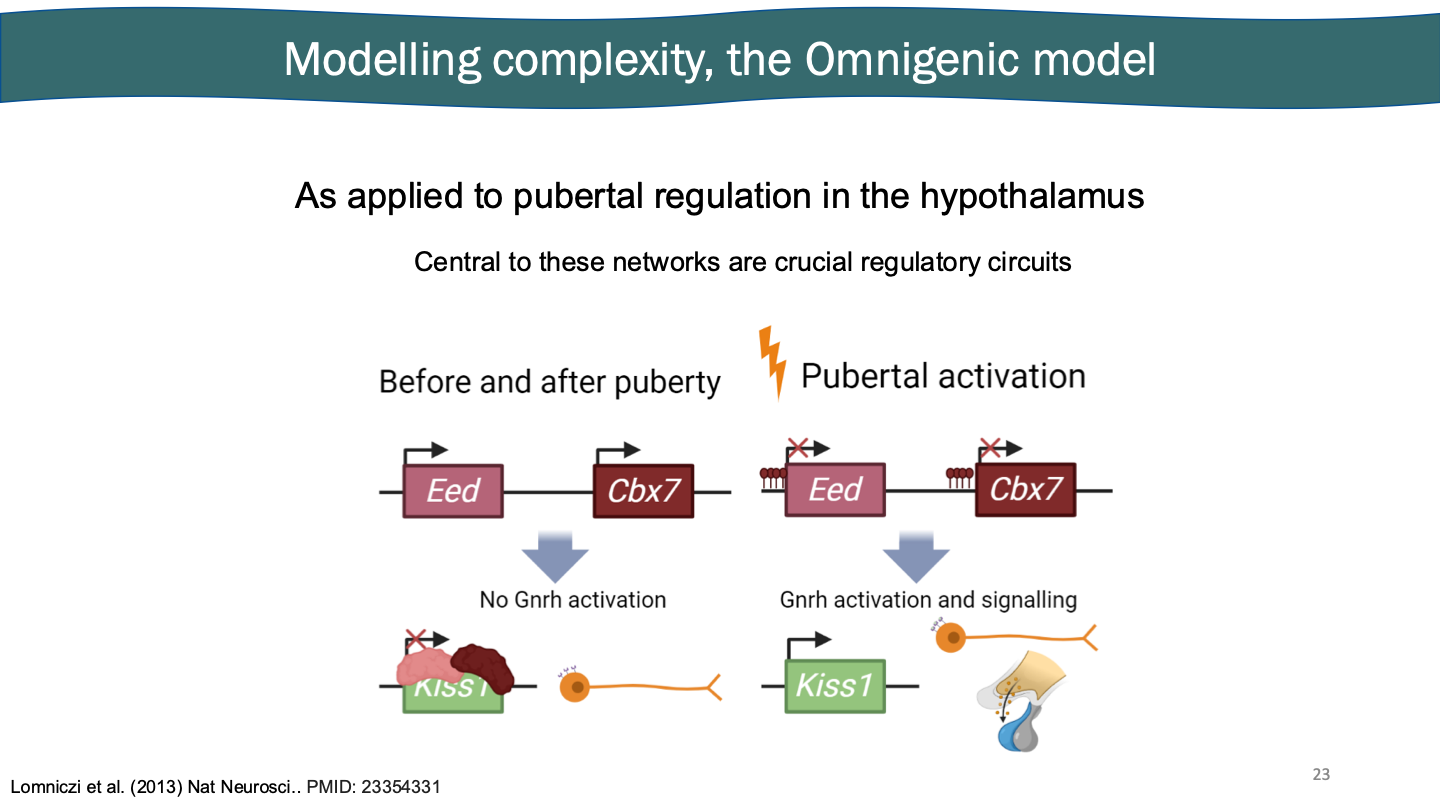
Core: the genes that make the stuff for your trait. puberty - Gnrh1
Regulatory: the genes that control the expression of your core genes. puberty - TF that binds to Gnrh1
Peripheral: genes that function downstream of core- and regulatory- genes. puberty - another gene influenced by (2)
Modelling complexity, the omnigenic model
As applied to pubertal regulation in the hypothalamus: hormones work as a signal and receptor. Might be considered a core gene (estrogen). There are three steps before puberty gets started. Found in female rat
Multinomics
In omnigenics you have to measure a ton of things to truly understand. You can order things into a slightly less complicated structure. Through all the uses, humans may have 22-55k genes. Still very complicated. We can sequence more than just DNA.
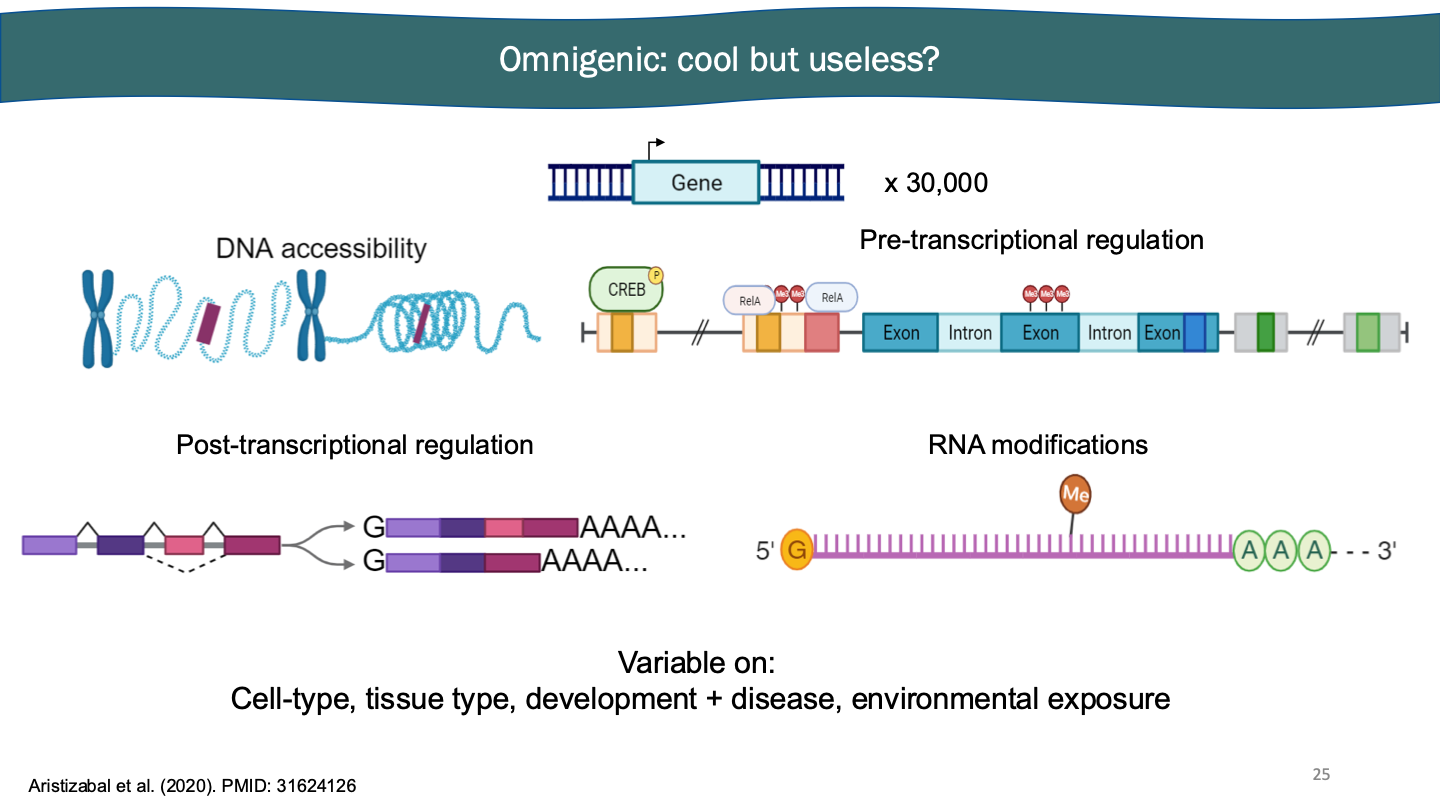
In multinomics we can sequence more than just a whole gene - really finding the focus. Sequencing technology that has been created. Pull out transcription factor with sequencing. It’s DNA sequencing. But instead of doing everything, you’re choosing specific area.
What is XXX-seq?
Selecting DNA (or RNA) molecules that you are interested in before performing a sequencing protocol you have learned
Illumina sequencing (most common), nanopore sequencing, PacBio sequencing
How much RNA abundance is there, use reverse transcriptase. Amplify, then normal sequence. Put in cells, open chromatin. Primers attracted to transposase. Pull out DNA and then sequence.
RNA-seq genome-wide transcriptional abundance, ATAC-seq, ChIP-seq.
Multi-omics: we can sequence more than the whole genome. Main histone consistent, but tails have different covalently bonded molecules. Enhancers on own, promoters on own. You can get promoters to act as enhancer. Cut and run you pull out and eat it. ChIP-seq you eat it. Covalent bonds attached to histones are extremely highly correlated with how that part of the genome is regulated.
Working on integrating to create the image. Reverse engineer to see what is most important.
Multiomics - measure and evaluate each layer, then integrate to recreate the main image
Genomics of extreme adaptation
Elephant has more cells than mouse, but elephants live an extremely long time and don’t get cancer. Their cells aren’t more likely to die or whatever since there’s more. Annotate genomes (need to know where repetitive genes sare). Adaptations in biologically relevant genes and pathways. Protocadherin gene copy expansion.
Sugargliders are able to glide. Flying lemur lost or gained ability to fly independently. Distill trait from the species. Compare bats to other rodents.
Big cross species - find a hit, look within. EMX2. Shot them up with gene, and showed a different in traits and how it developed. Within-species design. Mouse transgenic design → develop limbs and lose them.
They were able to take comparative, multi-omic approach to find a candidate gene involved in wing development
They used gene-treatments in their animal models to show they found a “core” gene in their species, they used used transgenics to show that the gene is likely “core” in mice, an independent species.
While a massive success story, but their study was limited to coding genes.
Orientation is very sorted out. Elephants have whatever - you need long assemblies.
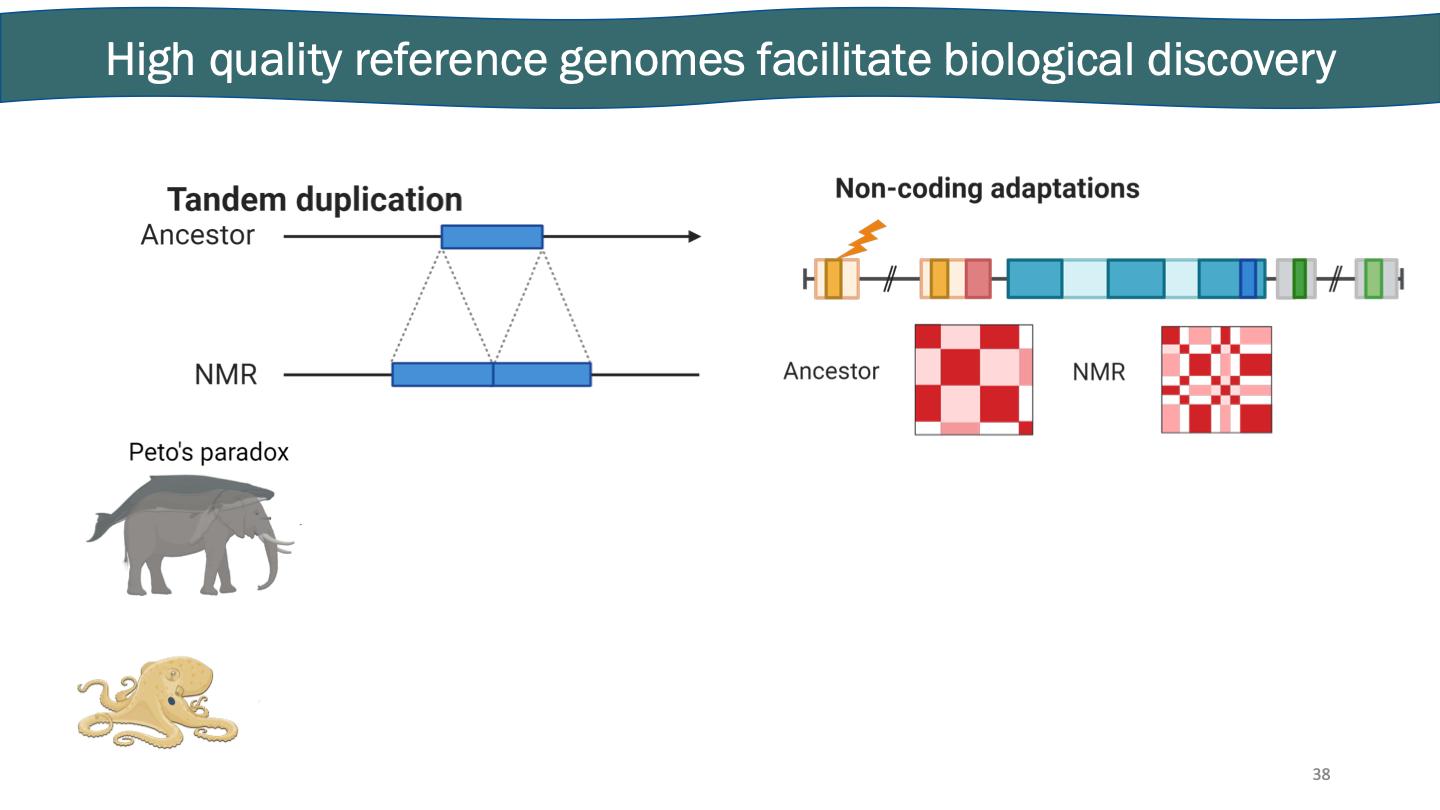
Genome assembly technologies recently made a massive leap. The human genome was finished 2 years ago. Ultra long protocol goes through once and gets tired. HiFi reads can help show the preciseness.
These technologies are generalizable to any species. ONT ultra-long reads and PacBio HiFi reads.
The naked mole-rat
They have the potential to go through puberty, but may just not. Puberty is delayed indefinitely. If you put an attractive female nearby, puberty speeds up. Only a few traits have a specific identified trait. Quality of genome of naked mole rat.
NMR has 4000 contigs. Human DNA is about 50 percent repetitive, as is NMR. 6000 bp long. 20% of your DNA is a specific repeat. Insanely difficult to do shit.
NMR genome assembly was done with long reads and trio-binning. Genome assembly and fazing. Individuals (parents) all have 1000s of structural variations in genome. Queen has 6kB. Genome assebmly craft will get very confused. Both are true - in reality the sequencing must have screwed up, and they start doing weird things.
FISH karyotype → allows us to anchor our assembly to physical regions on chromosomes.
In silico karyotype → Fluorescent karyotype. In metaphase, genetic markers tag. Most likely to get rearranged are coloured. Structural origin of our shit compared to theirs.
The naked DNA sequence. Must annotate a genome. Analyze DNA sequence, collecting functional data. You don’t know if it’s gene B, but it probably is just based on how evolution works.
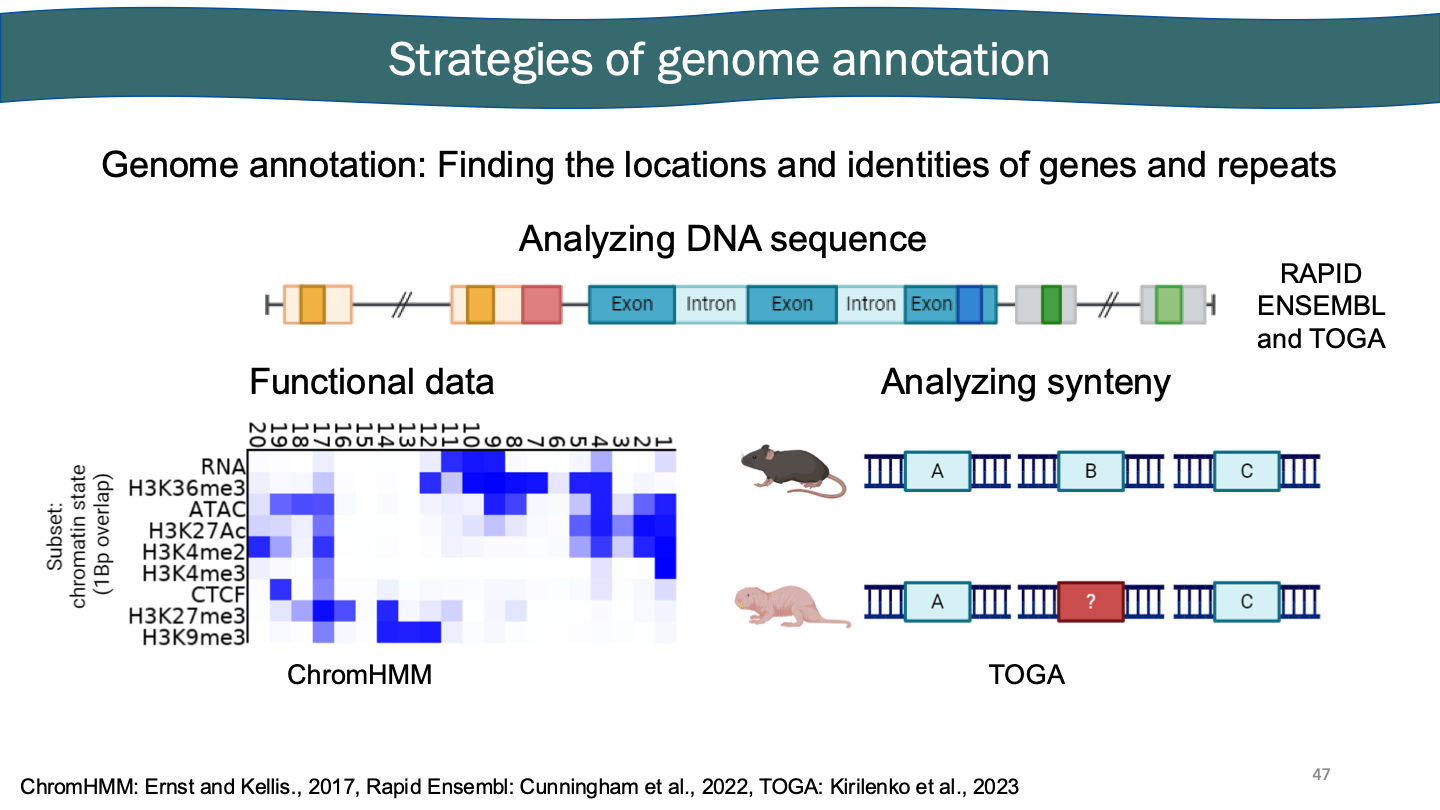
Many genes are important - between species that don’t disappear. RNA sequencing. RNA maps to genomes. Probably a gene. Go back to DNA sequence. Could they possibly make a protein. Have we seen it before, is it species specific?
Gene actively being turned on or off. Taking all this time to activate in different cells. Genome helps us do functional experiments.
Having a good genome can reduce errors in multi-omic experiments → maternal and paternal long read assembly. Tiny bit of DNA showing there’s similar, big repeat couldn’t be resolved in short read, so all DNA aligned to genome ends up in weird area. Often quite responsible for changes and add up. May see peaks, but it’s just messed up genome.
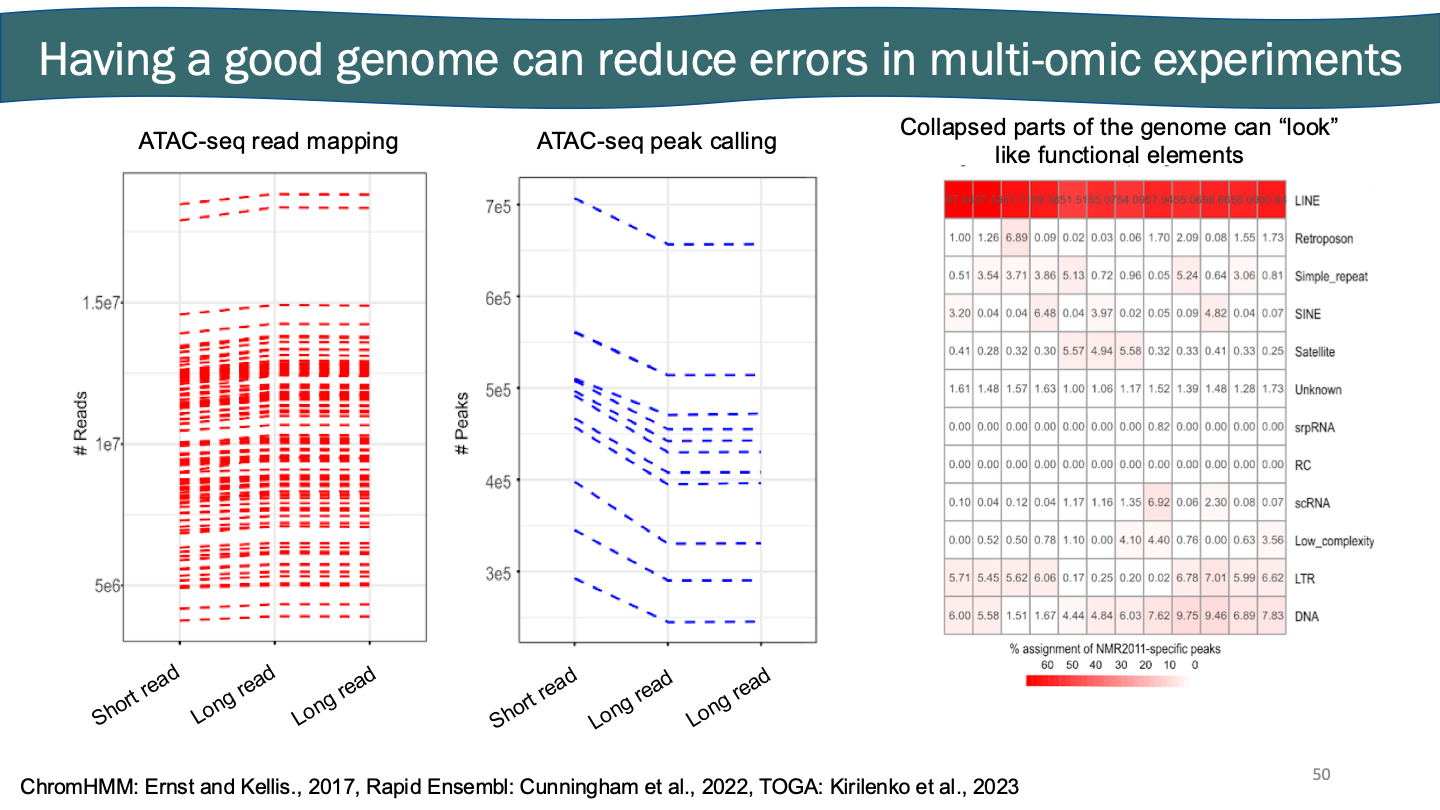
Multi-omic experiment + good genome → helps find tandem duplications
Interesting genes with multiple copies. Tandem duplications make the species unique. Green tracks are duplications. You can take genome wide data.
TINF2 → member of the shelterin complex, protects telomeres. Helps protects cells in the NMR brain in low-oxygen environments, telomeres play a crucial role in aging and cancer.
Kyat1 → makes Kynurenic acid (KYNA), which is neuroprotective. NMR have high levels compared to mouse. Role in cancer is ambiguous, linked to aging and fertility.