Recombination and Transformation
Creating Recombinant Plasmids
Restrictions enzymes: cut DNA. Each restriction enzyme is very specific to a particular recognition site, which typically has a size-based palindromic sequence.
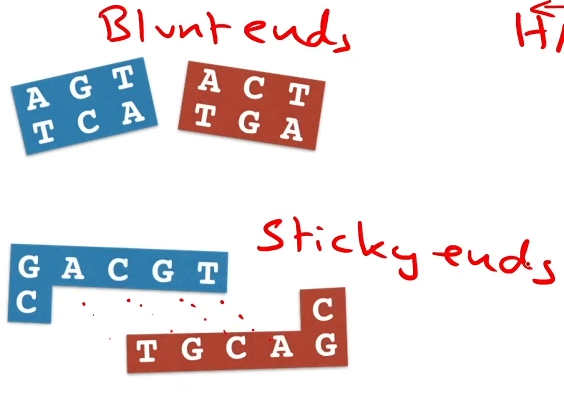
Some restriction enzymes cut at the same place on each strand, leaving ‘blunt ends’; others cut at different positions, leaving ‘sticky end ends.’
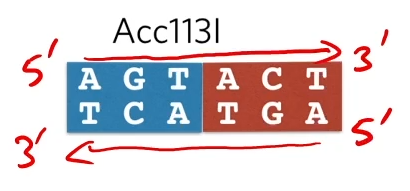 Defining a palindromic restriction site:
Defining a palindromic restriction site:
It’s a region of six bases that reads the same in a 5’ to 3’ direction on both strands
Some restriction enzymes cut down the middle and the result it ‘blunt ends’
while other restriction enzymes’ complementary overheads (which are attracted to each other)and we call those ‘stick ends’
Step 1: Choosing a restriction enzymes
An endonuclease (an enzyme that cleaves a polynucleotide chain through the separating of nucleotides) is chosen to cut upstream and downstream of the gene, leaving sticky ends
if you want to make a lot of copies of [___] all you need to do is find a palindrome upstream that is a the 5’ and downstream of that sequence that we want to make copies
 Then you need to get a restriction endonuclease that cuts at that restriction site (need to choose the correct enzyme for DNA [___] as some enzymes like XX1 cut leaving sticky ends while XX2 cuts leaving blunt ends and rn you need the sticky ends)
Then you need to get a restriction endonuclease that cuts at that restriction site (need to choose the correct enzyme for DNA [___] as some enzymes like XX1 cut leaving sticky ends while XX2 cuts leaving blunt ends and rn you need the sticky ends)

Step 2:
A plasmid ic chosen that has two genes that each encode observable traits. Of these genes, one must contain the restriction site of the restriction endonuclease being used.
Next, we have to find a plasmid. Plasmids are small extra-chromosomal (not part of their chromosome) loops of DNA that come naturally from bacteria
We want to choose a plasmid that has two genes that both confer on a bacteria an observable trait. That is, any bacteria that make up this plasmid will have two genes in it and will be able to tell it’s got those two genes because the bacteria will look different/behave differently in some way.
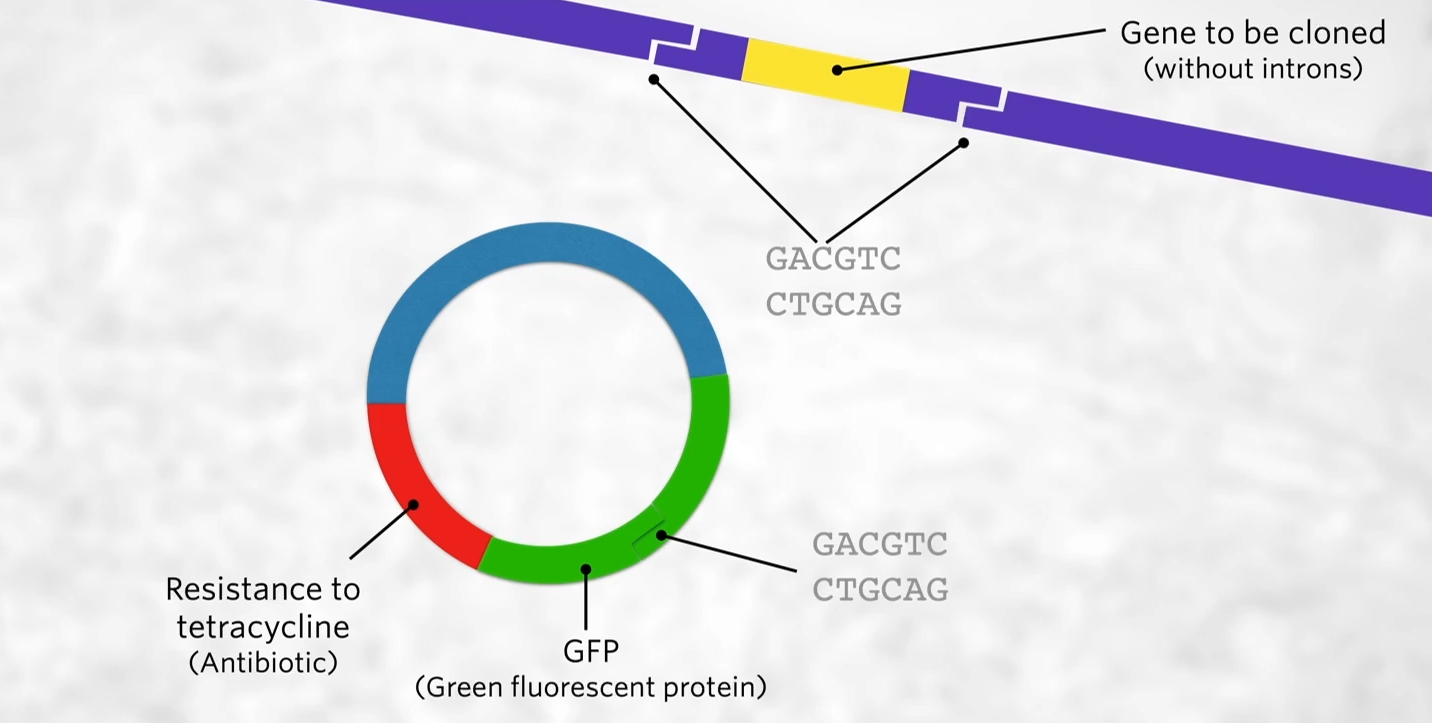
DNA one of those genes needs to have the restriction site for the restriction enzyme that we’ve used to cut the human DNA to get that yellow gene out of the DNA. We want to use an enzyme with the same restriction site so that the sticky ends are complementary to each other, BUT we want the restriction enzyme to be in one but not both of those genes.
Notice that the gene in yellow doesn’t have any introns, that’s because bacteria don’t have introns in their DNA so they don’t know what to do with introns while in our cells, the RNA polymerase transcribes the DNA, exons, and introns, and then during RNA processing the introns get cut out just leaving the exons but that doesn’t happen in bacteria. So if we put a human gene that has introns it won’t work. Thus we need to use a human gene that doesn’t have introns which can be done is through making the gene synthetically or cDNA (copy DNA works using reverse transcriptase the transcribed messenger RNA backward to DNA)
Step 3: Using the restriction endonuclease
The same restriction enzyme is used to cut both the source gene and the plasmid. As a result, the source gene and the plasmid have complementary sticky ends.
 Havingcut the plasmid open and having cut that human gene out of the cDNA we now have a cut upoen plasmid, a piece of DNA with our gene on it with stick ends. What we’re hoping that is that human DNA will stick into the plasmid like this and it should because we’ve got sticky ends.
Havingcut the plasmid open and having cut that human gene out of the cDNA we now have a cut upoen plasmid, a piece of DNA with our gene on it with stick ends. What we’re hoping that is that human DNA will stick into the plasmid like this and it should because we’ve got sticky ends.
Step 4: Making a recombinant plasmid
When the source gene and the plasmids (containing foreing ‘passenger’ DNA) are mixed, sometimes the source gene will be incorporated into the plasmid, creating a recombinant. The bond is completed by adding ligase.
 Need to complete those sugar-phosphate bonds(phosphodiester bonds between sugar and phosphate along the backbone of the DNA)(bc rn weak hydrogen bonds hold it between adenine & thymine and cytosine & guanine) and that’s what DNA ligase does.
Need to complete those sugar-phosphate bonds(phosphodiester bonds between sugar and phosphate along the backbone of the DNA)(bc rn weak hydrogen bonds hold it between adenine & thymine and cytosine & guanine) and that’s what DNA ligase does.
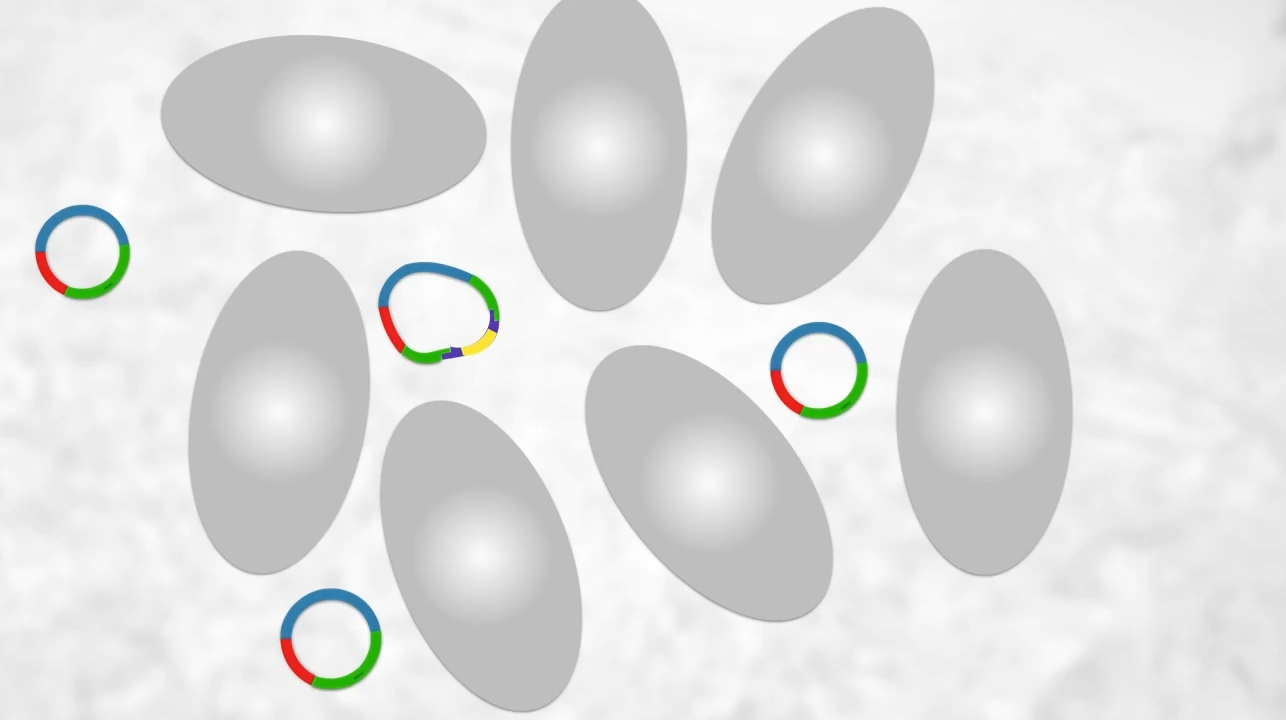
 (btw it’s more common for the plasmid to be cut open by restriction enzymes but since the ends are sticky it will just join back up again) By the end of this process, we will have recombinant plasmids (left) and regular plasmids (left) so how do we find these recombinant plasmids??
(btw it’s more common for the plasmid to be cut open by restriction enzymes but since the ends are sticky it will just join back up again) By the end of this process, we will have recombinant plasmids (left) and regular plasmids (left) so how do we find these recombinant plasmids??
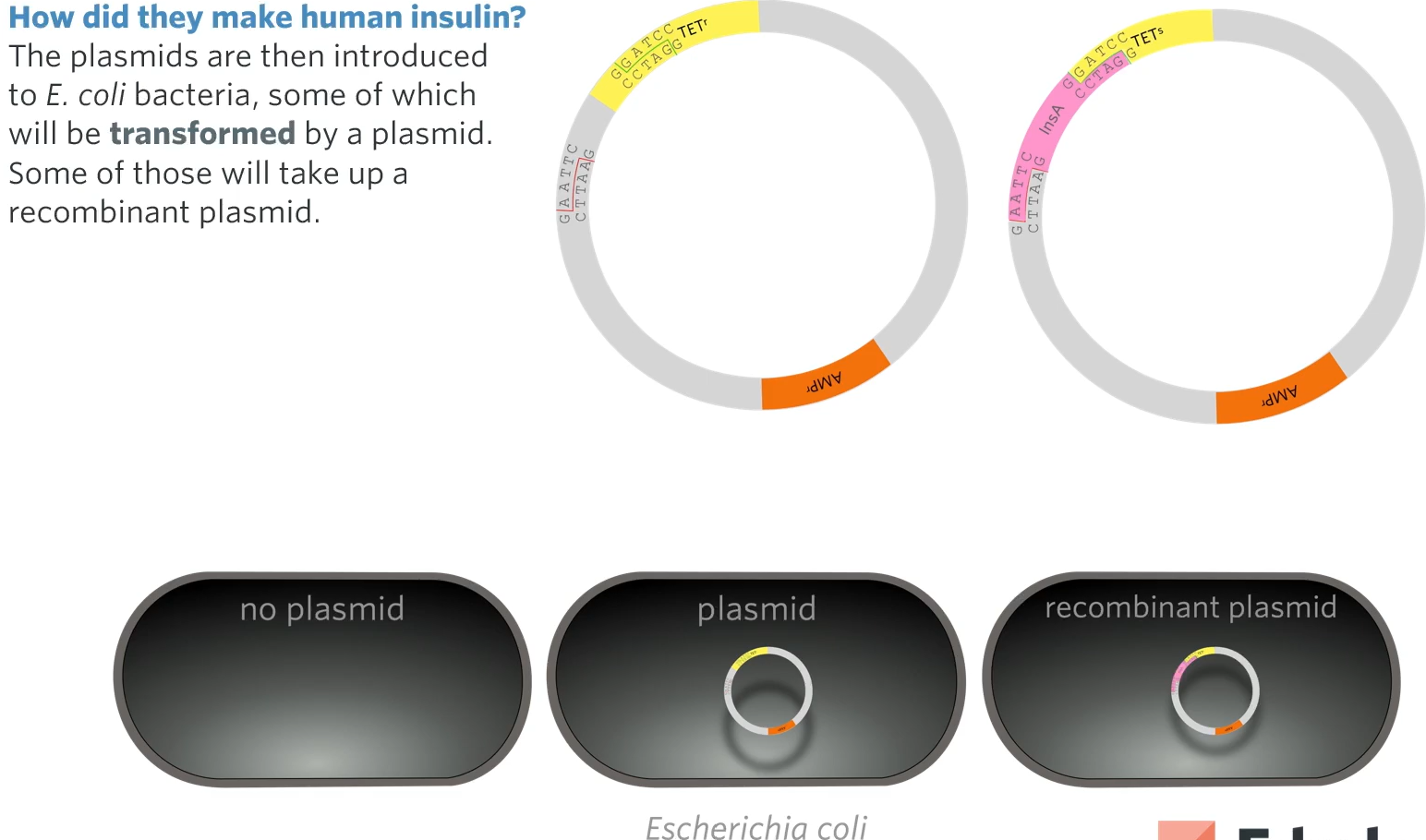
Step 5: Transformaiton
Bacteria (such as E.Coli) are made competent to take up plasmid. Only some bacteria will take up a plasmid. Only some of those will take up the recombinant plasmid.
 What we do is mix them with bacteria which will naturally take up plasmids from their environments (scientists can do this to make the bacteria more competent in taking up plasmids such as passing electricity through the solution, heat shocking them, heating and then cooling down)
What we do is mix them with bacteria which will naturally take up plasmids from their environments (scientists can do this to make the bacteria more competent in taking up plasmids such as passing electricity through the solution, heat shocking them, heating and then cooling down)
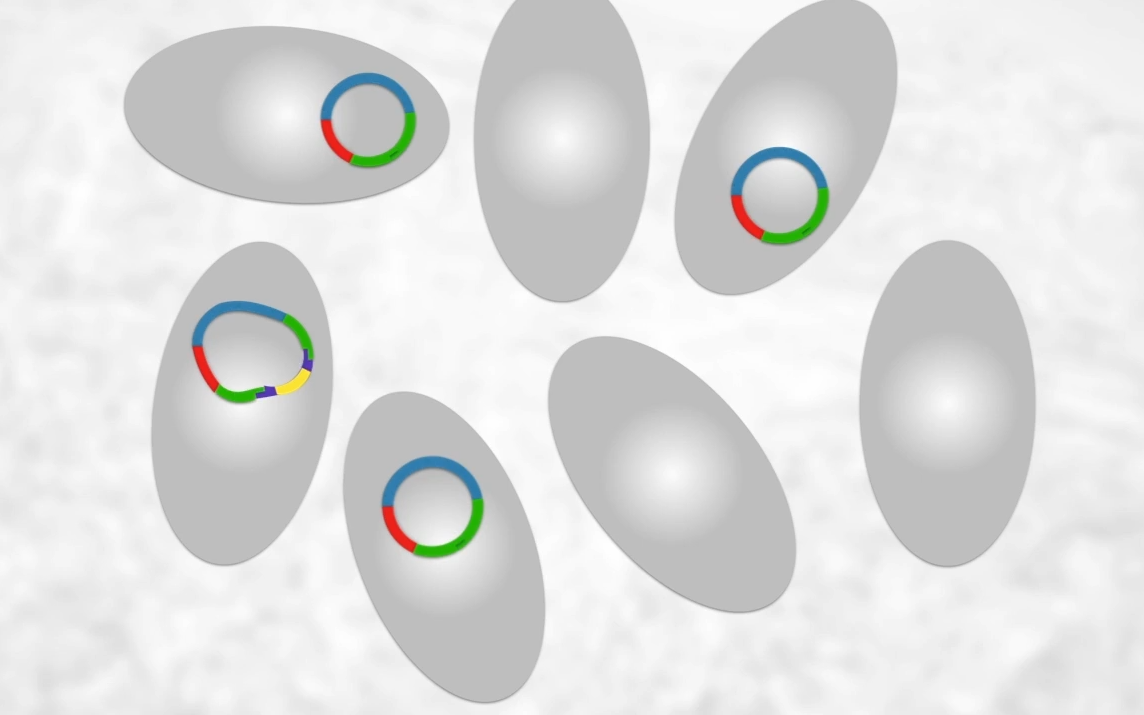 As seen only some will have our recombinant plasmid though many will not so how do we find the bacteria?
As seen only some will have our recombinant plasmid though many will not so how do we find the bacteria?
Step 6: Identifying and culturing bacteria transformed by a recombinant plasmid.
The bacteria that have taken up a plasmid can easily be identified by growing the bacteria on an agar plate containing the antibiotic. Bacteria that have not taken up a plasmid will lack the gene for resistance to the antibiotic and will not be able to grow.
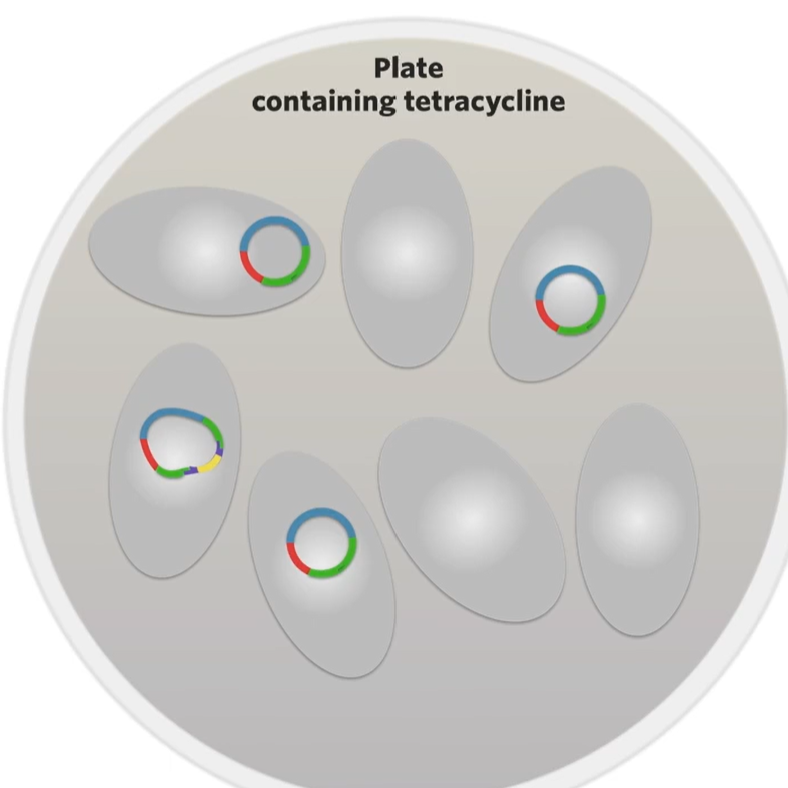 First thing to do is culture those bacteria on an agar plate that contains the antibiotic used (ex. tetracycline) so that by the end of culturing everything that has grown on the plate must be a transformed bacteria (a bacteria that has taken up a plasmid). Some will become transformed bacteria by a recombinant plasmid while a regular plasmid will transform some but they all will have a plasmid) So how do we find the bacteria that the recominant plasmid has transformed?
First thing to do is culture those bacteria on an agar plate that contains the antibiotic used (ex. tetracycline) so that by the end of culturing everything that has grown on the plate must be a transformed bacteria (a bacteria that has taken up a plasmid). Some will become transformed bacteria by a recombinant plasmid while a regular plasmid will transform some but they all will have a plasmid) So how do we find the bacteria that the recominant plasmid has transformed?
The difference is the one that’s been transformed by a recombinant plasmid now has a broken green fluorescence protein gene. That’s why we wanted to make sure that the restriction site for AAT2 was in the green glourescne gene to make sure that the restriction site for our restriction enzyme would interrupt on of those two genes that give a bacteria an observable trait.
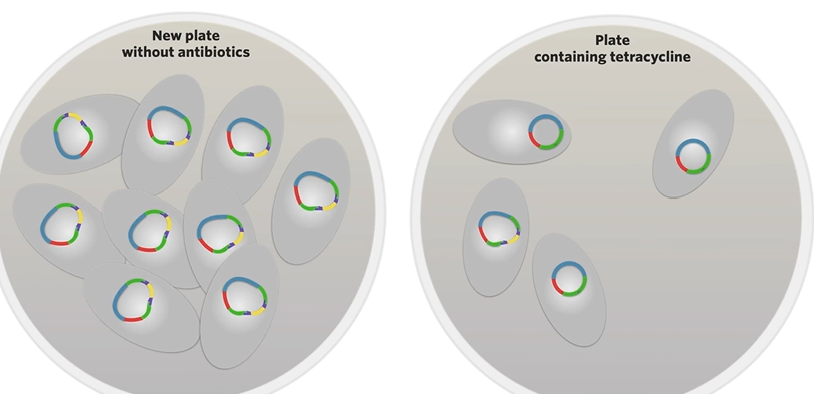
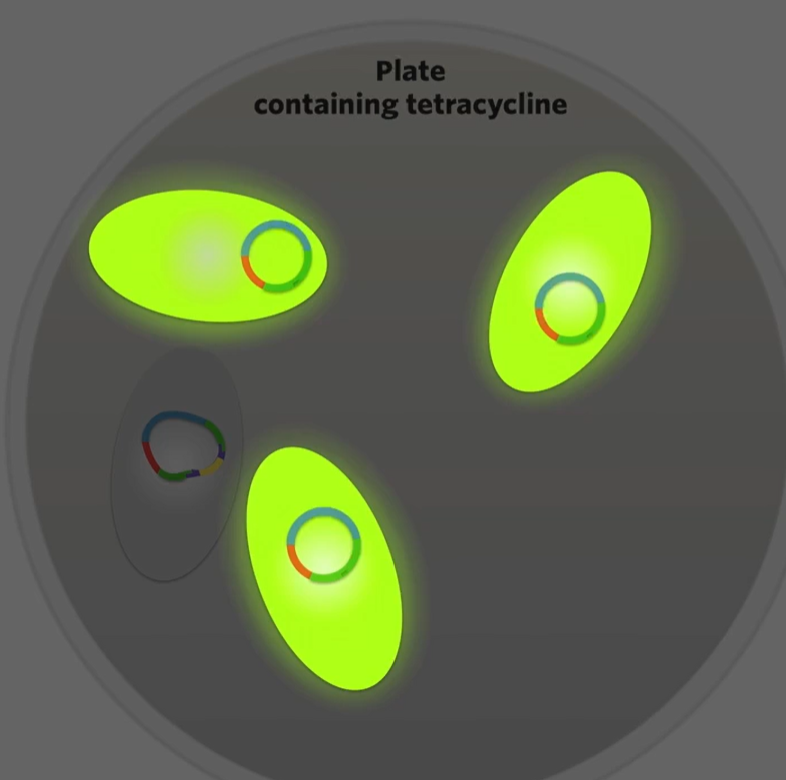 then like scape the bacteria that doesn’t glow into a plate without antibiotics and as they divide by binary fission all the daughter cells that are produced will contain those recombinant plasmids ginvg you a culture of bacteria containing recombinant plasmids
then like scape the bacteria that doesn’t glow into a plate without antibiotics and as they divide by binary fission all the daughter cells that are produced will contain those recombinant plasmids ginvg you a culture of bacteria containing recombinant plasmids
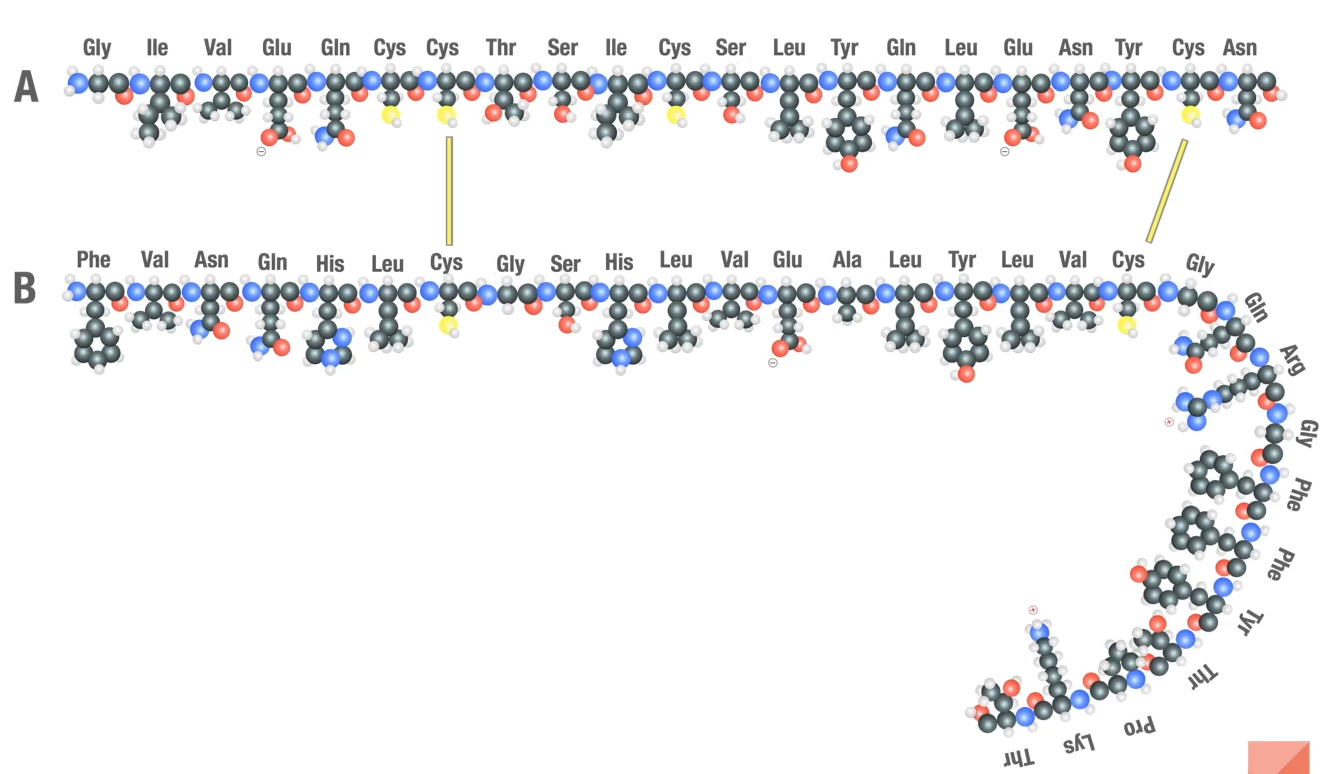
Human Insulin
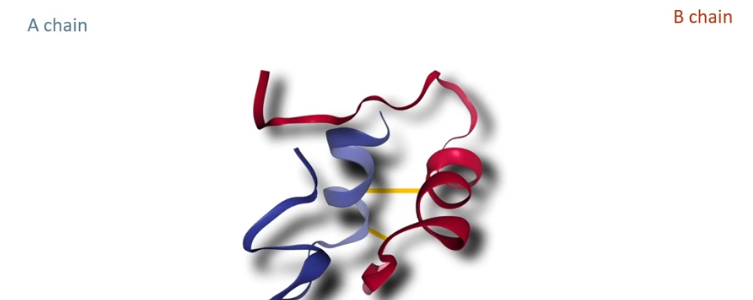
Insulin: a small protein with just 51 amino acids in two separate chains called the A chain (21 amino acids long) and the B chain (38 amino acids long) which are held together by 2 disulfide bonds between cysteine a minor acids residues
How did they make human insulin?
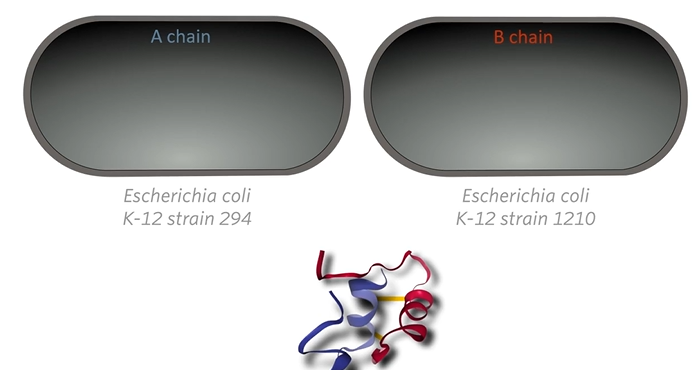
Genentech cloned each other polypeptides separately in a different strain of E.coli bacteria and then combined them in vitro to make insulin.
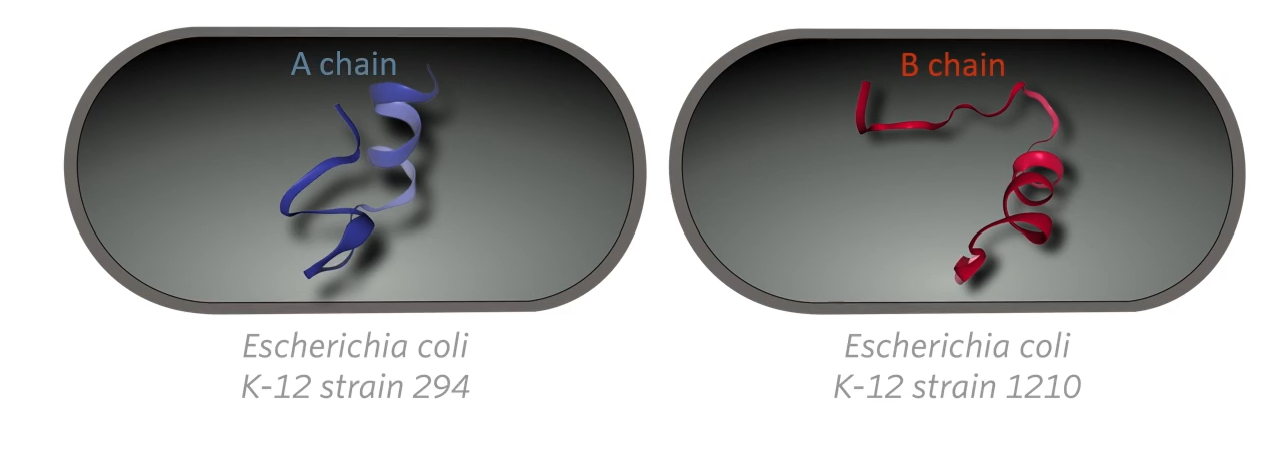
They cloned the A chain, the A peptide in one strain of E. Coli bacteria, and the B chain was cloned in a different strain then they extracted the peptide from the two different strains of bacteria and combined them in vitro that is in a glass outside of the bacteria. So there would never a point in time when any bacteria cell would have human insulin in it. At any point in time, any bacterial cell would only have half a human insulin molecule then they would be combined later outside the bacteria mostly because of a safety protocol.
How did they make human insulin?
→Focus on A chain
Step 1: Make a plasmid that contains a human insulin A chain gene (without intron bc bacteria don’t have introns and if you put a human gene with intron the bacteria isn’t going to know what to do with it and it won’t work).
 Remember the genetic code is redundant meaning that there are several different codons that code for most amino acids (ex. CUG codes for leucine but so does CUC)
Remember the genetic code is redundant meaning that there are several different codons that code for most amino acids (ex. CUG codes for leucine but so does CUC)
The made a gene that would code for the correct sequence of amino acids but as it turns out it was a completely different sequence of nucleotides than the sequence of nucleotides in the human insulin gene in our cells but it doesn’t matter because it’s still codied for the same sequence of amino acids.
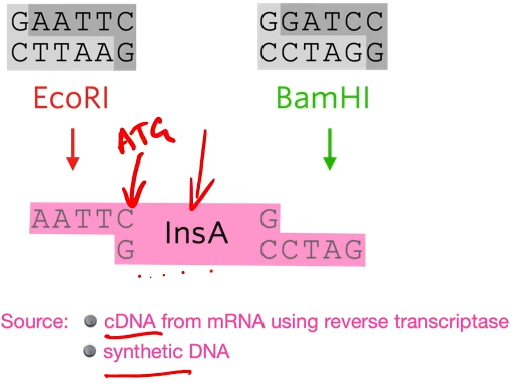 they put an extra codon on the front, an extra ATG that codes for methionine.
they put an extra codon on the front, an extra ATG that codes for methionine.
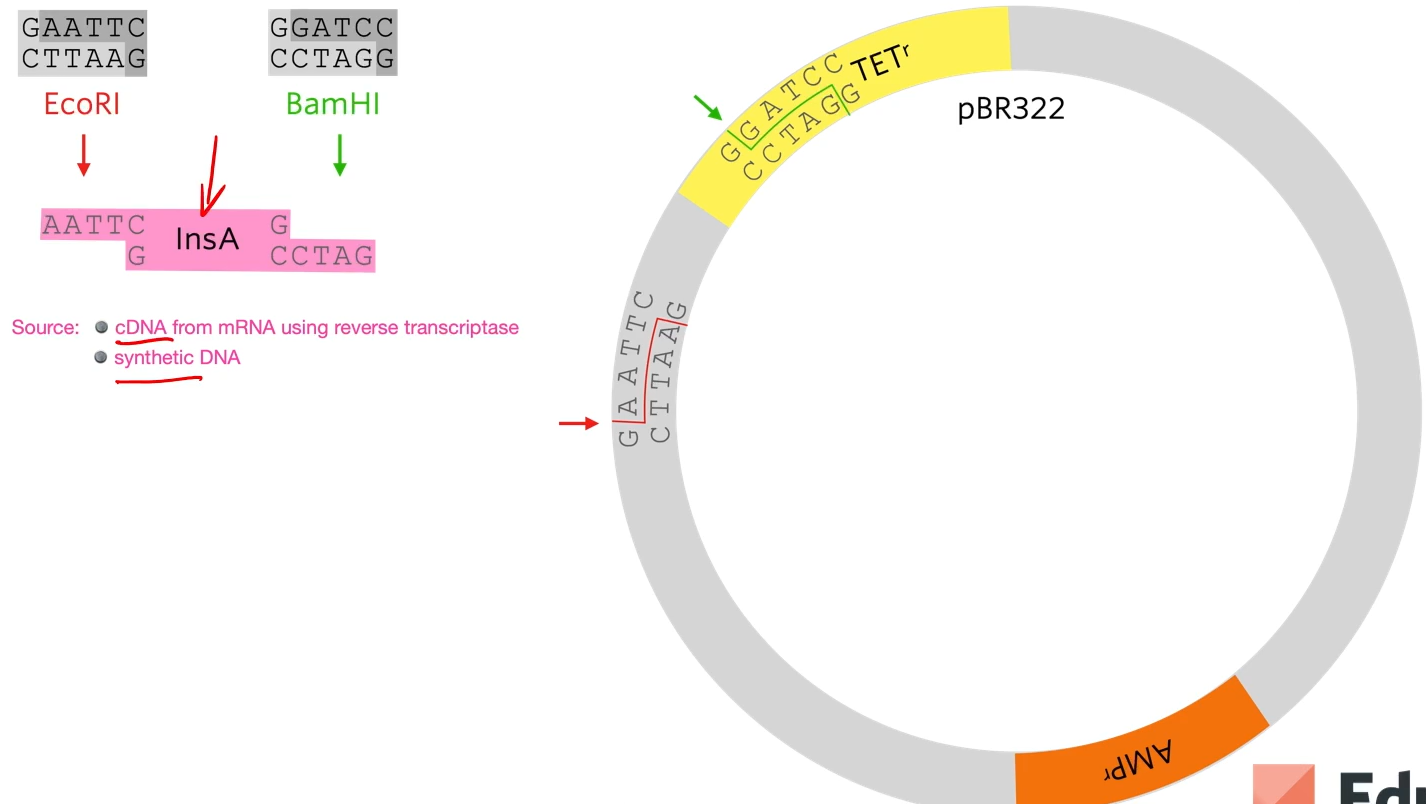
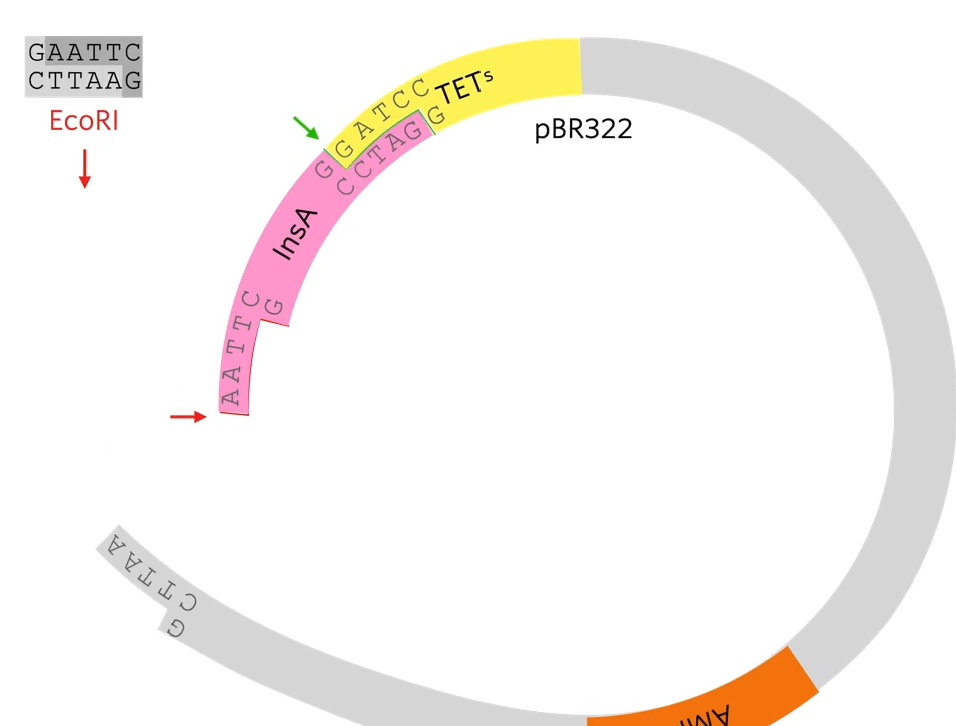
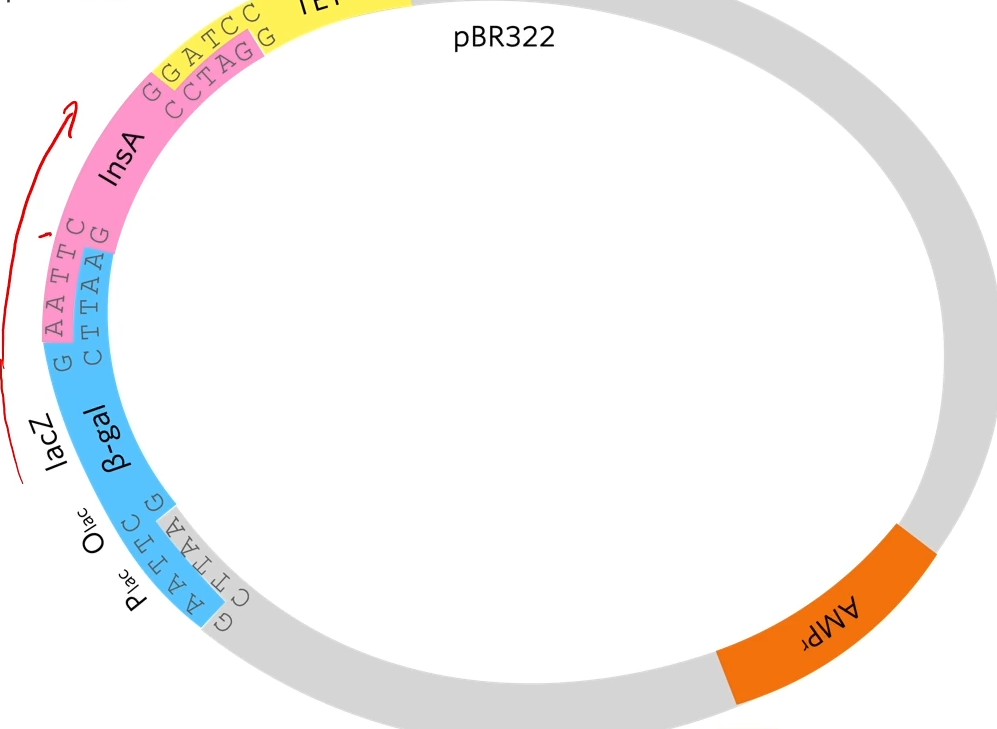
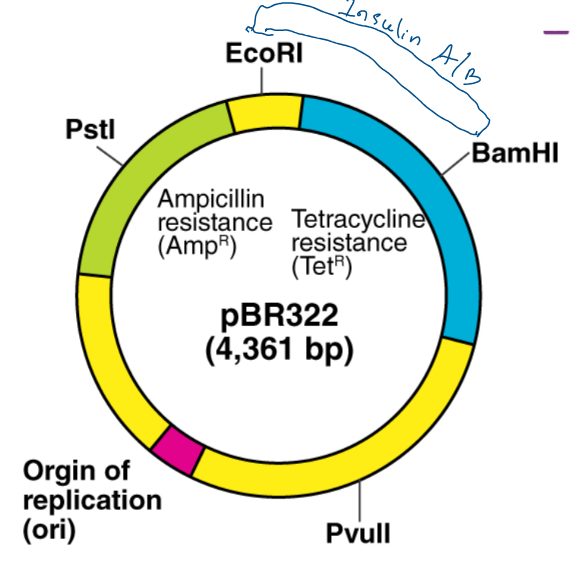
EcoRi and BamHI are restriction enzymes to cut out different sticky ends of the DNA . They sued the same two restriction enzymes to cut the plasmid that they chose.
Looking at the plasmid, first of all a gene for resistance to ampicillin (antibiotic) and a gene for resistance for tetracycline but as said before, the restriction site for the restriction enzyme needs to be within one of those genes (they chose tetracycline) So the restriction site for BAMH1 is in the tetracycline gene so if the restriction enzyme cuts that and a bit of DNA gets added there the tetracycline gene won’t work properly anymore.
A sa result of cutting out using two diffrente restriction enzymes is a chunk of the plasmid to be cut out but the sticky ends are not complementary to each other.
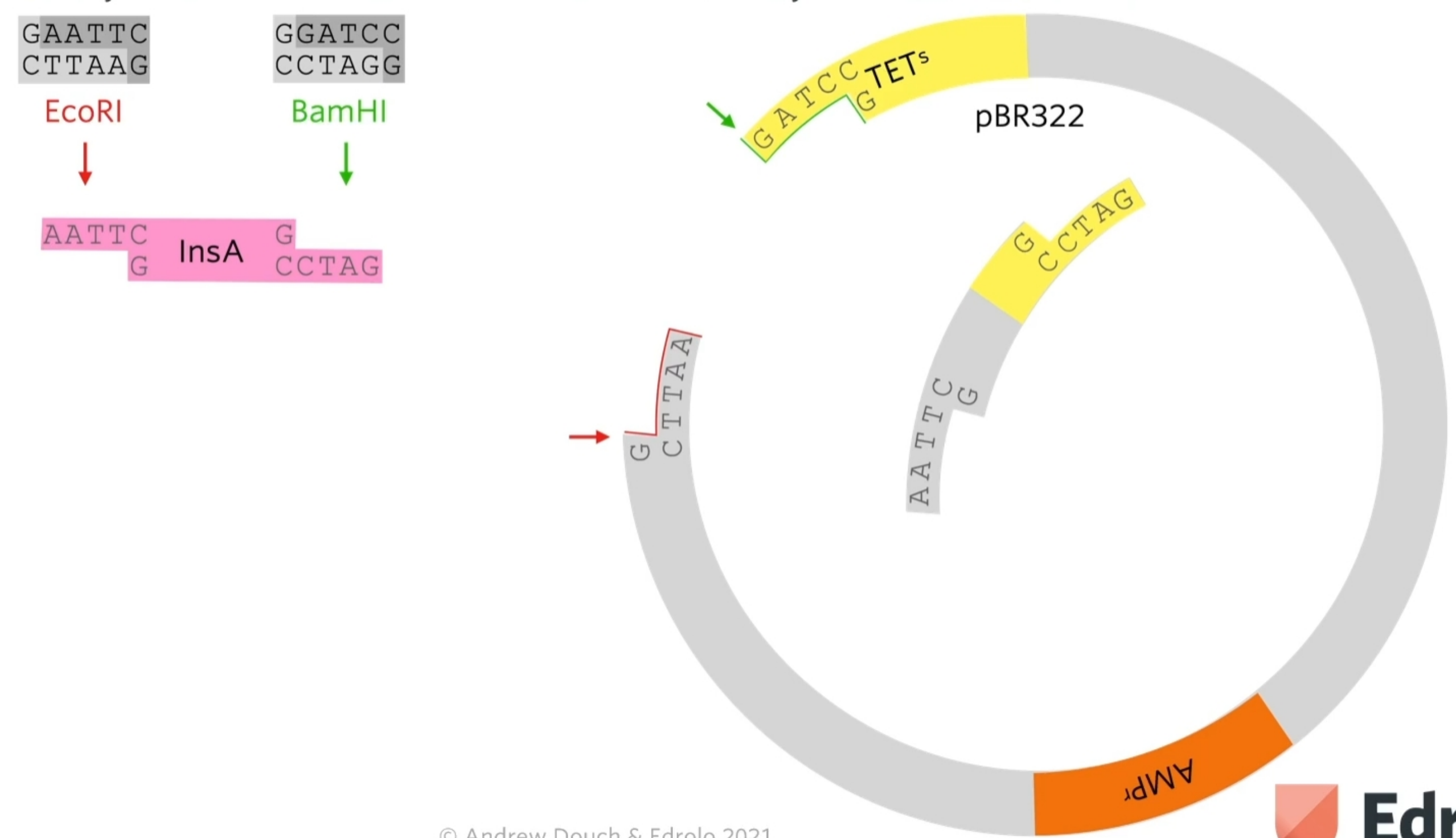 What we’re hoping is that human insulin A gene, which has complementary stick ends to the two halves of that plasmid will join in there like that. By using two different restriction enzymes, it guarantees that the human insulin A gene joins in with the right orientation.
What we’re hoping is that human insulin A gene, which has complementary stick ends to the two halves of that plasmid will join in there like that. By using two different restriction enzymes, it guarantees that the human insulin A gene joins in with the right orientation.
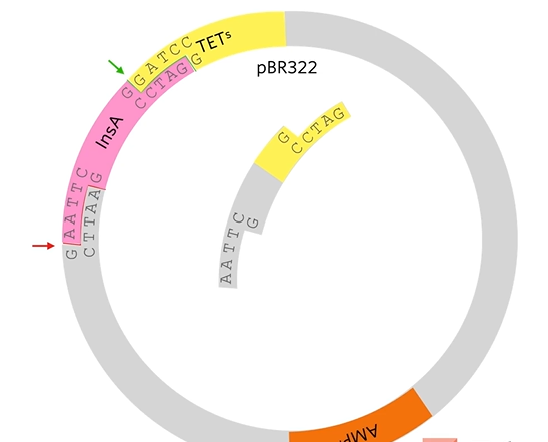 The reason that that plasmid won’t just open and rejoin again is that these stick ends are not complementary as they’ve been cut with different restriction enzymes. I't’s important because if they could cut open and just rejoined then we would end up with a plasmid that gives resistance to ampicillin but doesn’t give resistance to tetracyclin and doesn’t have the human gene in it
The reason that that plasmid won’t just open and rejoin again is that these stick ends are not complementary as they’ve been cut with different restriction enzymes. I't’s important because if they could cut open and just rejoined then we would end up with a plasmid that gives resistance to ampicillin but doesn’t give resistance to tetracyclin and doesn’t have the human gene in it
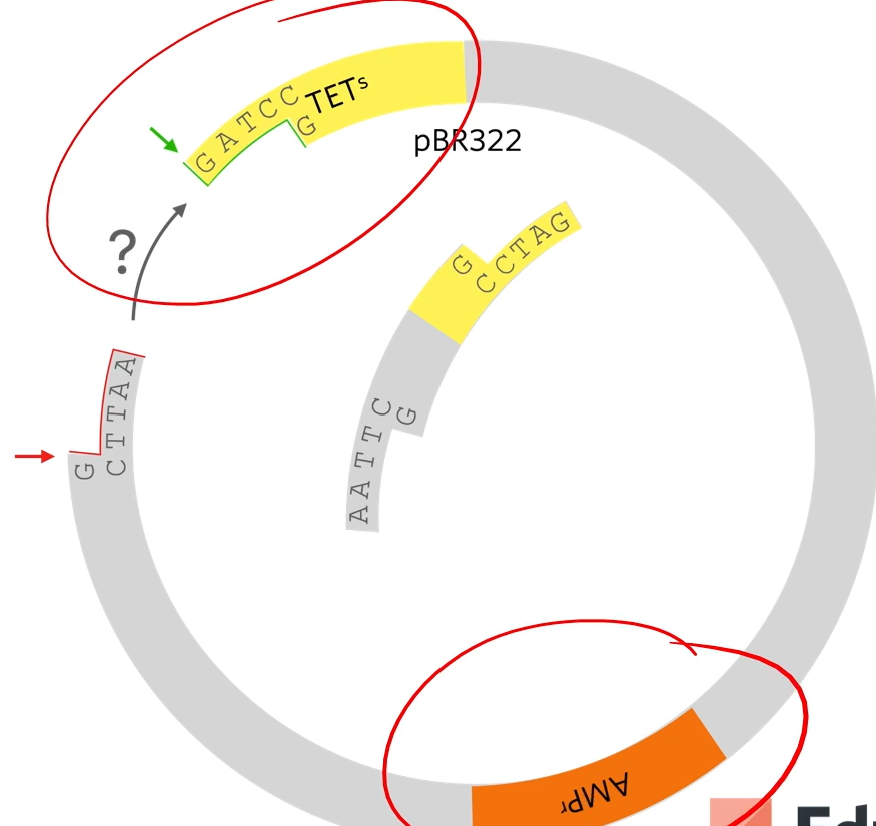
So afterward what we do is mix those plasmids with E.Coli bacteria (three combinations with no plasmid, plasmid, and recombinant plasmid)
how do we know which did and which didn’t?
same as before
Two antibiotic plates are used to identify E. coli colonies transformed by recombinant plasmids. First, ampicillin selects transformed from non-transformed bacteria (bc any bacteria that don’t have a plasmid at all won’t grow on this plate what we don’t know is why of those colonies have a plasmid that contains the human insulin A gene, we figure that out by using another agar plate). Second, a plate containing tetracycline is used to identify which transformed bacteria have recombinant plasmids (because the plasmids with human insulin A gene won’t be resistant to this. We’ll wait until the colonies start growing).

They then get the plasmids out of those bacteria by lysing the bacteria to split them open and purify the plasmids out of them leaving them with a bunch of plasmids which are the ones that they’ve started with.
Then they add another gene to the plasmid ( it’s an added complication but VCAA says you need to learn this).
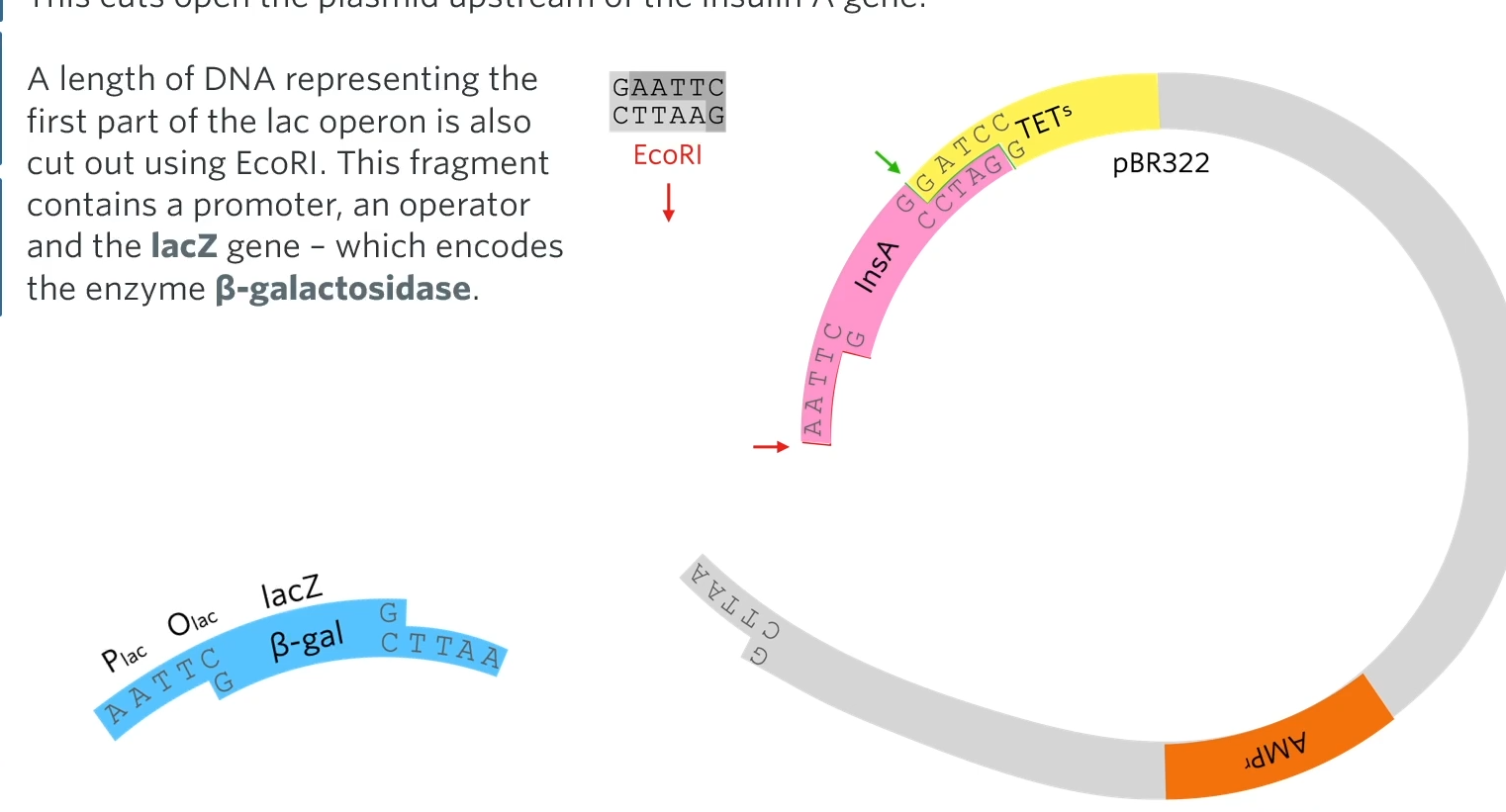
Next, the recombinant plasmids are treated with EcoRI (again)
They’re going to use the restriction enzyme Echo R1 which won’t cut the restriction site for BAMH1 but the spot for the restriction site for Echo R1 so that it will cut out the plasmid like this:
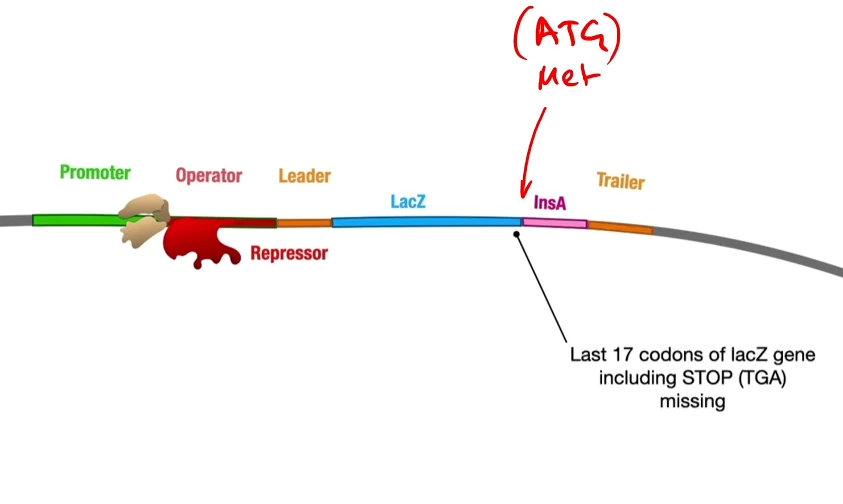
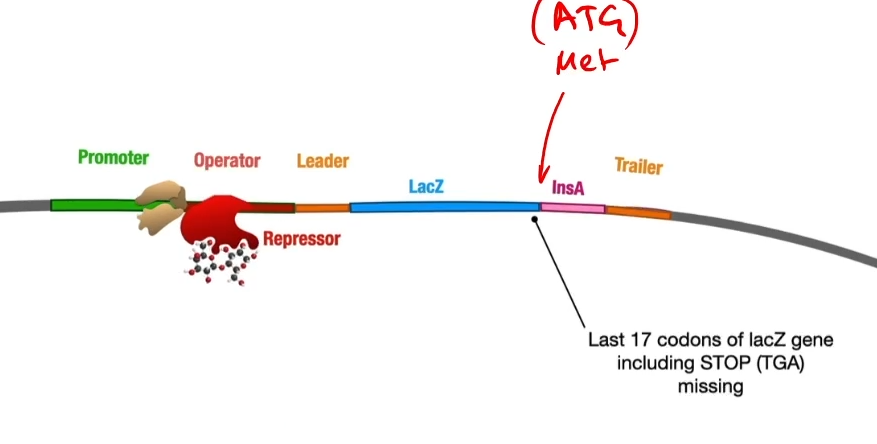
 This cuts open the plasmid upstream of the insulin A gene. A length of DNA representing the first part of the lack operon is also cut out using EcoRI. This fragment contains a promoter, an operator and the lacZ gene-which encodes the enzyme Beta-galactosidase.
This cuts open the plasmid upstream of the insulin A gene. A length of DNA representing the first part of the lack operon is also cut out using EcoRI. This fragment contains a promoter, an operator and the lacZ gene-which encodes the enzyme Beta-galactosidase.
Then they introduce a piece of DNA, a gene called LacZ together with a promoter and an operator. So that piece of DNA though has been cut out using Echo R1 so it’s got complementary sticky ends to both sides of the ctu in that plasmid. So when they mix together, some of the plasmids will take up the Beta-gal gene at the front of the insulin A gene.
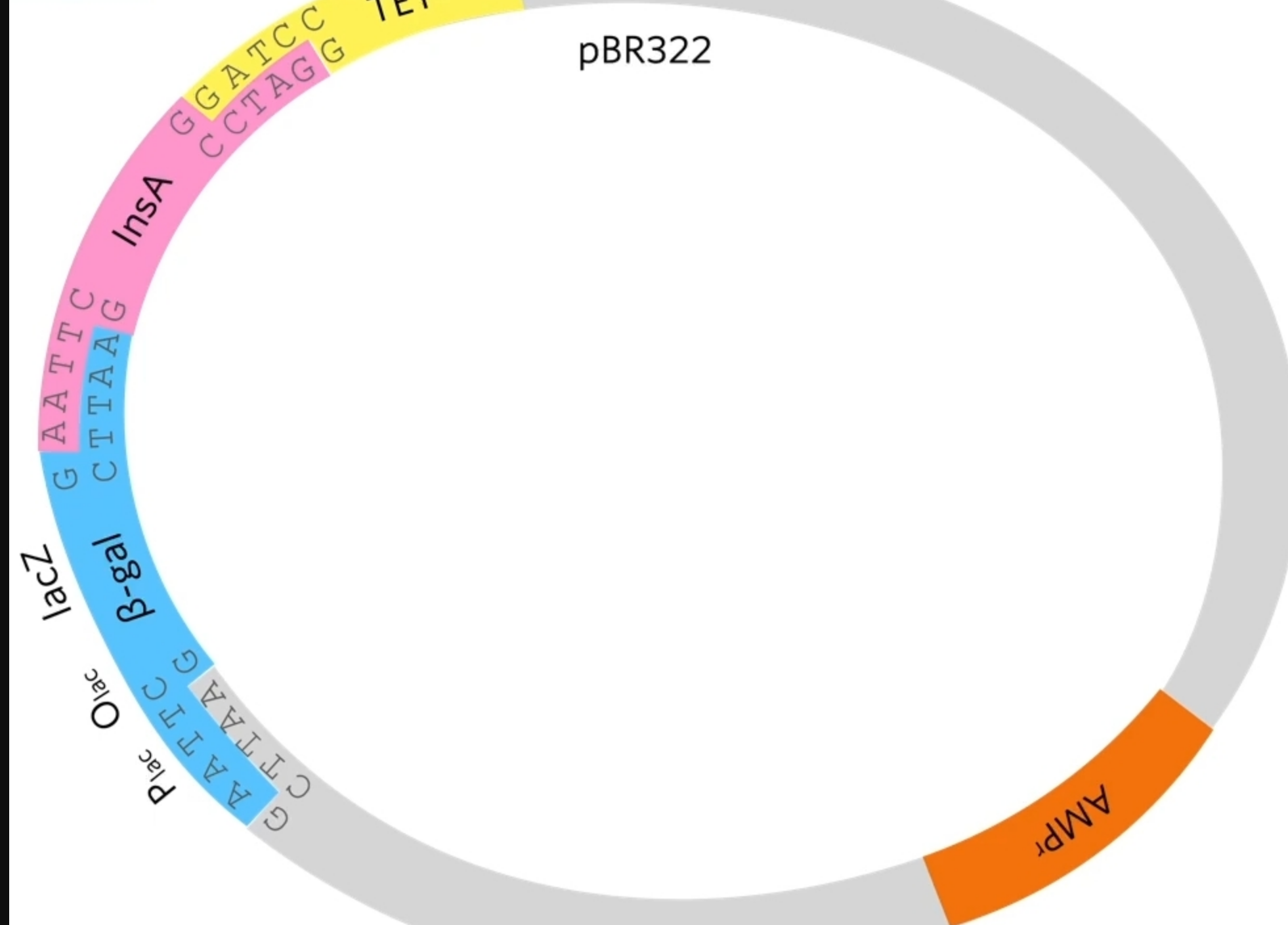 By cutting of the last bit of the gene you lose the stop codon. This means that RNA polymerase, when it transcribes this it’s going to read around and then when it gets to the end of beta-gal it’s just going to keep going so essentially the insulin A protein will be part of the beta-galactosidase protein.
By cutting of the last bit of the gene you lose the stop codon. This means that RNA polymerase, when it transcribes this it’s going to read around and then when it gets to the end of beta-gal it’s just going to keep going so essentially the insulin A protein will be part of the beta-galactosidase protein.
 Again the plasmids are mixed with E.Colid but this time the possible combinations are no, recombinant plasmid with insulin and recombinant plasmid with insulin and lacZ
Again the plasmids are mixed with E.Colid but this time the possible combinations are no, recombinant plasmid with insulin and recombinant plasmid with insulin and lacZ
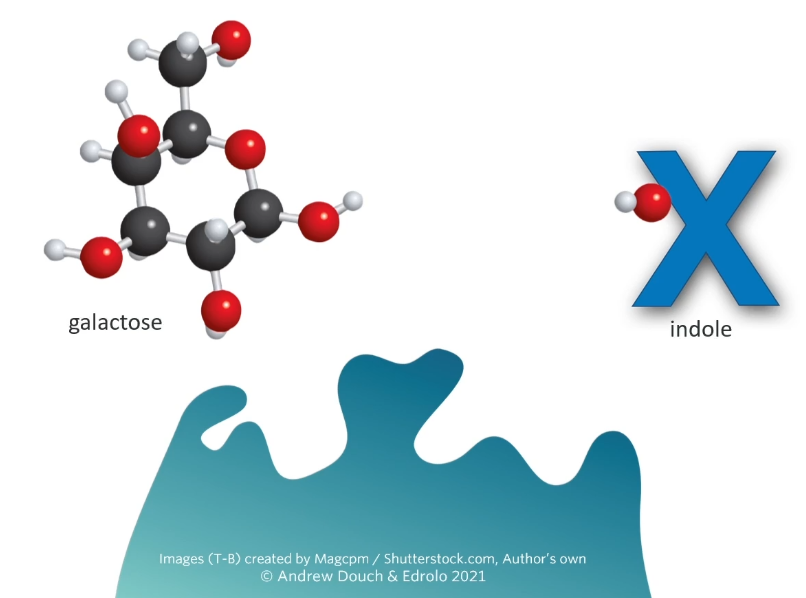
B-galactosidase hydrolysis lactose to glucose and galactose
These genes produce a protein or some sort of effect. The beta-galactosidase gene produces beta-galactosidase (represented by the blue enzyme. What beta-gal normal does in a cell is it grabs lactose (disaccharide) and it splits(digests?) that lactose into glucose and galactose. But scientists have made this extra compound this special compound called X-gal.
B-gala will also cut Xgal into glucose and an indole which turns blue
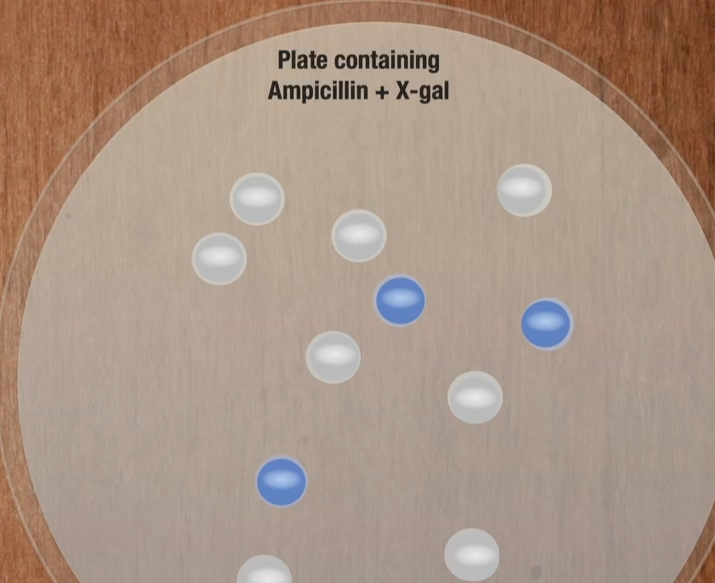
 Any cell that has a beta-galactosidase gene in it will produce the enzyme beta-galactosidase and if you grow in in the presence of X-gal, those bacteria are going to turn blue. Remember what we’re trying to do is find the bacteria which has taken up the plasmid.
Any cell that has a beta-galactosidase gene in it will produce the enzyme beta-galactosidase and if you grow in in the presence of X-gal, those bacteria are going to turn blue. Remember what we’re trying to do is find the bacteria which has taken up the plasmid.
The E.Coli are grown on a plate containing X-gal. This makes it easy to identify colonies that contain the recombinant plasmid because they produce B-Gala and will be blue in color
Why is the lacZ gene used?
the promoter is prokaryotic so can be used by RNA polymerase to initiate transcription
the operator is useful because it makes it easy to stimulate transcription using allolactose or IPTG
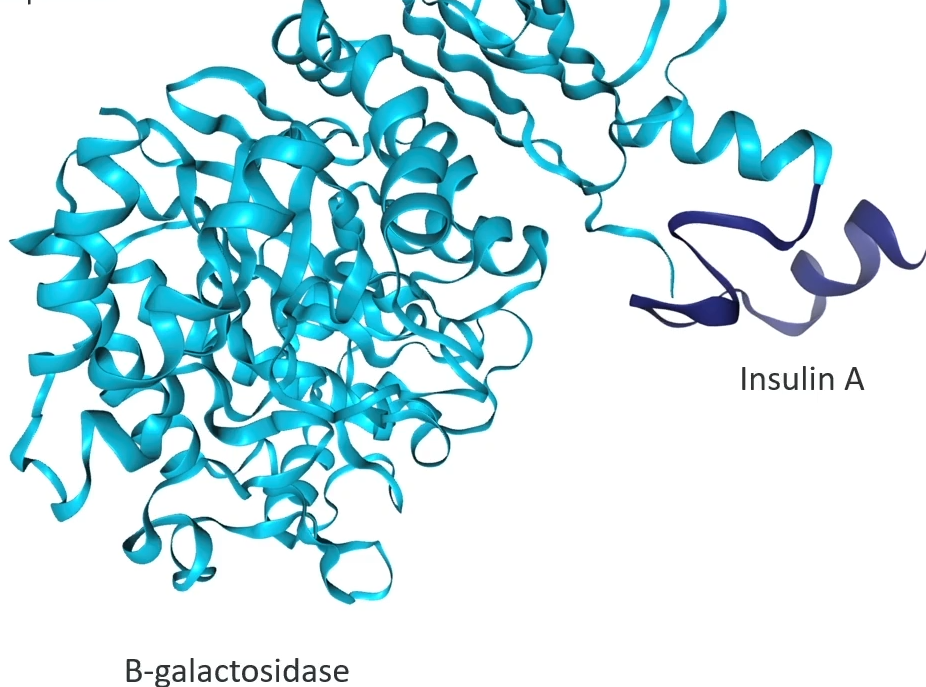
it creates a fusion protein
The reasons is that a bacterial cell will digest small unfolded peptides so if the cell was just producing human insulin A, that 21 amino acid peptide, the enzymes in the bacteria would just digest it right away and it would be useless. However, the enzymes in bacteria don’t do that to big proteins. So you make a fusion protein.
 (remember that there’s no stop codon)
(remember that there’s no stop codon)
Like any gene expression in an operon, the RNA polymerase will assemble at the promoter but be blocked by a repressor and be stuck by the repressor on the operator.
The advantage of using the start part of the operon to do this is because of the operator with a repressor stuck on it so that geneticists are able to turn this on at will. You turn it on by adding some allolactose which will stick onto the repressor protein and change its shape turning the gene on and unblocking that operator
 So then RNA polymerase will transcribe messenger RNA, a ribosome will translate into a fusion protein of beta
So then RNA polymerase will transcribe messenger RNA, a ribosome will translate into a fusion protein of beta-galactosidase and the insulin A peptide tacked onto the end of it because there was no stop codon so that it keeps on adding the amino acids from the insulin A peptide(if there was a stop codon it would make beta-galactosidase and then stop) 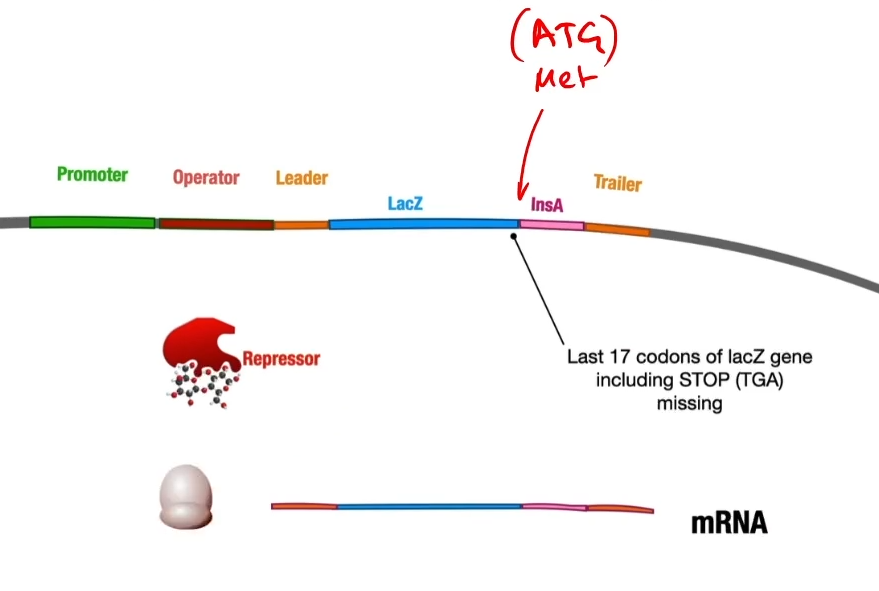
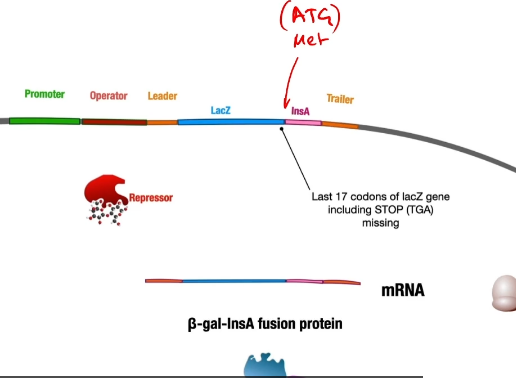 The fusion protein is necessary to protect the insulin A chain from enzymes in E. coli that rapidly degrade small unfolded peptides.
The fusion protein is necessary to protect the insulin A chain from enzymes in E. coli that rapidly degrade small unfolded peptides.
 vocab real quick:
vocab real quick:
Fusion gene: two genes that share a single proter and are transcribed and translated together because there's no stop codon between them.
A methionine was added to the start of the insulin A gene, which makes it possible to separate the fusion protein once it is out of the cell.
Now you have a bacterial cell with lots of fusion proteins in it that are a combination of beta-galactosidase and insulin. All that needs to be done then is we split those bacteria open, lyse them, extract those fusion proteins from the bacteria, and then separate the insulin A from the beta-galactosidase protein and you can do that because of the methionine that was added.
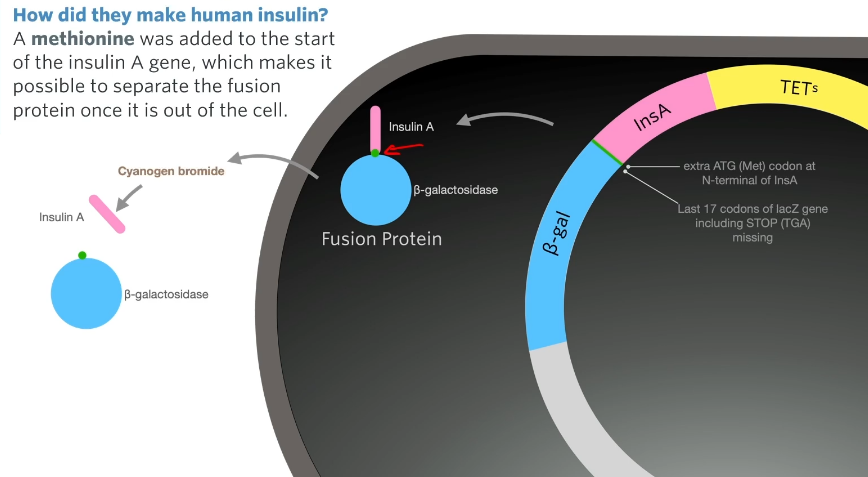 There is no methionine in the insulin A protein or in the insulin B chain (i.e no methionine in insulin) so the methionine acts as a spot to separate the A chain from beta-galactosidase with a chemical that breaks down cyanogen bromide.
There is no methionine in the insulin A protein or in the insulin B chain (i.e no methionine in insulin) so the methionine acts as a spot to separate the A chain from beta-galactosidase with a chemical that breaks down cyanogen bromide.
This entire procedure was done for A chain and B chain now you just need to mix them together and they will spontaneously fold into three-dimensional shapes and form the sulfide bonds between cysteines.
Class Questions 🗣
 Video Notes:
Video Notes:
production of human insulin
Production of Recombinant plasmids
First DNA encoding human insulin is obtained from a human pancreas
 Then the restriction enzyme is used to cut the DNA
Then the restriction enzyme is used to cut the DNA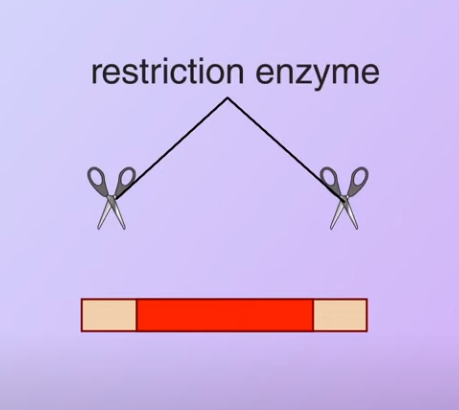 Meanwhile, a plasmid with antibiotic-resistance genes is obtained from a bacterium
Meanwhile, a plasmid with antibiotic-resistance genes is obtained from a bacterium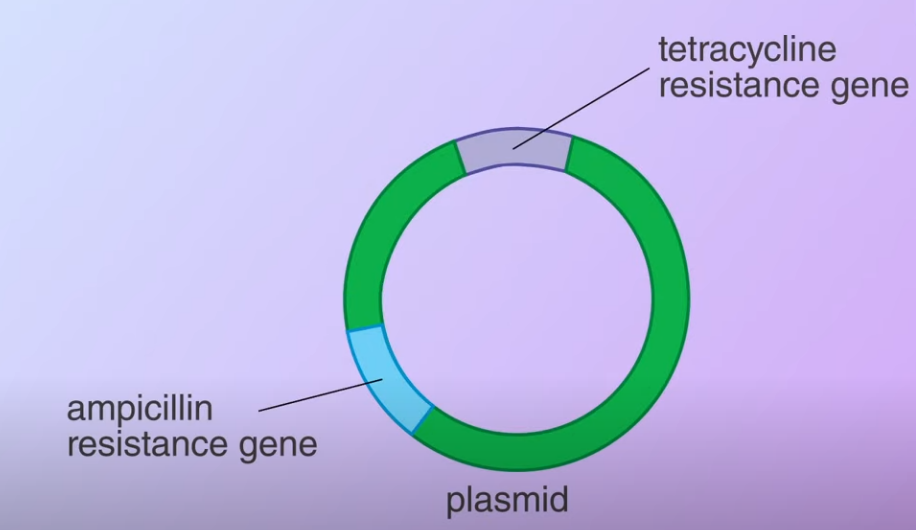 The plasmid is cut open using the same restriction enzyme. When the DNA fragemnets and the plasmid mix together (Transformed) some of the plasmids bind to the DNA fragments and with the help of DNA ligase (an enzyme that facilitates the joining of DNA strands together) forming recombinant plasmids
The plasmid is cut open using the same restriction enzyme. When the DNA fragemnets and the plasmid mix together (Transformed) some of the plasmids bind to the DNA fragments and with the help of DNA ligase (an enzyme that facilitates the joining of DNA strands together) forming recombinant plasmids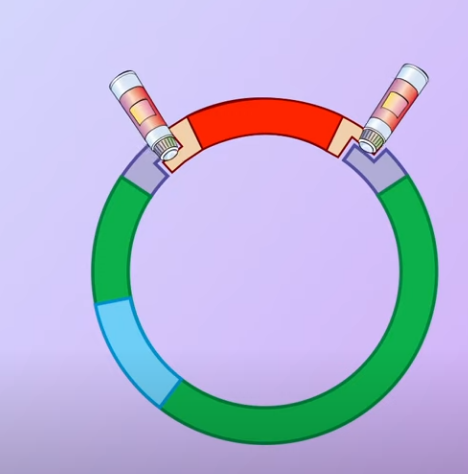 Note that some of the open plasmids do not pick up the DNA fragments and join by themselves. These are called nonrecombinant plasmids
Note that some of the open plasmids do not pick up the DNA fragments and join by themselves. These are called nonrecombinant plasmidstransformation and selection of bacteria
These plasmids are mixed with the bacteria E.Coli. This is used to introduce the plasmids to the host cells
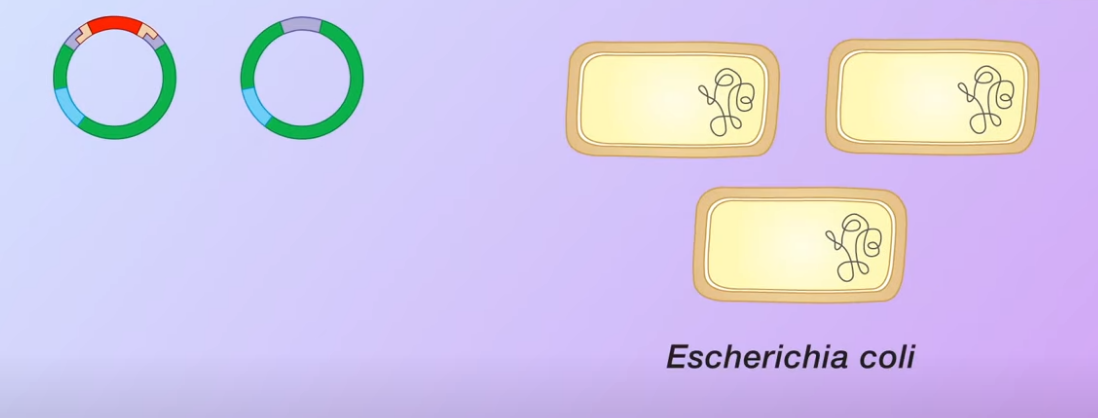 However only some of the bacteria ha
However only some of the bacteria havepicked up the plasmids, these are said to be transformed bacteria.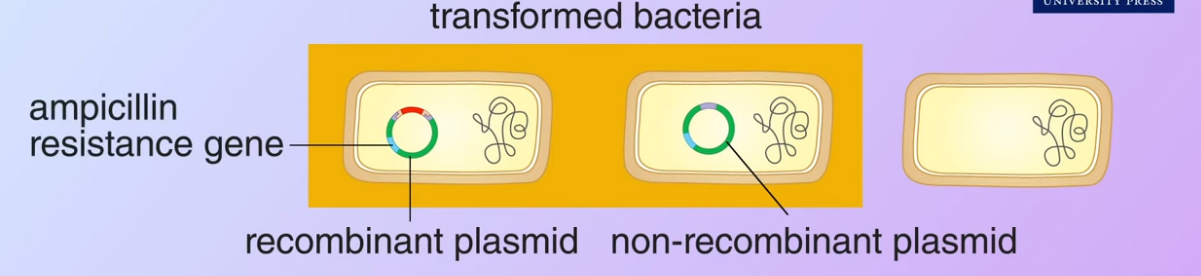 However, only some of the bacteria have picked up the plasmids which are said to be transformed and only some of the transformed contain a recombinant plasmid. The remaining transformed bacteria have the non-recombinant plasmid. Meanwhile, some of the bacteria do not pick up any plasmids at all. In this example
However, only some of the bacteria have picked up the plasmids which are said to be transformed and only some of the transformed contain a recombinant plasmid. The remaining transformed bacteria have the non-recombinant plasmid. Meanwhile, some of the bacteria do not pick up any plasmids at all. In this examplethe plasmids we use carry a resistance gene for the antibiotic ampicillin.
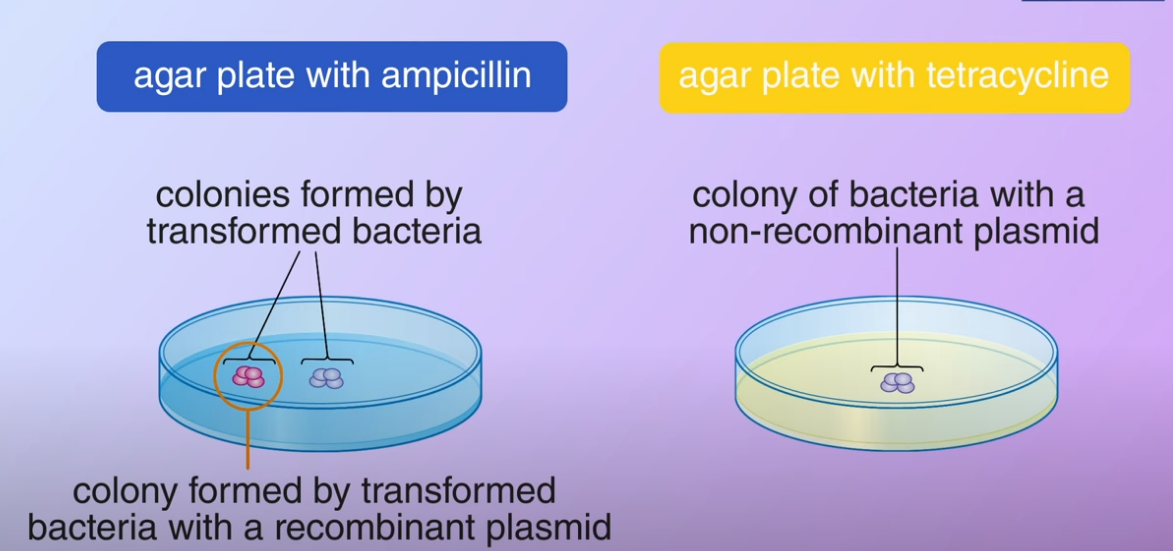
To select the transformed bacteria we first culture the bacteria on an agar plate containing ampicillin.
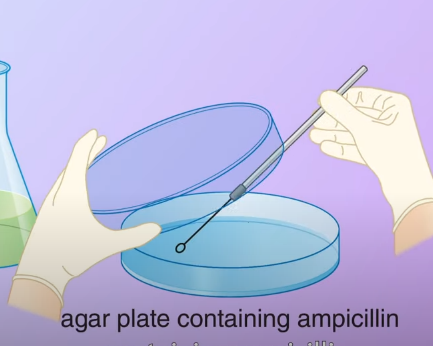 The ampicillin resistance gene in the plasmid enables the transformed bacteria to survive. They then divided and formed colonies, the bacteria that didn’t pick up any plasmid could not survive
The ampicillin resistance gene in the plasmid enables the transformed bacteria to survive. They then divided and formed colonies, the bacteria that didn’t pick up any plasmid could not survive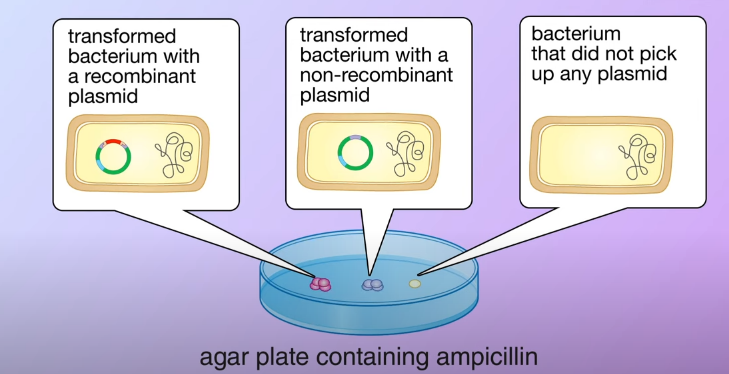 After that, its transferred a portion of bacteria from each colony to a second agar plate containing tetracycline as the transformed bacteria, and with the non-recombinant carrying the tetracycline resistance gene they survived and divided to form colonies. On the other hand in the transformed bacteria with the recombinant plasmid, the DNA fragment inserted made the tetracycline resistance nonfunctional therefore the bacteria cannot survive on the plate.
After that, its transferred a portion of bacteria from each colony to a second agar plate containing tetracycline as the transformed bacteria, and with the non-recombinant carrying the tetracycline resistance gene they survived and divided to form colonies. On the other hand in the transformed bacteria with the recombinant plasmid, the DNA fragment inserted made the tetracycline resistance nonfunctional therefore the bacteria cannot survive on the plate.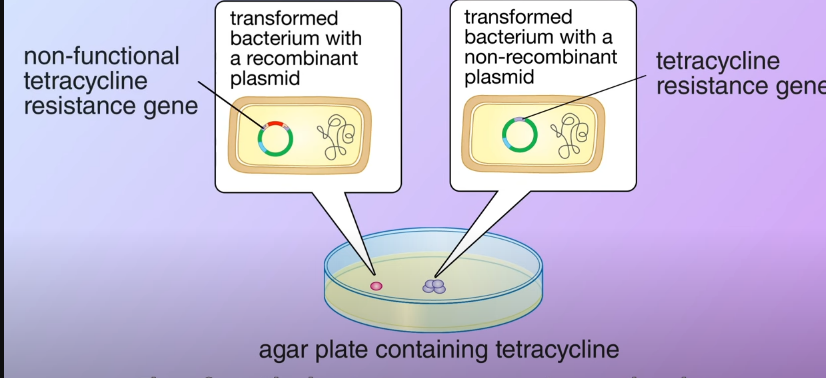
Manufacture of human insulin
The transformed bacteria are then cultures in fermenters. In each bacterium, the recombinant plasmid replicates independently of the bacterial chromosome. When the bacteria divide, the plasmid is also copied to the daughter cells so that many copies of the DNA encoding human insulin are produced
 Finally, gene expression is induced in the bacteria to produce polypeptides. The polypeptides are extracted from the bacteria and processed into functional human insulin.
Finally, gene expression is induced in the bacteria to produce polypeptides. The polypeptides are extracted from the bacteria and processed into functional human insulin.