Pathology Final SG
Cellular response to stress and noxious stimuli
Types of necrosis
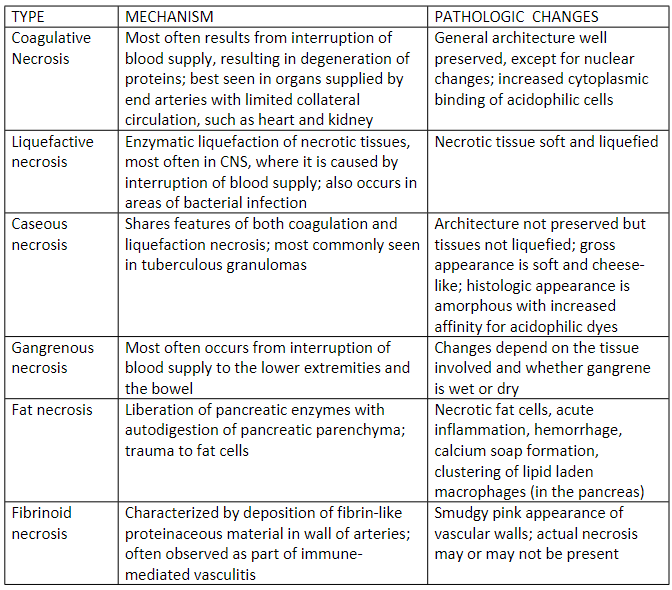
Coagulative necrosis:
Architecture of dead tissues is preserved for a span at least of some days; affected tissue exhibits a firm texture; the injury denatures not only the structural proteins but also enzymes and so blocks the proteolysis of dead cells; necrotic cells are ultimately removed by phagocytosis by infiltrating leukocytes and digestion of the dead cells by the enzymes of leukocytes; Ischemia caused by obstruction in a vessel may lead to coagulative necrosis of the suppled tissue in all organs except for the brain. A localized area of coagulative necrosis is called an infarct
Liquefactive necrosis:
Digestion of dead cells resulting in transformation of the tissues into a liquid viscous mass. It is seen in focal bacterial or, occasionally, fungal infections, because microbes stimulate the accumulation of leukocytes and the liberation if enzymes from these cells. The necrotic material is frequently creamy yellow because of the presence of dead leukocytes and is called pus
Gangrenous necrosis:
A term usually applied to a limb, generally lower leg, that has lost its blood supply and has undergone necrosis (usually coagulative necrosis) involving multiple tissue planes. When bacterial infection is superimposed there is more liquefactive necrosis because of the actions of degradative enzymes in the bacteria and the attracted leukocytes (giving rise to the term wet gangrene)
Caseous necrosis:
Is encountered most often in foci of tuberculous necrosis. The term caseous (cheese-like) is derived from the febrile white appearance of the area of necrosis.
Fat necrosis:
It refers to focal area of fat destruction, typically resulting from release activated pancreatic lipases into the substance of the pancreas and peritoneal cavity This occurs in calamitous abdominal emergency known as acute pancreatitis
Fibrinoid necrosis:
a special form of necrosis usually seen in immune reactions involving blood vessels. This typically occurs when complexes of antigens and antibodies are deposited in the wall of the arteries. Deposits of these ‘immune complexes’ together with fibrin that has leaked out of vessels, result in a bright pink and amorphous appearance under the microscope
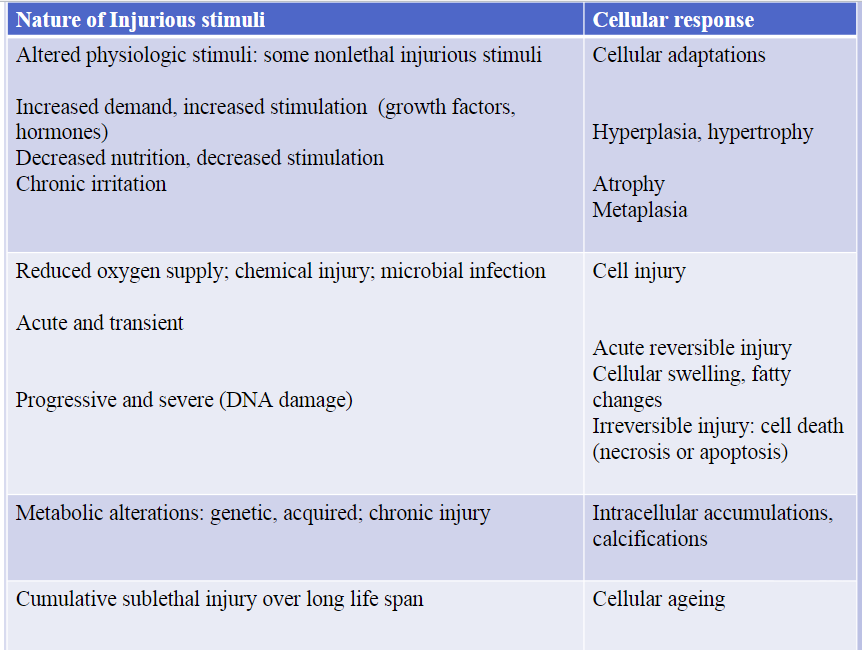
Morphogenic alterations in cell injury
REVERSIBLE INJURY
2 features recognized under light microscope:
Cellular swelling
Fatty changes
Cellular swelling appears whenever cells are unable to maintain ionic and fluid homeostasis and is a result of failure of energy-dependent ionic pumps in the plasma membrane
Fatty changes occur in hypoxic injury and various forms of toxic or metabolic injuries
IRREVERSIBLE INJURY: NECROSIS
Morphologic appearance is the result of:
Denaturation of intracellular proteins
Enzymatic digestion of the lethally injured cells
Cells unable to maintain membrane integrity and contents leak out Elicit inflammation in surrounding tissues
Necrosis
The morphologic appearance of necrosis as well as necroptosis is a result of denaturation of intracellular proteins and enzymatic digestion of the lethally injured cells
The enzymes that digest the necrotic cells are derived from lysosomes of the dying cells themselves
NUCLEAR CHANGES
Appears due to nonspecific breakdown of DNA
Pyknosis: characterized by nuclear shrinkage and increased basophilia; the chromatin condenses into solid , shrunken basophilic mass
Karyorrhexis: pyknotic nucleus undergoes fragmentation
Karyolysis: the basophilia of chromatin may fade, a change that presumably reflects loss of DNA because of enzymatic degradation by endo nucleases
Pathologic Calcifications
Dystrophic calcification: Deposition of calcium at sites of cell injury and necrosis
dead or dying tissues its called this
Metastatic calcifications: Deposition of calcium in normal tissues, caused by hypercalcemia (usually a consequence of parathyroid hormone excess)
in normal, vital tissues
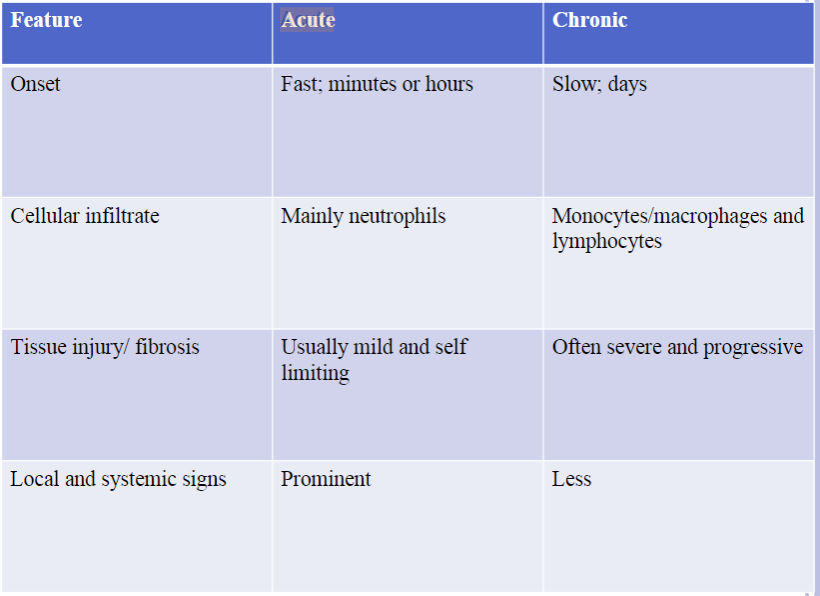
Acute and Chronic Inflammation
Acute Inflammation: the initial rapid response to infections and tissue damage
Develops within minutes to hours and is of short duration, lasting for several hours or a few days
Main characteristics include exudation of fluids and plasma proteins (edema) and emigration of leukocytes, predominantly neutrophils
When desired goals are achieved, the reaction subsides; if response fails, the reaction progresses to chronic phase
Host defense seen in innate immunity
Three major components:
1. Dilatation of small blood vessels leading to an increase in blood flow
2. Increased permeability of microvasculature enabling plasma proteins and leukocytes to leave the circulation
3. Emigration of leukocytes from microcirculation, their accumulation in the focus of
injury, and their activation to eliminate the offending agent
Five classic signs of acute inflammation
Rubor: rednes
Tumor: swelling
Calor: warmth
Dolor: pain
Functio laesa: loss of
function
CHRONIC INFLAMMATION
Is of longer duration
Associated:
More tissue destruction
Presence of lymphocytes and macrophages
Proliferation of blood vessels
Deposition of connective tissue
This reaction seen in adaptive immunity
Causes of Edema and effusion
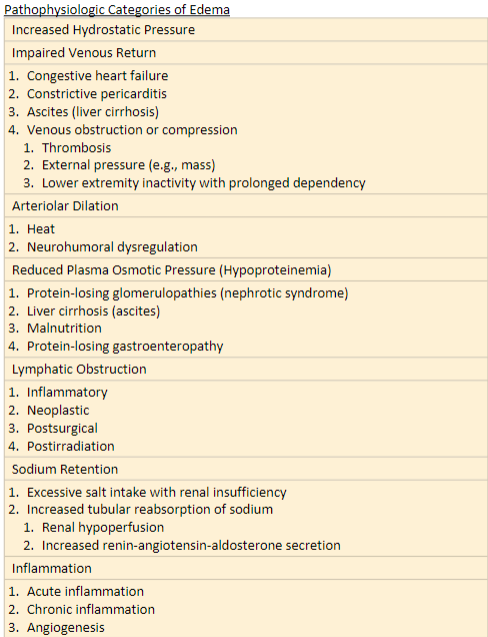
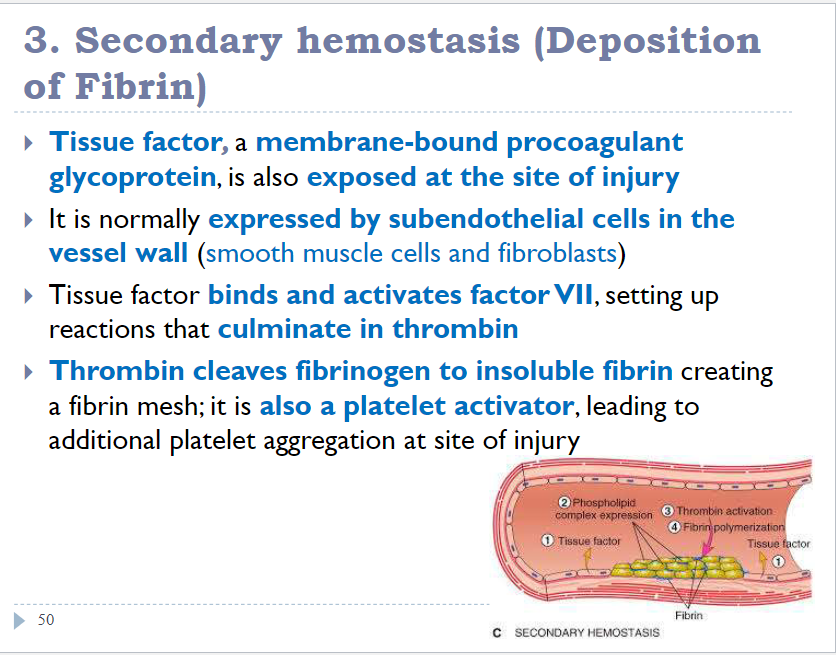
 Hemostasis and thrombosis
Hemostasis and thrombosis
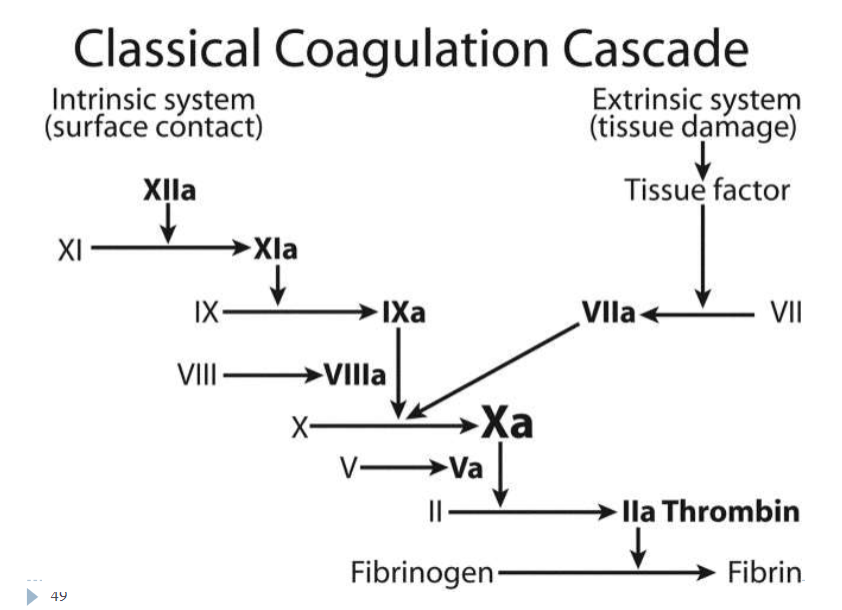
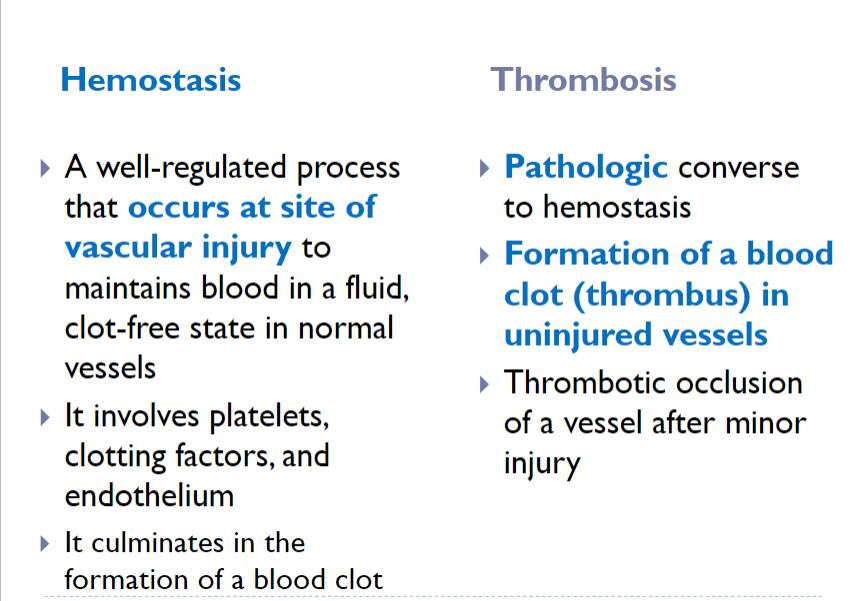 Role of Coagulation in hemostasis
Role of Coagulation in hemostasis
Marfan Syndrome (Ch 6. 43-50)
Disorder of connective tissue
manifested principally by changes in the skeletal, eyes, and cardiovascular system
1 in 5000 people
70-80% of cases are familial and transmitted by AD inheritance
remainder are sporadic and arise from new mutations
Caused by mutation in the FBN1 gene encoding fibrillin, which is required for structural integrity of connective tissues
clinical features:
tall stature
long fingers
bilateral subluxation of lens
mitral valve prolapse
aortic aneurysm
aortic dissection
Oral features
lips marked incompetent
retrognathia
a narrow, highly arched palate with crowding of the teeth
significantly greater risk for dental caries, higher pulpal calcifications and gingival inflammation, and TMJ subluxation
Ehlers-Danlos Syndrome (Ch 6. 52-58)
Comprise a clinically and genetically heterogenous group of disorders that result from some mutations in the genes that encode collagen, enzymes that modify collagen, and less commonly other proteins present in the extracellular matrix
Several variants of EDS, all characterized by defects in collagen synthesis or assembly
Each of the variants is caused by a distinct mutation involving one of the several collagen genes or genes that encode other ECM protein
Clinical features:
fragile hyperextensible skin vulnerable to trauma
hypermobile joints
ruptures involving the colon, cornea and large arteries
Wound healing is poor
Oral Manifestations
marked periodontal disease; seen at a relatively early age
easy bruising and bleeding during minor manipulations of the oral mucosa
recurrent subluxation of the TMJ
most patients have normal teeth
Ability of 50% of patients to touch the tip of their nose with their tongue (Gorlin sign)
seen in less than 10% of general population
Treatment:
accurate diagnosis and genetic counselling is very important
Familial Hypercholesterolemia (FHCL) (Ch 6. 60-66)
Caused by mutations in genes encoding the:
LDL (85%)
ApoB protein (5-10%)
activating mutations of PCSK9 (1-2%)
It affects how the body regulates and removes cholesterol
Autosomal Dominant disorder
Patients develop hypercholesterolemia because of impaired transportation of LDL into the cells
In heterozygotes for mutations in the LDL gene, elevated serum cholesterol greatly increases the risk of atherosclerosis and resultant coronary artery disease
homozygotes have an even greater increase in serum cholesterol and a higher frequency of ischemic heart disease
cholesterol also deposits along tendon sheaths to produce xanthomas (fatty deposits that build up under the skin)
Clinical Features
Tendinous xanthomas
Corneal arcus (deposit of cholesterol in arc around cornea of eye)
coronary artery disease
the aortic root is prone to develop atherosclerotic plaque at an early age
Treatment
Heterozygous FHCL: usually treated by statin therapy
Homozygous FHCL is harder to treat:
high dose of statin
LDL cholesterol apheresis (therapy that removes LDL from patient blood)
gene therapy
McArdle and Von Gierke disease (Ch 6. 80-82)
Glycogen storage diseases (glycogenosis)
inherited deficiency of enzyme involved in glycogen metabolism can result in storage of normal or abnormal forms of glycogen, predominantly in liver or muscles, but also in other tissues as well
Autosomal recessive
Von Gierke Disease
most common hepatic form
liver cells store glycogen
because of lack of hepatic glucose-6-phosphatase; enlarged liver and patients have hypoglycemia
McArdle Disease
myopathic form
lack of muscle phosphorylase giving rise to storage in skeletal muscles and cramps after exercise
Down Syndrome (Ch 6. 84-88)
Associated with an extra copy of genes on chromosome 21, most commonly due to trisomy 21 and less frequently from translocation of extra chromosomal material from chromosome 21 to other chromosomes
Maternal age has strong influence on the incidence of trisomy 21
Patients have severe intellectual disability, flat facial profile, epicanthic fold, cardiac malformations, higher rise of leukemia, infections, and premature development of Alzheimer disease
oral manifestations
open mouth with tendency of tongue protrusion
fissured and furrowed tongue
Mouth breathing with drooling
chapped lower lip and angular cheilitis
increased periodontal disease
Klinefelter Syndrome (Ch 6. 97-99)
HAPPENS IN MEN
is a sex chromosome disorder in boys and men that result from the presence of an extra X chromosome in cells (46XXY)
at least 2 X and one or more Y chromosomes
Patients have testicular atrophy, sterility, reduced body hair, gynecomastia (enlargement of breast tissue in males) and eunuchoid body habitus (body shape characterized by long limbs, reduced muscle mass, narrow shoulders, and a wider-than-usual pelvis)
most common cause of male sterility
Turner Syndrome (Ch 6. 100-102)
HAPPENS IN FEMALES
Monosomy X chromosomal abnormality occurs as a random event during the formation of reproductive cells (eggs and sperm) in the affected person’s parent; most cases not inherited
chromosomal affects development in females
short stature, webbing of the neck, cardiovascular malformations, amenorrhea, lack of secondary sex characteristic, and fibrotic covaries (lack of estrogen)
Most common is short stature, which becomes evident by about age 5
Systemic Lupus Erythematosus (Ch 4. 125-136)
Most common connective tissue diseases in the US
Type III hypersensitivity
Serious multisystem disease
exhibits a variety of cutaneous and oral manifestations
very difficult to diagnose in early stages of the disease (nonspecific and vague pattern)
Women affected 8-10x more frequently than males
average age is 31 years
Clinical features
common findings include fever, weight loss, arthritis, fatigue, and general malaise
in 40-50% of patients, a characteristic butterfly rash (Malar rash) is seen (over the molar area and nose, typically sparing the nasolabial folds)
sunlight makes the lesions worse
Arthritis- multiple joints (polyarthritis)\
photosensitivity
Kidneys are affected 40-50% of patients; may ultimately lead to kidney failure, thus it is typically the most significant aspect of the disease
Cardiac involvement is common; pericarditis is the most frequent involvement
at autopsy nearly 50% of patients display warty vegetation affecting the heart valves (Libman-Sacks endocarditis)
some patients may develop superimposed bacterial endocarditis
Oral lesions
Develop in 5-25% of patients
Usually affect palate, buccal mucosa, and gingiva
may be lichenoid or granular or nonspecific
Lupus cheilitis: involvement of the vermillion border of the lower lip is sometimes seen
Varying degree of ulcerations, pain, erythema, and hyperkeratosis may be seen
HIV/ AIDs (Ch 4. 208-224)
Secondary immunodeficiencies
encountered in individuals with cancer, diabetes, and other metabolic diseases, malnutrition, chronic infections, and in persons receiving chemotherapy or radiation therapy for cancer or immunosuppressive drugs to prevent GVHD or to treat autoimmune disease
HIV infection/AIDS
a virus that attacks the body’s immune system; if HIV is not treated it can lead to AIDS (acquired immunodeficiency syndrome)
AIDS is the late stage of HIV infection that occurs when the body’s immune system is badly damaged because of the virus
A person with HIV is considered to have progressed to AIDS when:
the # of their CD4 cells fall below 200 cells per cubic millimeter of blood (220 cells/mm3 ); in someone with a healthy immune system, CD4 counts are between 500 and 1,600 cells/mm3 )
OR
they develop one or more opportunistic infections regardless of their CD4 count
Caused by the retrovirus human immunodeficiency virus (HIV) and is characterized by profound immunosuppression that leads to opportunistic infections, secondary neoplasms, and neurological manifestations
In the USA, it is the:
2nd leading cause of death in men between 25 and 44 years of age
3rd leading cause of death in women between 25 and 44 years of age
Retrovirus
after infecting a cell, a retrovirus uses an enzyme called reverse transcriptase to convert its RNA into DNA
the retrovirus then integrates its viral DNA into the DNA of the host cell, which allows the retrovirus to replicate
HIV, the virus that causes AIDS is a retrovirus
HIV epidemiology
5 group of adults at high risk for developing AIDS:
men who have sex with men
heterosexual transmission, chiefly due to contact with members of other high risk groups contacts of members of another high-risk group
IV drug users
Hemophiliacs, especially those who received large amounts of factor VIII or factor IX concentrates before 1985
Recipients of blood and blood components infected with HIV
HIV infection of the newborn
3 major routes of transmission
sexual contact
parental inoculation
passage of the virus from infected mother to their newborn
The propertied of HIV
a human retrovirus belonging to the lentivirus family
2 genetically different but related forms of HIV have been isolated from patients with AIDS
HIV-1: most common in USA, Europe and central Africa
Spherical shape, electron dense core containing RNA, surrounded by lipid envelope
HIV-2: principally in west Africa and India
Pathogenesis and course of HIV infection and AIDS
virus entry into cells: requires CD 4 and coreceptors; main cellular targets are CD4+ helper cells, macrophages and dendritic cells
Viral replication: the virus genome integrates into host cell DNA
Mechanism of immune deficiency:
Loss of CD4+ T cells; CD4 falls below 200 Cells per cubic mm of blood
relentless and profound depletion of CD4+ T cells (1-2 billion CD4+ cells die each day)
Normal CD4/CD8 ratio is 2:1 (AIDs CD4/CD8 ratio is <0.5
Defective macrophage and dendritic cell functions
Destruction of architecture of lymphoid tissue (late)
B-Cell system in HIV infection
patients with AIDS are unable to mount an antibody response to a new antigen
Progression of infection
Acute infection of mucosal T cells and dendritic cells
Viremia and dissemination of virus
Latent infection of cells in lymphoid tissue
Continuing viral replication and progressive loss of CD4+ T cells
Clinical manifestations
Opportunistic infections
tumors (B cell lymphoma)
CNS abnormalities
Primary Immunodeficiency Diseases (Ch 4. 197-205)
Caused by genetic (inherited) defects that affect the innate immunity (phagocytes, NK cells or complement) or acquired immunity (B and T cells)
most of these are detected in infancy, between 6 months- 2 years of life
X-Linked Agammaglobulinemia (Bruton Agammaglobulinemia)
characterized by failure of B cell precursors to develop into mature B cells
X-linked inheritance
Almost seen entirely in males; symptomatic; apparent at about 6 months of age as maternal immunoglobins are depleted
light chains are not synthesized, therefore complete immunoglobulins are absent
Recurrent bacterial infections
H. influenzae, S. pneumoniae, S. aureus
Isolated IGA Deficiency
Common immunodeficiencies caused by impaired differentiation of naïve B cell to IgA-producing plasma cells
Patients have an extremely low levels of both serum and secretory IgA; most are asymptomatic
1 in 600
Because IgA is the major antibody in mucosal secretions, mucosal defenses are weakened and infections occur in the respiratory, GI, and urogenital tracts
Symptomatic patients present with recurrent sinopulmonary infections and diarrhea
Thymic Hypoplasia (Digeorge syndrome)
T-cell deficiency that results from failure of development of the thymus
low number of T cells in the blood and lymphoid tissue and poor defense against certain viral and fungal infections
90% cases cause by a small germline deletion that maps to chromosome 22q11
part of the CATCH 22 syndrome
cardiac defects, abnormal facial features, thymic hypoplasia, cleft palate, hypocalcemia
Systemic Sclerosis (Scleroderma) (Ch 4. 172-193)
Characterized by:
Chronic inflammation thought to be result of autoimmunity
Widespread damage to small blood vessels
progressive interstitial and perivascular fibrosis in the skin and multiple organs
The skin is most affected, but the GI tract, kidneys, heart, muscles, and lungs also are frequently involved
Pathogenesis
cause is unknown
Disease likely results from 3 interrelated processes- autoimmune responses, vascular damage, and collagen deposition
Autoimmunity: CD4+ cells respond to an unknown antigen, accumulate in skin and release cytokines that activate inflammatory cells and fibroblasts; several of these cytokines stimulate transcription of genes encoding collagen and other extracellular matrix proteins
Vascular damage
microvasculature disease is present in the early course of the disease; this could be the result of chronic inflammation; eventually widespread narrowing of the microvasculature leads to ischemic injury and scarring
Fibrosis:
fibroblasts of these patients have a intrinsic abnormality that causes them to produce excessive amount of collagen
Clinical Features:
Women are 3-5x more frequently affected than men
50-60 year age group
although SS shares many features with SLE, RA, and polymyositis, its distinctive features are the striking skin changes
insidious onset, with the cutaneous changes often responsible for bringing the problem to the patients attention
Raynaud phenomenon
manifested as numbness and tingling of fingers and toes caused by episodic vasoconstriction of arteries and arterioles, is seen in virtually all patients
often first sign of the disease
vasoconstrictive event triggered by emotional distress or exposure to cold
not specific to SS; can also be seen in healthy individuals and other autoimmune diseases
Dysphagia
attributed to esophageal fibrosis and its resultant hypomobility is present in more than 50% of patients; eventually destruction of the esophageal wall leads to atony and dilation, especially at its lower end
Respiratory difficulty
caused by pulmonary fibrosis may result in right-sided cardiac dysfunction
Myocardial fibrosis
may cause cardiac failure
Skin
develops a diffuse and hard texture, and its surface is usually smooth
Facial skin
subcutaneous collagen depositions results in characteristic smooth, taut, masklike facies; nasal alae become atrophied
Hands
fingers may be fixed in a claw-like position with resorption of terminal phalanges (acro osteolysis)
Internal organ fibrosis and/or vascular damage
Oral features:
occur in varying degrees
Microstomia
develops as a result of deposition of collagen in perioral tissues
seen in 70% of patients and results in limited mouth opening
Characteristic furrows radiating from the mouth produce a “purse string” appearance
loss of attached gingival mucosa and multiple areas of gingival recession may occur
Dental radiographs
diffuse widening of the periodontal ligament space is often present throughout the dentition
extent of widening may vary
Varying degrees of resorption of the posterior ramus of the mandible, the coronoid process, the chin, and the condyle may be detected on panoramic radiographs (seen in 10-20% if patients
Limited cutaneous systemic sclerosis
was preciously referred to as CREST syndrome to denote key features
C-calcinosis
R-Raynaud phenomenon
E-esophageal dysmotility
S-sclerodactyly
T-telangiectasias
Laboratory studies
virtually all patients have that react to a variety of nuclear antigens (ANAs)
two ANA strongly associate with sytemic sclerosis have been established
one of these directed against DNA topoisomerase I (anti-scl 70), is highly specific, it is present 10%-20% of patients with diffuse systemic sclerosis
The other, anticentromere antibody is found in 20-30% of patients, who tend to have CREST Syndrome
Treatment
difficult; natural waxing and waning course of the disease makes it difficult to assess the effectiveness of a given treatment in an open trial
Systemic medications such as penicillamine, are prescribed to inhibit collagen production
other management strategies are directed at controlling symptoms
dental symptoms: collapsible dentures
Sjogren Syndrome (Ch 4. 160-171)
Chronic disease characterized by dry eyes (keratoconjunctivitis sicca) and dry mouth (xerostomia) resulting from immunologically mediated destruction of the lacrimal and salivary glands
occurs as an isolated disorder (primary form) also known as sicca syndrome or more often in association with another autoimmune disease (secondary form)
among the associated disorders, rheumatoid arthritis is the most common, but some patients may have SLE, Scleroderma, vasculitis, or thyroiditis
Pathogenesis
the characteristic decrease in tears and saliva (sicca syndrome) is the result of lymphocytic infiltration and fibrosis of the lacrimal and salivary glans
the infiltrate contain activated CD4+ helper T-cells and some B cells, including plasma cells
initiating trigger may be a viral infection of the salivary glands, which causes local cell death and release of tissue self-antigens
75% of patients have rheumatoid factor whether co-existing RA is present or not
decreased saliva and tears result from inflammatory destruction of exocrine glands
Lymphatic infiltration (predominantly CD4+ T cells and some plasma cells) and fibrosis of lacrimal and salivary glands
Clinical Features:
mostly commonly occurs in women between 50-60 years of age; rare examples have been described in children
female ration is 9:1
keratoconjunctivitis produces blurring of vision, burning and itching, and thick secretions
Xerostomia results in difficulty in swallowing solid food, a decrease in the ability to tast, cracks and fissures in the tongue, and dryness of oral mucosa
parotid gland enlargement seen in ½ of patients
the severity of xerostomia can vary widely from patient to patient
Saliva may appear frothy, with lack of usual pooling of saliva in the floor of the mouth
tongue often becomes fissured and exhibits atrophy of the papillae
Oral mucosa is red and tender and usually results in secondary candidiasis
Radiographic features of SS
often reveal punctate sialectasis and lack of normal arborization of the ductal system, typically demonstrating a “fruit-laden, branching tree”
Lab values
a variety of autoantibodies can be produced, and their presence can be helpful clue in diagnosis
a positive rheumatoid factor (RF) is found in approx. 60% of patients, regardless whether the patient has RA
Antinuclear antibodies (ANAs) are present 75-85% of patients
2 particular nuclear antibodies- antibody SS-A (anti Ro) and anti SS-B (anti-La) may be found, especially in SS patients
Treatment
mostly supportive
dry eyes are best managed by periodic used of artificial tears
artificial saliva and sugarless gun or candy can help keep mouth moist
NOTE
about 5% patients with Sjogren syndrome develop lymphoma, an incident that is 40-fold greater than normal
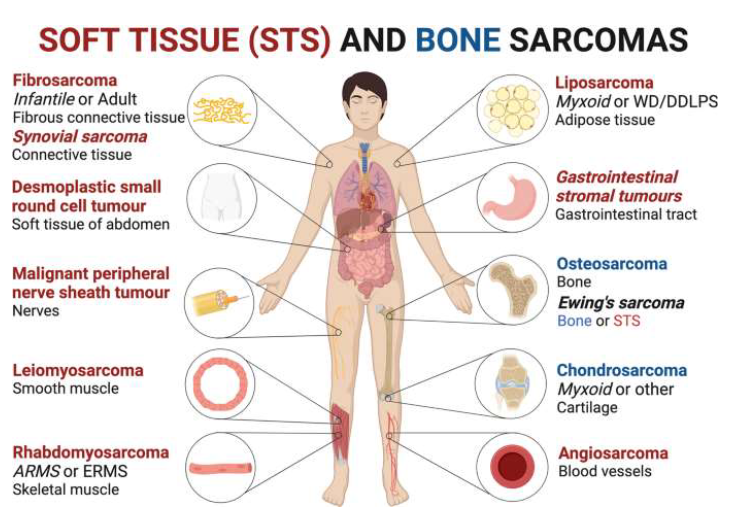
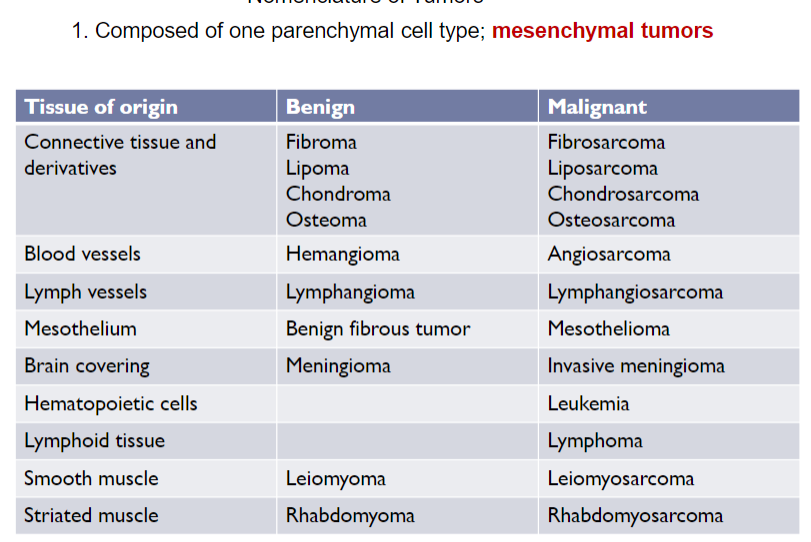
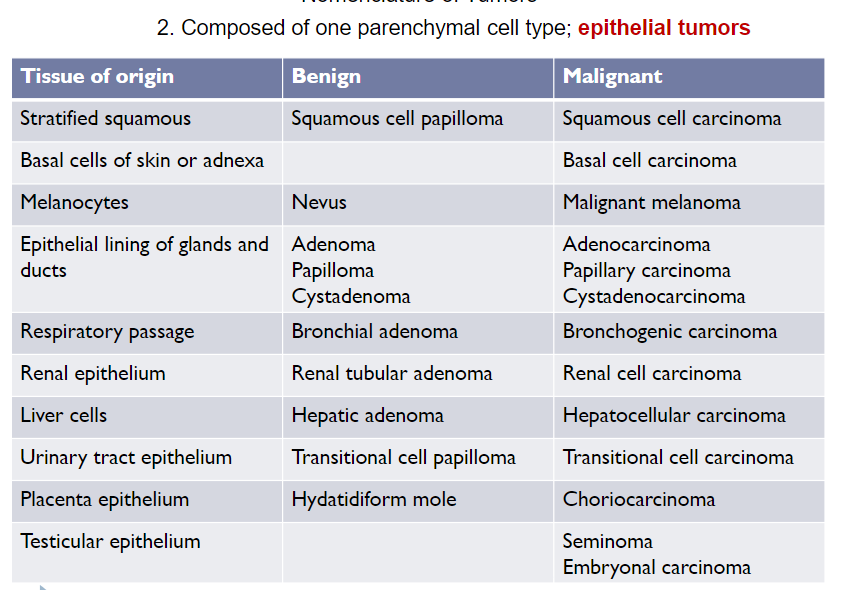
Nomenclature of Tumors (Ch 5. 5-16)
Benign Tumors
Remain localized at their site of origin and are generally amenable to surgical removal
Patient generally survives
Exceptions arise when benign tumors occur in vulnerable locations such as the brain; here, even "benign" tumors may cause significant morbidity and are sometimes even fatal
Naming of benign tumor of mesenchymal cells: "oma" is attached to the name of the cell type from which the tumor arises
Chondroma, adenoma, papilloma
Malignant Tumor
Can invade and destroyed adjacent structures and spread to distant sites (metastasize)
Collectively called "cancers"
Not all cancers peruse a deadly course; some are discovered at early stages that allow for surgical excision, and others are cured with systemically administered drugs or therapeutics antibodies
Malignant tumors arising in epithelial cell origin: carcinoma
Malignant tumors arising in solid mesenchymal tissues: sarcoma
Malignant tumors arising in blood-forming cells: leukemia
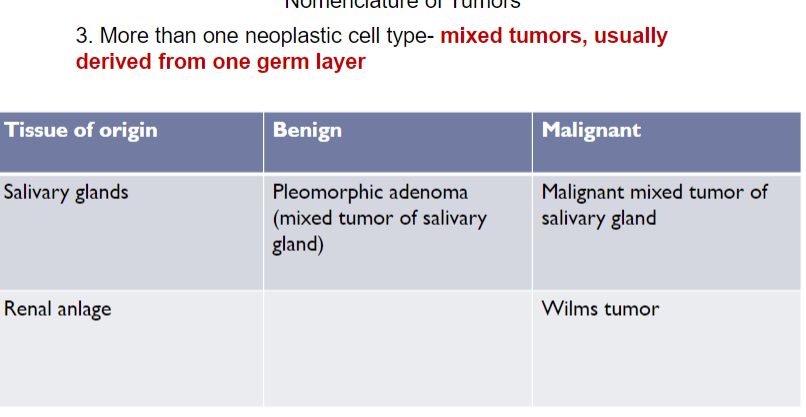
Mixed tumors
in most neoplasms, all parenchyma cells closely resemble on another, but some types of tumors more than 1 line of differentiation is evident, creating distinct subpopulation of cells
Classic example: mixed tumor of salivary glands (pleomorphic adenoma), which contains epithelial components scattered within myxoid stroma that may contain islands of cartilage and bone
Kawasaki Disease (Ch 8. 65-67)
Acute, febrile, usually self-limiting illness of infancy and childhood associated with large- to medium-sized vessel arteritis
80% < 4-years of age
Clinical significance stems from involvement of coronary arteries
Coronary arteritis can result in aneurysms that rupture or thrombose, causing myocardial infarction
In genetically susceptible people, a variety of infectious agents (mostly virus) have been positioned to trigger the disease
Presents with conjunctival and oral erythema, blistering edema of hands and feet, erythema of palms and soles, desquamative rash, cervical LN enlargement
Approx. 20% of untreated patients develop cardiovascular signs and symptoms resulting in asymptomatic coronary arteritis, to giant coronary artery aneurysms
These aneurysms are associated with rupture, thrombosis, MI, or sudden death
With IV immunoglobulin therapy and aspirin, the rate of symptomatic disease is < 4%
Diagnostic Features:
Red eyes
Coronary artery aneurysms
Swollen lymph nodes
Peeling of skin
Red, dry, cracked lips around fingernails/toenails and inflamed tongue
Widespread rash
Fever (for more than 5 days)
Swelling and/or erythema of palms/soles
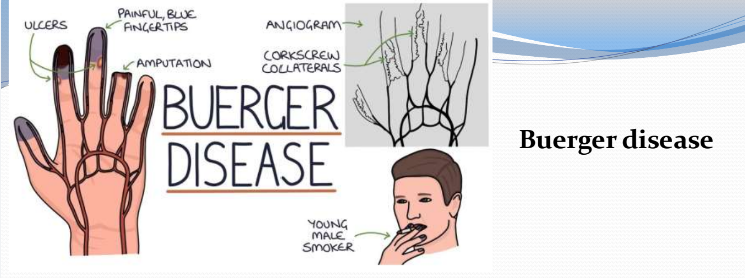
Buerger Disease (Thromboaniitis Obliterans) (Ch 8. 71-73)
Characterized by segmental, thrombosing, acute and chronic inflammation of medium- and small-sized arteries, especially the tibial and radial arteries, that often lead to vascular insufficiencies, typically of the extremities
Occurs almost exclusively in heavy cigarette smokers
Usually before the age of 35
Early manifestations include Raynaud phenomenon, instep foot pain induced by exercise, and a superficial nodular phlebitis
Vascular insufficiencies tend to be accompanied by severe pain, even at rest, due to neural involvement
Chronic extremity ulcerations can develop, progressing over time to frank gangrene
Smoking abstinence in the early stage of the disease can prevent further attacks
Once established, vascular lesions do not respond to smoking abstinence
Granulomatosis with Polyangiitis (Ch 8. 68-70)
Previously called Wegener granulomatosis
Necrotizing vasculitis characterized by a triad of acute necrotizing granulomas of the upper respiratory tract or the lower respiratory tract or both, necrotizing or granulomatous vasculitis affecting small-to-medium-sized vessels, and focal necrotizing, often crescentic, glomerulonephritis
More common in males; average age of 40 years
Classic features include bilateral pneumonitis, chronic sinusitis, mucosal ulcerations of the nasopharynx, and renal disease
Rashes, myalgia, articular involvement, neuritis, and fever can also occur
If left untreated, the disease is rapidly fatal with 80% mortality within 1 year
Strawberry gingivitis
Raynaud Phenomenon (Ch 8. 76-80)
Results from exaggerated vasoconstriction of arteries and arterioles in response to cold or emotion
It most commonly affects the extremities, particularly fingers and toes
Occasionally the nose, earlobes, or lips are affected
Restricted blood flow induces paroxysmal pallor and even cyanosis in severe cases
Involved digits show "red, white, and blue" color changes
Primary Raynaud phenomenon affects 3%-5% of the general population
in young women
occurs symmetrically in the extremities
severity and extent of involvement does not progress
Secondary Raynaud phenomenon refers to vascular insufficiencies due to arterial disease caused by other entities (SLE, scleroderma, Buerger disease, or even atherosclerosis)
Asymmetric involvement of extremities
progressively worsens over time
RP may be the first indication of immune-mediated vasculitides, any patient with symptoms should be evaluated
10% patients manifest an underlying disorder
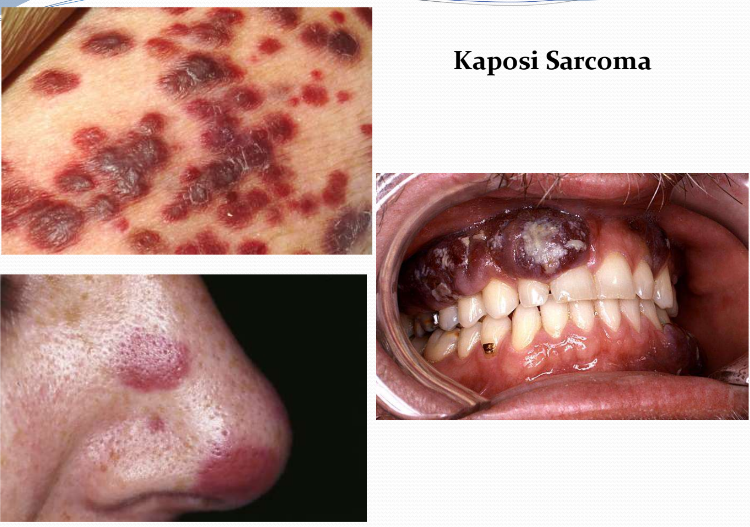
Kaposi Sarcoma (Ch 8. 98-100)
Malignant neoplasm of endothelial cells caused by human herpesvirus 8 (HHV 8; AKA KS herpesvirus)
It is common in patients with AIDS; its presence is used as a criterion for diagnosis of AIDS
4 clinical presentations:
Classic
Endemic (African)
Epidemic (AIDS-related)
Iatrogenic (transplant-associated)
Clinically progress through 3 stages: Patches (red-purple macules), raised plaques and eventually nodular lesions
 Atherosclerosis (Ch 8. 34-42)
Atherosclerosis (Ch 8. 34-42)
Intimal based lesion composed of a fibrous cap and a atheromatous core
The constituents of the plaque include smooth muscle cells, extracellular matrix, inflammatory cells, calcifications, lipids, and necrotic debris
Plaques develop and grow slowly over decades
Atherogenesis is driven by an interplay of vessel wall injury and inflammation
stable plaques can produce symptoms related to chronic ischemia by narrowing vessel lumens
Unstable plaques can produce fatal ischemic complications related to acute plaque rupture and embolism
Underlies the pathogenesis of coronary, cerebral, and peripheral vascular disease
Causes more morbidity and mortality (roughly 1/2 of all death) in the western world than any other disorder
Major risk factors:
Nonmodifiable:
genetic abnormalities, family history, increasing age, male gender
Modifiable
hyperlipidemia, hypertension, cigarette smoking, diabetes, inflammation
Consequences
Major consequences
myocardial infarction (heart attack)
cerebral infarction (stroke)
aortic aneurysm
peripheral vascular disease (gangrene of the legs)
Large elastic arteries (aorta, carotid, and iliac arteries) and large and medium sized muscular arteries (coronary and popliteal arteries) are the major targets
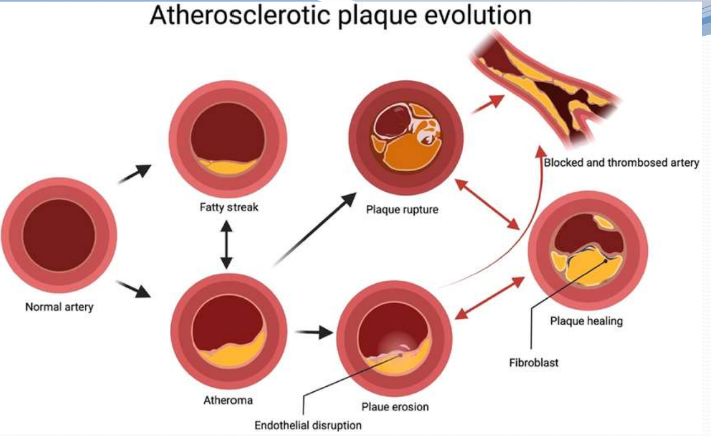
Formation and evolution of atherosclerotic plaques
the atherosclerotic plaques progress from a fatty streak to a classic atheroma leading to either an erosion or rupture of thin-capped fibroatheroma
Atheroma cycles between healing, thrombosis and finally blockage of the concerned artery
there could by multiple cycles of healing and rupture before an artery is blocked
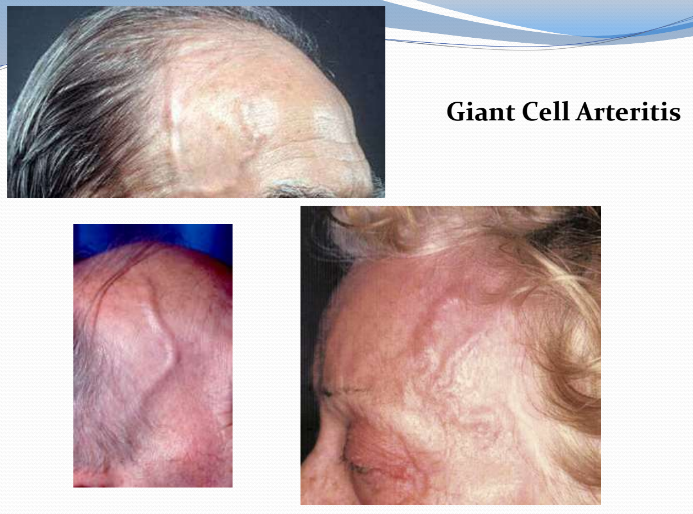
 Giant Cell (temporal) Arteritis (GCA) (Ch 8. 53-57)
Giant Cell (temporal) Arteritis (GCA) (Ch 8. 53-57)
Chronic, classically granulomatous inflammation of large to small sized arteries that principally affect arteries in the head (T-cell response)
It is the most common form of vasculitis among elderly adults in the Us and Europe (rare before the age of 50)
Ophthalmic, vertebral and aorta may be involved
ocular symptoms abruptly appear in about 50% patients ( diplopia and blindness); early diagnosis and treatment is vital
Temporal arteries are not particularly vulnerable but their name is added to the disorder since they are the most readily biopsied in making diagnosis
Symptoms are vague and constructional (fever, fatigue, weight-loss) or may involve facial pain and head ache, most intense along the course of superficial temporal artery, which may be painful to palpation
Diagnosis is based on biopsy and histologic confirmation
GCA can be extremely focal within an artery
adequate biopsy requires at least 1-cm segment, even then, a negative biopsy result does not include the diagnosis
Treatment: corticosteroids and anti-TNF therapies are effective
Lingual necrosis is a rare manifestation of GCA
Tongue infarction at second day
initial auto-amputation of necrotic tongue at 5th day
tongue at 20th day presenting full epithelization
 Deep Vein Thrombosis (Ch 8. 87)
Deep Vein Thrombosis (Ch 8. 87)
Also known as thrombophlebitis and phlebothrombosis
deep vein thrombosis account for 90% of cases
Decreased blood flow in the setting of prolonged immobilization is the most common cause of lower extremity deep vein thrombosis (DVT)
Can occur with extended bed-rest or sitting during a long airplane ride or automobile excursion
additional risk factors include pregnancy, oral contraceptive use, obesity, CHF, and malignancy
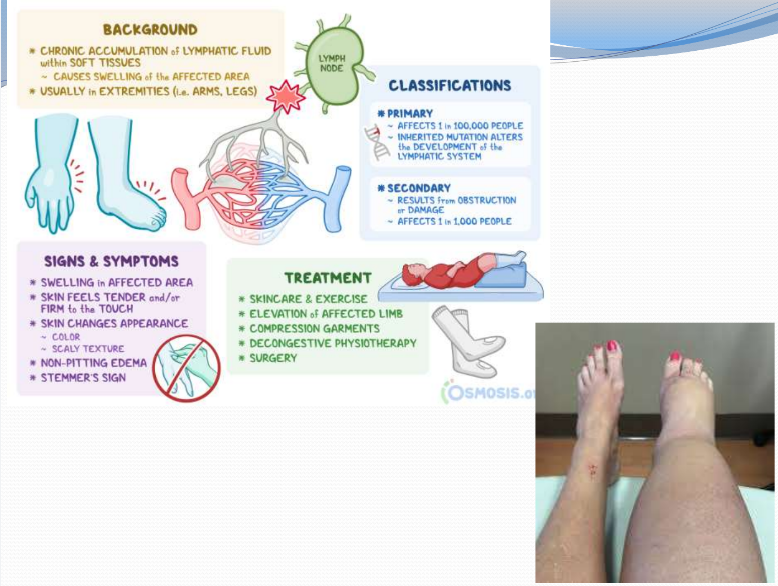
Lymphedema (Ch 8. 91-92)
Build-up of fluid in soft tissues when the lymph system is damaged or blocked
Secondary causes include:
Tumors
Surgical procedures
Post radiation fibrosis
Filariasis
Post inflammatory thrombosis or scarring
 Freckles (Ephelis) (Ch 7. 10)
Freckles (Ephelis) (Ch 7. 10)
Most common pigmented lesions of childhood in lightly pigmented individuals
Small (one to several mm), tan-red to light brown macules (flat lesions) that appear after sun exposure
Results from increased amounts of melanin pigment within basal keratinocytes
Number of melanocytes remain the same
Once present, they fade and darken in a cyclic fashion during winter and summer, respectively
Melanocytic Nevus (Moles) (Ch 7. 12)
Benign neoplasms caused in most cases by acquired activating mutations in components of the BRAF or less often, RAS signaling pathways
Tan to brown, uniformly pigmented, small (< 6mm) relatively flat macules or elevated papules (small round topped elevated lesion) with well-defined, rounded borders
Nevus cells are rounded cells
They are from neural crest origin
Considered to be cousins of melanocytes
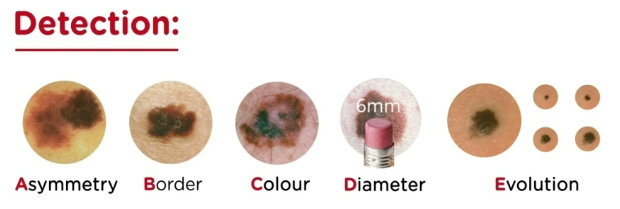
Melanoma (Ch 7. 15)
Most deadly of all skin cancers; highly aggressive; strongly linked to acquired BRAF mutations caused by exposure to UV radiation in sunlight
In 10%-15% of patients, the risk of melanoma is inherited as an AD trait with variable penetrance
The A,B,C,D, and E of melanoma
Asymmetry: lesions are asymmetric
Borders: irregular and often notched
Color is variable and not uniform
Diameter larger than nevi and is increasing (> 6mm)
Evolving; lesions are constantly enlarging, either
superficially or in a nodular fashion
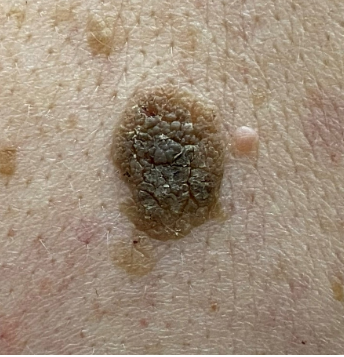
Seborrheic Keratosis (Ch 7. 18-19)
Common; occurs most frequently in middle-aged or older individuals
Arise spontaneously; particularly numerous on trunk
In people of color, multiple small SK on the face are termed dermatosis papulose nigra (seen in 35% of African-American adults)
May appear suddenly in large numbers as part of paraneoplastic syndrome (Leser-Trelat sign)
Round, elevated, coin-like waxy plaque that varies in diameter from mm to several cm; "pasted on" look
Uniformly tan to dark brown; velvety to granular surface
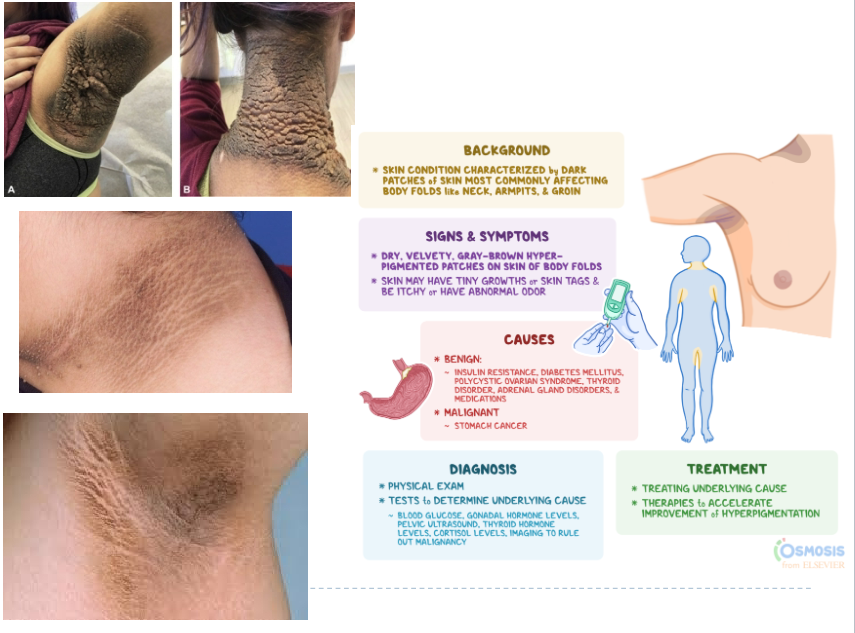
Acanthosis nigricans (Ch 7. 20-21)
Thickened, hyperpigmented skin with a velvet-like texture
Most commonly appears in the flexural areas (axillae, skin folds of the neck, groin-area where upper thighs meets the lowest part of abdomen, and anogenital areas)
2 types:
In at least 80% cases it is associated with benign conditions and develop gradually; usually during childhood and puberty
In 20% of cases, it is associated with cancers; most commonly gastrointestinal adenocarcinoma; usually in middle-aged or older individuals
Can be an important cutaneous sign of several underlying benign and malignant conditions
Fibroepithelial Polyp (Skin Tag) (Ch 7. 22)
AKA: acrochordon
One of the most common cutaneous lesions
In middle-aged or older individuals; on the neck, trunk, and face
Soft, flesh-colored, bag-like tumors
Often attached to the surrounding skin by a slender stalk
Not uncommon or them to undergo ischemia due to torsion which maycause pain and precipitate their removal
Squamous Cell Carcinoma (Ch 7. 31-32)
Second most common tumor arising on sun-exposed sites in older people, exceeded only by basal cell carcinoma
> men (except for lesions of lower legs)
Usually discovered while small and respectable; < 5% metastasize to regional lymph nodes
Most common cause is DNA damage induced by exposure of UV light; second most common association is chronic immunosuppression
Other risk factors include tobacco, betel nut chewing, industrial carcinogens, ionizing radiations, chronic ulcers, and old burn scars
Stems from multiple driver mutations: TP53, CDKN2A, PIK3CA, KMT2D, and NOTCH1
Basal Cell Carcinoma (Ch 7. 33)
Distinctive, locally aggressive cutaneous tumor associated with mutations in PTCH1, PTCH2, SMO and SUFU genes
Most common locally invasive cancers in humans (1 million cases/ year in the US)
Slow growing; rarely metastasize
Vast majority recognized in early stages
Occurs on sun exposed skin; incidence increases in the setting of immunosuppression
Pearly papules
Telangiectasia present on papules
Urticaria (Hives) (Ch 7. 45-46)
Common hypersensitivity reaction
Usually caused by localized degranulation of mast cells and is uniformly associated with dermal microvasculature hyperpermeability
Combination of these effects produce pruritic edematous plaques called wheals; Small pruritic papules to large erythematous plaques
> between ages 20 and 40; all ages are susceptible; > in areas exposed to pressure (trunk, distal extremities, and ears)
Individual lesions develop and fade within hours (usually, 24 hours); episodes may last for days or persist for months
Treatment: antihistamines; oral corticosteroids
Psoriasis (Ch 7. 51-52)
Chronic inflammatory dermatosis that appears to have an autoimmune basis; 1%-2% of US population affected
Environmental and genetic factors at play
Persons of all ages affected; approx. 15% of patients have associated arthritis; mild or severe
> skin of knees, elbows, scalp, lumbosacral areas, intergluteal clefts and glans penis
Well-demarcated, pink to salmon-colored plaques covered by loosely adherent silver-colored
scales
Koebner phenomenon: local trauma can induce lesion in susceptible individuals
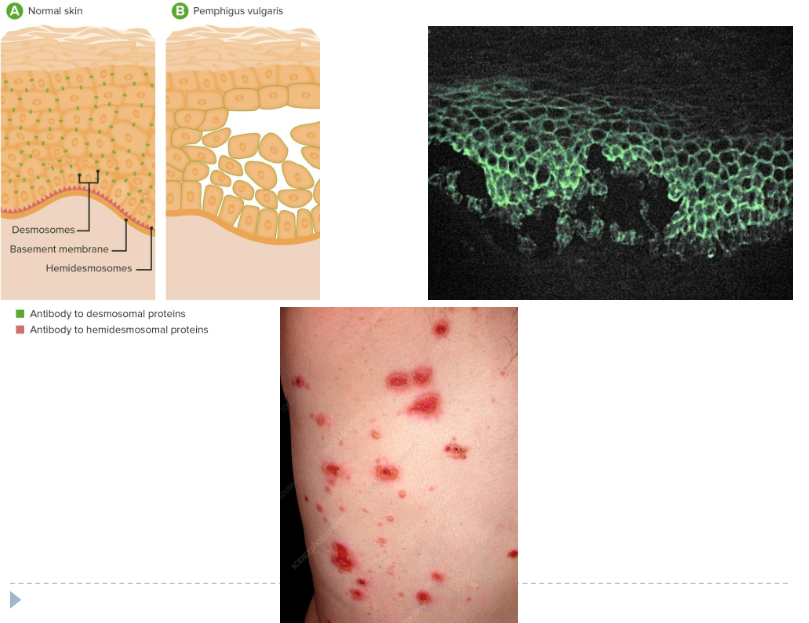
Pemphigus (Ch 7. 59-60)
Blistering disorder caused by autoantibodies (IgG) that result in the dissolution of intercellular attachments (desmoglein) within the epidermis and mucosal epithelium
> 4-6th decade of life; M = F
Pemphigus vulgaris most common variant; involves skin and mucosa
Oral ulcers may persist for years before skin lesion appear
Primary lesions are vesicles or bullae that rupture easily, leaving shallow erosions
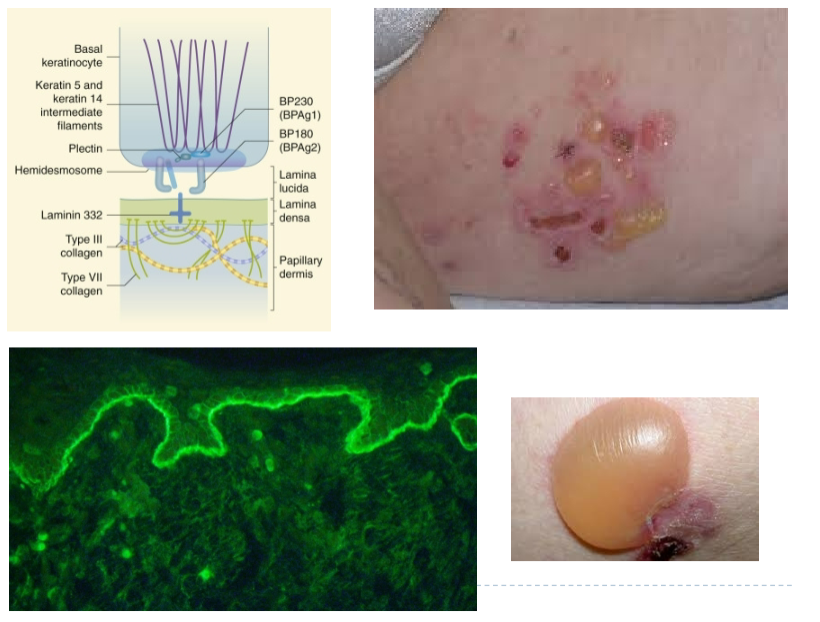
Bullous Pemphigoid (Ch 7. 61-62)
Caused by autoantibodies that bind to the proteins (hemidesmosomes) that are required for adherence of basal keratinocytes to the basement membrane
Antibodies deposition occur in a continuous linear pattern at the dermoepidermal junction
Generally, affects elderly individuals; 10%-15% show oral lesions
Tense bullae filled with clear fluid involving erythematous or normal appearing skin; , 2 cm in diameter; do not rupture easily
Heal without scarring
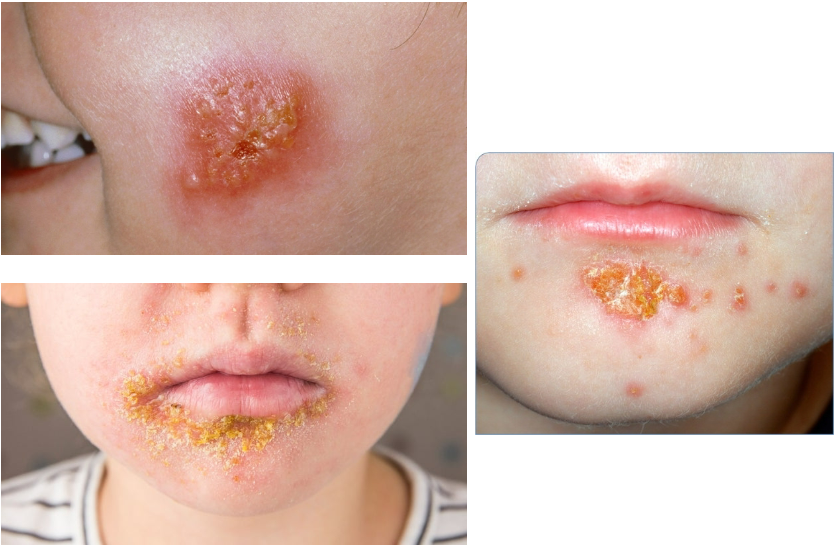
Impetigo (Ch 7. 79-80)
Common superficial bacterial infection of skin
Highly contagious; frequently seen in otherwise healthy children as well as occasionally in adults in poor health
Usually involves exposed skin, particularly that of the face and hands
Caused by S. aureus
Erythematous macules with multiple small pustules; as pustules break, shallow erosions form, covered with drying serum giving the appearance of honey colored crust
If crust not removed, new lesions form on the periphery of the crust
Osteogenesis Imperfecta (brittle bone disease (Ch 9. 12-13)
Defects in extracellular structural protein
Most common inherited disorder of connective tissue
AD; caused by mutations in genes encoding Type 1 collagen
Phenotypically heterogenic disorder caused by deficiencies of type 1 collagen synthesis
Principally affects bone but also impacts other tissues rich in type 1 collagen (joints, skin, teeth, eyes, and ears)
Characterized by extreme skeletal fragility, blue sclera, hearing loss, and small misshaped, and blue-yellow teeth (secondary to dentin deficiency)
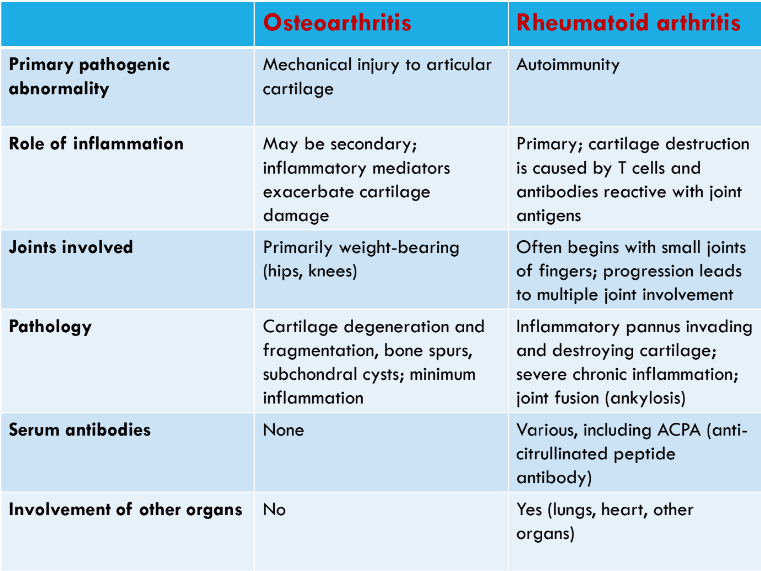
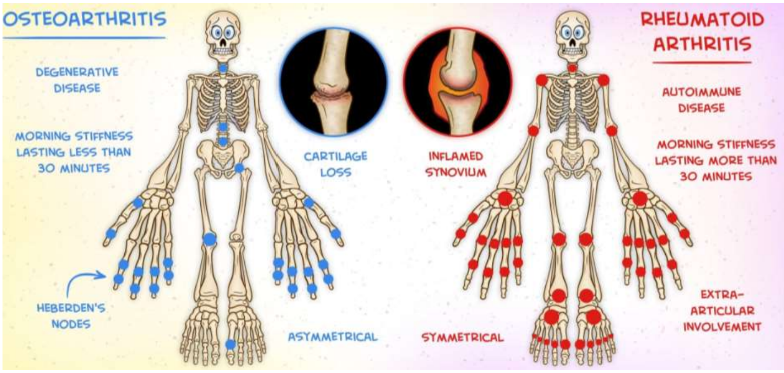
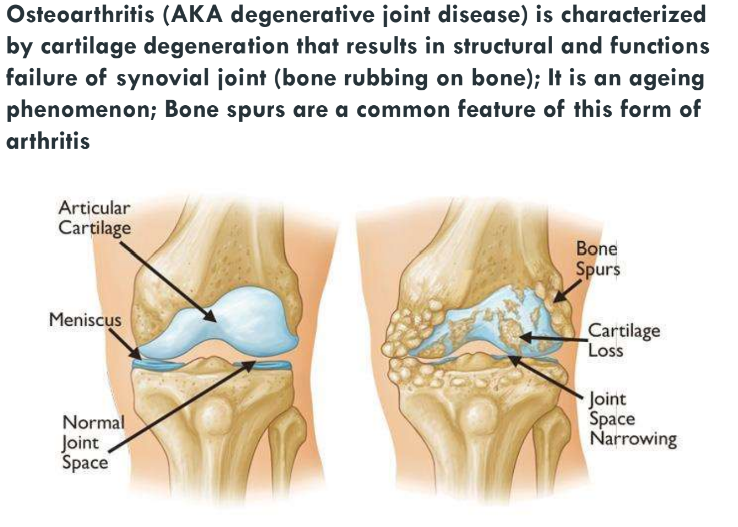
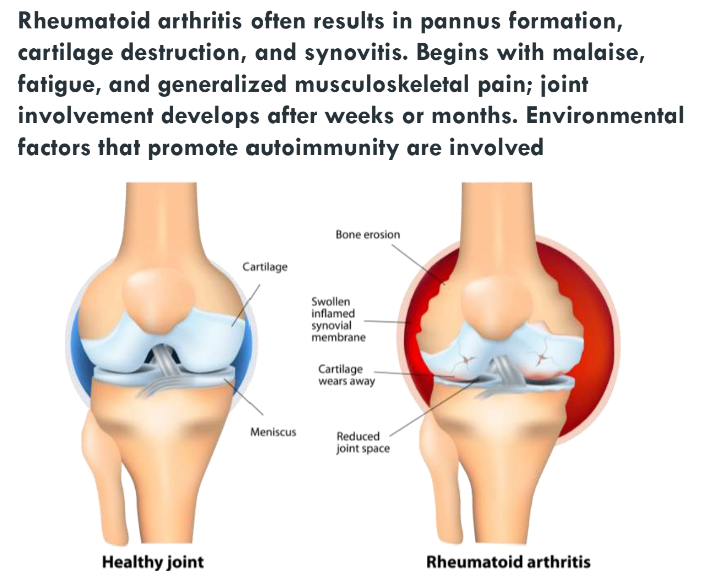
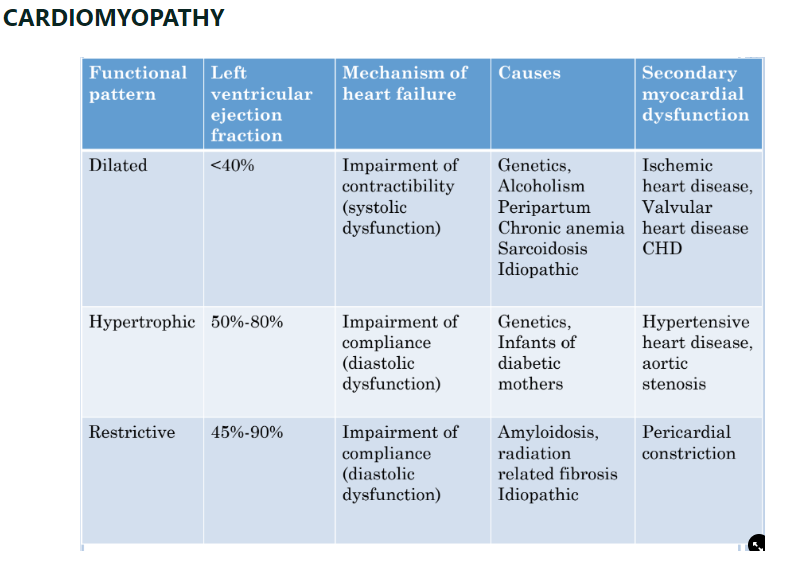
Osteoarthritis and Rheumatoid arthritis (Ch 9. 88-92)
Arthritis is inflammation of the joints
clinically, most important forms are:
Osteoarthritis
Rheumatoid arthritis
Achondroplasia (Ch 9. 10-11)
Defects in hormones and signal transduction proteins
the most common skeletal dysplasia and a major cause of dwarfism
AD disorder
Gain-of-function in the FGFR3 gene
Characterized by retarded cartilage growth resulting in shortened proximal extremities, an enlarged head and budging forehead and depression of the root of the nose, and a trunk of relatively normal length
Adrenal cortex hyperfunction/ Cushing syndrome/ Cushing Disease (Ch 11. 13-24, 146-149)
Corticotropic Adenomas
Excess production of the ACTH leads to adrenal hypersecretion of cortisol and the development of hypercortisolism
Cushing Disease signs and symptoms
Moon facies
Buffalo hump
Truncal obesity
Violaceous striae
Hirsutism
Hypertension
Glucose intolerance or diabetes mellitus
Visual symptoms if adenoma large
Cushing Syndrome
Disorder caused by conditions that produce elevated glucocorticoid levels
Can be broadly divided into:
Exogenous: due to corticosteroid therapy prescribed for other medical purposes
Endogenous: caused production of adrenocorticotropic hormone by an adrenal adenoma (10%-20% cases) and carcinoma (5% cases)
NOTE: If pituitary adenoma is responsible, it is called Cushing disease (70% cases)
4W: 1M
Ectopic ACTH may be secreted by small-cell lung Ca
Clinical Features
Usually develops slowly and onset is subtle
The most consistent feature is weight gain, particularly in the central area of the body and hypertension
Accumulation of fat in the dorsocervical spine region results in a buffalo hump appearance
Fat accumulation in the facial region results in the characteristic rounded facial appearance known as moon facies
Other common findings include:
Red-purple abdominal striae
Hirsutism
Mood changes
Osteoporosis
Hypertension
Hyperglycemia with thirst and polyuria
Muscle wasting
Diagnosis
1. Person receiving large amounts of corticosteroids (greater than the equivalent 20 mg of prednisone) daily for several months
2. Elevated levels of cortisol and low levels of ACTH (ACTH independent CS)

Adrenal Cortex hypofunction/Addison Disease
Insufficient production of adrenal corticosteroid hormone caused by the destruction of adrenal cortex
Causes include
Autoimmune destruction (most common cause in Western countries)
Infections (TB, deep fungal infections, particularly in AIDS patients)
Rarely, metastatic tumors, sarcoidosis, amyloidosis, or hemochromatosis
Clinical features do not begin to appear until at least 90% of the glandular tissue has been destroyed
With gradual destruction of the adrenal cortex, insidious onset of fatigue, weakness, irritability, depression, and hypotension is noted over period of months
Generalized hyperpigmentation of skin occurs (bronzing)
More prominent in sun-exposed skin and over pressure points
Caused by elevated levels of pro- opiomelanocortin (POMC), derived from anterior pituitary and is a precursor of both ACTH and melanocyte stimulating hormone (MSH)
Oral Manifestations include:
Diffuse or patchy, brown, macular pigmentation of the oral mucosa
Often the oral mucosal changes are the first manifestation of the disease, with the skin pigmentation occurring afterwards
In primary hypoadrenocorticism, plasma levels of ACTH are high (>100 ng/L)
In secondary hypoadrenocorticism the levels are normal (9 to 52 ng/L) or low (due to decreased production of ACTH by the pituitary gland)
Treatment includes hormone replacement therapy
Physiologic dose of glucocorticosteroids is approximately 30-45 mg of hydrocortisone or its equivalent, per day, in divided doses
Under stressful conditions, the body needs additional hormone
NOTE: this adjustment is generally not required for dental procedures performed using LA and lasting less than 1 hour
Hypothyroidism+ Hashimoto thyroiditis
Hypothyroidism:
Condition caused by a structural or functional derangement that interferes with the production of thyroid hormone
Prevalence increases with age
> women (10W;1M)
Primary Hypothyroidism
Accounts for a vast majority of cases
May be:
Congenital
Autoimmune
Idiopathic
CRETINISM
Refers to hypothyroidism that develops in infancy and early childhood
Clinical Features:
Severe intellectual disability
Short stature
Coarse facial features
Protruding tongue
Umbilical hernia
MYXEDEMA
Hypothyroidism development in the older child or adult
Clinical findings
Early symptoms include generalized fatigue, apathy, and mental sluggishness which mimics depression
Speech and intellectual functions are slowed
Patients are listless, cold intolerance, and frequently overweight
Skin is cold
Reduced cardiac output
HYPOTHYROIDISM LAB FINDINGS
Lab values play a vital role in diagnosis of suspected hypothyroidism because of nonspecific nature of symptoms
TSH levels are increased in primary hypothyroidism
TSH levels are not increased if hypothyroidism takes place at the pituitary or hypothalamus levels
Measurement of serum TSH levels is the most sensitive screening test for this disorder
Hashimoto Thyroiditis
Autoimmune disease
Results in destruction of the thyroid gland and gradual and progressive thyroid failure
Prevalence between 45 and 65 years of age
> women (10:1)
Pathogenesis
Caused by a breakdown in self-tolerance to thyroid autoantigens
Predisposition has a strong genetic component
Clinical Features
Painless enlargement of the thyroid gland associated with some degree of hypothyroidism
> in middle-aged women
Enlargement symmetric and diffuse
Incidences are at an increased risk for developing other autoimmune disease
They are also at an increased risk of B-cell lymphoma
Lab Values
Fall in serum levels to T3 and T4
Compensatory increased serum TSH


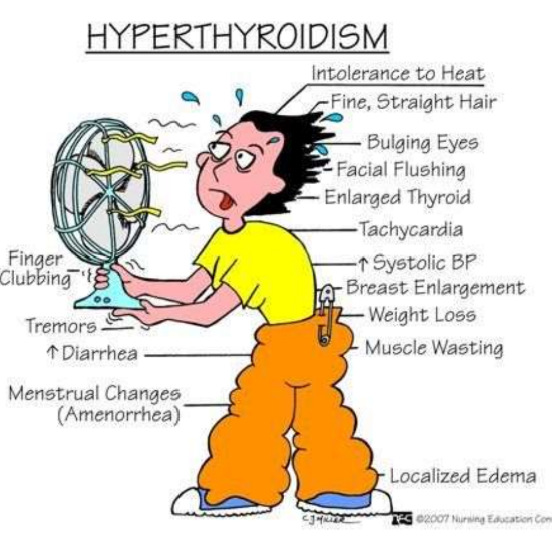
Hyperthyroidism/ Graves Disease
Lab Findings for hyperthyroidism
Low TSH values accompanied by increase in free T4 levels
NOTE: measurement of serum TSH concentration is the most useful single screening test for hyperthyroidism)
In occasional patients, hyperthyroidism results predominantly from increased circulatory levels of T3 (T3 toxicosis); in these cases, free T4 levels may be decreased
Treatment
1. Beta blockers to control symptoms
2. Thionamide to block new hormone synthesis
3. An iodine solution to block the release of thyroid hormone
4. Radioactive iodine, which is incorporated into thyroid tissues resulting in ablation of thyroid function over a period of 6 to 18 weeks
Graves Disease
Autoimmune disorder
Peak incidence between 20 and 40 years of age
> women (10:1)
Characterized by the production of autoantibodies against multiple thyroid proteins most importantly the TSH receptors
Most common cause of endogenous hyperthyroidism
Characterized by a triad of clinical findings
Hyperthyroidism associated with diffuse enlargement of the gland
Infiltrative ophthalmopathy and resultant exophthalmos
Localized, infiltrative dermopathy, sometimes called pretibial myxedema, which is present in a minority of patients
Lab Values
Elevations in serum levels to T3 and T4
Decreased serum TSH
Treatment
Treated by beta blockers (dampened symptoms related to increased sympathetic nervous system activity)
Radioiodine ablation
Thyroidectomy

 Aphthous Ulcers
Aphthous Ulcers
Common, often recurrent, and painful
Cause is unknown; affect 40% of the population
Most frequent in the first two decades of life
Tend to be clustered within some families and may be associated with immunological disorders including celiac disease, inflammatory bowel disease, and Bechet syndrome
Lesion may be single or multiple, shallow, mucosal ulcerations covered by a thin exudate and rimmed by a narrow zone of erythema; seen on nonkeratinized mucosa
Lesions resolve spontaneously in 7-10 days but sometimes persist for weeks, particularly in immunocompromised patients
Fibroma
Submucosal nodular mass of fibrous connective tissue stroma
> on buccal mucosa along bite line or gingiva
Reactive process induced by repetitive trauma
Treatment is complete surgical excision
Pyogenic Granuloma (Pregnancy Tumor)
Exophytic inflammatory lesion
> gingiva (75%); > in children, young adults, and pregnant women
Red to purple in color and frequently ulcerated
In some cases rapid growth is alarming and elicit concern of malignancy
Histologically, they are highly vascularized proliferation of organizing granulation tissue
PG can regress, mature into dense fibrous masses, or develop into peripheral ossifying fibroma
Treatment is complete surgical excision
Peripheral Ossifying Granuloma
Reactive growth
Occurs exclusively on the gingiva
> young females (10-19 years of age)
Some arise in long-standing PG, and others develop de novo from cells of the periodontal ligament
POF appears as a red, ulcerated nodular lesion
Treatment is complete surgical excision down to the periosteum is required
Recurrence rate of 8% to 16%
Oral Hairy Leukoplakia
Distinctive oral lesion on the lateral border of the tongue caused by Epstein Barr Virus that usually occurs in immunocompromised patients
In patients infected with HIV, OHL may portend development of AIDs
OHL are increasingly seen in patients who are immunocompromised for other reasons including cancer therapy, transplant-associated immunosuppression and old age
Clinically, white linear lines or hyperkeratotic thickenings are seen on the lateral border of the tongue; they cannot be scraped off
Burkitt lymphoma
A B-cell neoplasm; two types:
Endemic
Sporadic
Immunodeficiency associated: occurs in HIV + patients and in organ transplant patients
It may be the fastest growing human neoplasm
Characterized by the translocation and deregulation of the MYC gene on chromosome 8; t(8;14)
Endemic BL:
Seen in parts of Africa
Often presents with massive involvement of maxilla and mandible
Common in children aged 4 to 7 who have malaria and Epstein- Barr virus
Sporadic BL:
Seen worldwide
Involves the abdomen area
60% over the age of 40 years; 40% children
Account for 1-2% of adult lymphoma cases
Despite its fast-growing nature, Burkitt lymphoma is one of the most curable forms of non-Hodgkin lymphoma
More than 90% of children with localized tumors and more than 85% with widespread disease are cured
Extra:
Jackie Kennedy Died of Non- Hodgkin Lymphoma
Multiple Myeloma
Uncommon, clonal and malignant plasma cell proliferative disorder
Characterized by the abnormal increase of monoclonal immunoglobulins
Consequences of undiagnosed disease are severe leading to:
Hypercalcemia
Renal dysfunction
Anemia
Bone pain accompanied by lytic lesions
Etiology unknown
Frequent alterations and translocations in the promoter genes, especially chromosome 14
Other oncogenes such as NRAS, KRAS, and BRAF may participate in plasma cell proliferation
Median age at diagnosis of about 70 years
Slightly more commonly seen in males than females
Increased incidence in African American and black populations by as much as two-fold compared to White
Clinical Features:
Quite variable; typically more subacute and insidious in onset
Can present with severe symptoms
Symptoms:
Hypercalcemia (C)
Renal dysfunction (R)
Anemia (A)
Bone pain with lytic lesions "punched out lesions" (B)
Cells produce abnormal clonal, complete or incomplete, immunoglobulins
Abnormal immunoglobulin, usually a light chain, was previously referred to as Bence-Jones protein
Dental Manifestations
Poor healing
Jawbone involvement
Bleeding tendency
Infections
Pain and paresthesia
Patients often on IV bisphosphonates- results in medication-related osteonecrosis of bone (MRONJ)
Prognosis depends on the stage of the disease
Hodgkin Lymphoma
A rare monoclonal B-cell lymphoid neoplasm characterized by the following four features:
Usually presents in young adults
Commonly arises in cervical lymph nodes
Involves scattered large mononuclear Hodgkin and multinucleated Reed-Sternberg cells on a background of non-neoplastic inflammatory cells
Characteristic neoplastic cells are often surrounded by T lymphocytes
Divided into two distinct categories that demonstrate different pathologic and clinical features:
Classical HL: accounts for approx. 95% of HL; further subdivided into 4 subgroups
Nodular sclerosis (NSHL)
Lymphocyte-rich (LRHL)
Mixed cellularity (MCHL)
Lymphocyte-depleted (LDHL)
Nodular lymphocyte-predominant HL (NLP- HL)
It has a bimodal distribution
Most of the affected patients are between ages 20 to 40 years
There is another peak from age 55 years and older
seen more in males
Oral manifestations
Patients treated with radiation may have dry mouth if submandibular and sublingual glands are in the field
Need prophylactic fluoride gel, varnish or toothpaste to prevent dental caries
Langerhans Cell Histiocytosis
Idiopathic condition
Characterized by proliferation of abnormal Langerhans (antigen-presenting) cells
Has characteristics of both an abnormal reactive process and a neoplastic process
Lower antigen presenting capabilities
Rare; 1 to 2 newborns per million per year
May occur at any age but is more likely to occur in those <15 years of age
Unknown etiology
Symptoms depend on organ involvement at the time of presentation
Rash is the most common presentation; single lesion or widespread involvement
Bony involvement occurs in about 78% of patients
Pulmonary lesions occur in 20% of patients
LN involved in 20% of patients; pulmonary symptoms or lymphadenopathy
Pituitary gland involvement causes diabetes insipidus
Prognostic Categories
Classification:
Single organ involvement
Unifocal disease
Multifocal disease
Multiorgan involvement
No organ dysfunction
Organ dysfunction
Low-risk (skin, LN, bone, and/or pituitary gland)
High risk (lung, liver, spleen, and/or bone marrow)
Traditional Clinical Classifications
Monostotic or polyostotic eosinophilic granuloma:
Solitary or multiple bone lesions without visceral involvement
Chronic disseminated histiocytosis (Hand-Schuller-Christian disease):
Involves skin, bone, and viscera
Acute disseminated histiocytosis (Letterer-Siwe disease):
Prominent cutaneous, visceral, and bone marrow involvement mainly in infants
Hand-Schuller-Christian Disease
Hemophilias
Factor VIII Deficiency
Factor IX deficiency
Von Willibrand Disease
Hemophilia
A medical condition in which the ability of the blood to clot is severely reduced, causing the patient to bleed severely from even a slight injury
The condition is typically caused by a hereditary lack of a coagulation factor, most often factor VIII
Factor VIII Hemophilia (Classic Hemophilia/ Hemophilia A)
Factor VIII is one of the essential blood proteins and plays a role in aiding the blood to clot in response to injury
Mutations of the F8 gene result in deficient levels of functional factor VIII
X-linked recessive trait; 30% are new mutations and do not have a family history
Accounts for 80% of hemophilias
Occurs in males; rarely, homozygous females affected
Called the Royal Disease since it appeared in one of Queen Victoria’s sons and was propagated in her descendants
Factor VIII Deficiency Clinical Features
Easy bruising
Massive hemorrhage after surgery or trauma
Hemorrhage into joints (hemarthroses) may eventually result in deformity
Petechiae and ecchymoses are characteristically absent
Treatment: factor VIII infusions after injury or before surgery
Factor IX Deficiency (Hemophilia B/ Christmas Disease)
X-linked recessive transmission
Clinically resembles Factor VIII deficiency
Much less common
Usually not as severe as Factor VIII deficiency
Von Willebrand Disease
A bleeding disorder caused by the qualitative or quantitative deficiency of the von Willebrand factor
Von Willebrand factor necessary for proper platelet adhesion to damaged blood vessels
It is a carrier for Factor VIII as well protecting it from degradation
Affected people may complain of:
Excessive bruising
Prolonged bleeding from mucosal surfaces
Prolonged bleeding after minor trauma
Clinical Features
Usually, mild symptoms
Women may have heavy menstrual periods
Often underdiagnosed
Most common inherited bleeding disorder
AD or AR
Thus, both men and women can have it in about equal
Angina Pectoris
Angina Pectoris is temporary chest pain or discomfort caused by the inability of diseased coronary arteries to deliver sufficient oxygen-laden blood to heart muscles
It is a sign of increased risk of heart attack
Triggered by emotional stress, physical exercise, exposure to very hot or cold temperatures, smoking, eating a heavy meal
Symptoms
pressure or pain in the center of the chest that may radiate to the shoulder, arm, back, neck, and jaws
some patients may describer them as like the sensation of having indigestion or gas
Types
stable angina
prinzmetal
Unstable angina
Stable angina pectoris
Stable Angina Pectoris is associated with fixed atherosclerotic narrowing of one or more coronary arteries
Discomfort in the chest described as deep, poorly localized pressure, squeezing, or burning sensation (like indigestion), but usually as pain
Produced by physical activity, emotional excitement, or physiological stress
Pain relieved by rest and/or nitroglycerin or calcium channel blockers
Prinzmetal angina pectoris
Prinzmetal Angina Pectoris is anginal pain occurring at rest or awakening patient from sleep
Usually associated with coronary artery spasm often adjacent to a site of atherosclerotic plaque
Mechanism underlying spasm poorly understood
Unstable Angina pectoris
increased frequency of angina episodes
Precipitated by progressively less exertion
Described as intense pain
More intense and lasts longer than stable angina (>20 minutes)
Induced by acute plaque change with a superimposed partial thrombosis or vasospasm or both
Angina pectoris Treatment
Nitroglycerin (oral medication classified as a vasodilator)
patients take nitroglycerin about 10 minutes before they begin doing an activity that will likely trigger symptoms
 Myocardial Infarction
Myocardial Infarction
Myocardial Infarction (heart attack) is the irreversible necrosis of heart muscle secondary to prolonged ischemia
Etiology: closely associated with coronary artery disease
Common symptoms include
chest discomfort
shortness of breath
discomfort in the upper body
Myocardial necrosis begins within 20-30 minutes of infarction and reaching full size within 3-6 hours
Begins in subendocardium extending toward epicardium
Thrombolytic agents (tissue plasminogen activator t-PA) may limit size of infarct during this time frame
Thrombolytic drugs: Eminase, retavase, streptase activase
Note: Aspirin is not thrombolytic, but may limit clot size- antiplatelet adhesion drug
Clinical complications of acute MI include
sudden death
cardiogenic shock
cardiac arrhythmias
cardiac tamponade due to cardiac rupture
Anatomic complications of MI include
valve insufficiency due to infarcted papillary muscle
ventricular aneurysms
mural thrombus giving rise to systemic arterial emboli
cardiac tamponade due to rupture of the infarcted area of the heart
Myocardial infarction treatment
reperfusion therapy is indicated in all patients with symptoms of ischemia of less than 12-hours duration
Nitrates: IV nitrates are more effective than sublingual nitrates (giving rise to nitric oxygen, which causes vasodilation-nitroglycerine)
Beta-blockers (reduces myocardial workload, and thus oxygen demant, via reduction in heart rate and BP)(olol’s- atenolol, bisoprolol)
lipid lowering treatment (statins)
Antithrombotic therapy (aspirin)
Pleural Tumor - Mesothelioma
Malignant mesothelioma
50% have a history of asbestos exposure
25-40 yrs. latent period
Asbestos not used since 1960
No direct link between smoking and mesothelioma
Essentially incurable unless detected at a limited stage
Median survival 11 months
Sarcoidosis
Systemic granulomatous disease of unknown cause that may involve many tissues and organs
Histologic feature: formation of noncaseating granulomas
Clinical manifestations include LN enlargement, eye involvement (dry eyes, iritis), skin lesions (erythema nodosum), and visceral (liver, skin, marrow) involvement
Lung involvement occurs in 90% of cases with formation of granulomas and interstitial fibrosis
Highest incidence in African American (10 X) and Scandinavians
Adults between 30 and 50 years of age
Papular sarcoidosis as a cutaneous manifestation seen on the upper back region. Multiple erythematous raised lesions are evident
Plaque sarcoidosis as a cutaneous manifestation seen on the upper arms. Multiple erythematous plaques are evident
Uveitis
Inflammation in the middle layer of the eye, called the uvea
Sarcoidosis Treatment
Two fundamental facts that exert heavy influence on the management of sarcoidosis
1. Frequently undergoes spontaneous regression without causing any permanent damage to the affected organs
2. Glucocorticoids, is the cornerstone for treatment (associated with several serious adverse effects)
Treatment is indicated only when symptoms are disabling and/or the granulomatous inflammation is relentlessly progressive, causing life- or organ-threatening disease
Sarcoidosis Oral Cavity
Lesions are a rare occurrence; may be the first presenting sign of the disease
May involve gingiva or other oral locations
Lesions present as:
Localized swelling
Nodules
Ulcers
Gingivitis
Gingival hyperplasia
Gingival recession
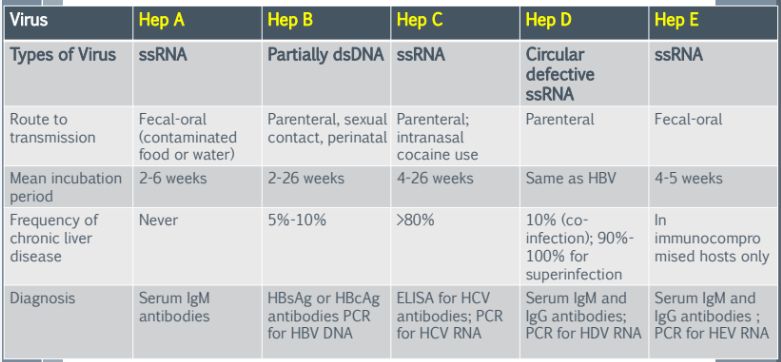
Hepatitis