PHAR2210 exam
paracetamol, ethanol or codeine/morphine pair
Drug absorption
Absorption: the movement of the drug from its site of administration to the systemic circulation
Drugs need to cross lipid membrane to move from one compartment to another, there are two ways of crossing:
Paracellular (through tight junctions): low molecular weight drugs
Transcellular (move across a cell): lipophilic drugs or via passive diffusion and/or active transport
Factors that affect oral absorption:
physiochemical properties of a drug
i. molecular size – oral absorption decreases as MW increases
ii. solubility – hydrophilicity and lipophilicity affect passive diffusion at membranes. can estimate solubility by using logP and polar surface area (PSA)
iii. charge – only unionised drug molecules can diffuse passively across lipid membranes
during aspirin overdose, can use activated charcoal or increase urine pH
various environments encountered during absorption eg.
ingested food
gut mobility
pH
microbiota
intestinal enzymes
transporters
solute carrier transporters (SLC) - facilitated diffusion and active transport
ATP-binding cassette transporters (ABC) - active transport
MUST KNOW transporters
solute carrier transporters (SLC)
Caco-2 cells - grow as model layer on flat surface. mimic eterocytes. first cells with differentiate and become polarised to form apical and basolateral membranes. express important efflux transporters, incl MDR1, BCRP and MRP2. used to investigate if drug undergoes intestinal efflux.
apical membrane
efflux transporters are on apical membrane, so are Caco-2 cells. can add cells to either side, to moniter movement of drugs.
can add drug to AM, then measure drug by taking sample from BM to calculate apparent permeability Papp(A-B) and vice versa. then with both can calculate efflux ratio ER.
if drug is poorly absorbed or undergoes extensive efflux, need to reduce this. if drug undergoes efflux, then need to consider if administer two drugs, does this affect efflux?
drug-drug interaction.
three ways to determine which drug transporter is involved.
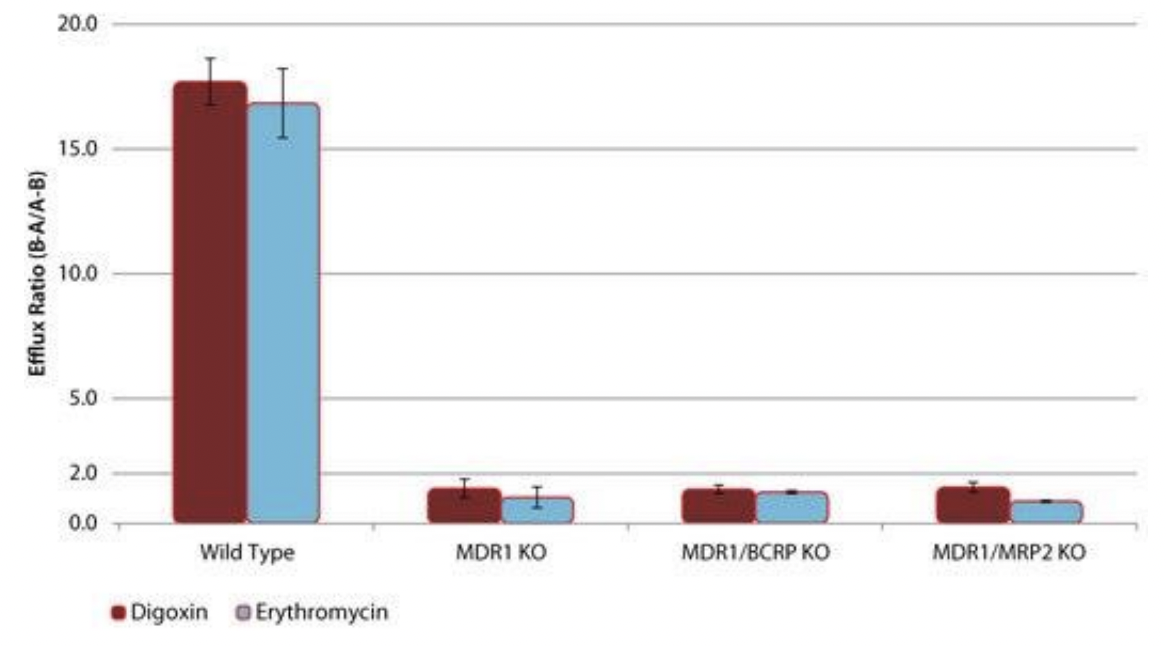
transporter knockout Caco-2 cell lines. can knock out one or two of the transporters. in picture below can see wild type ER is greater than 2, means two drugs undergo MDR1 mediated drug efflux. if KO MDR1, see ER goes below 2, which indicates these drugs no longer undergo efflux as transporter responsible is absent. purely mediated by MDR1, and not BCRP or MRP2
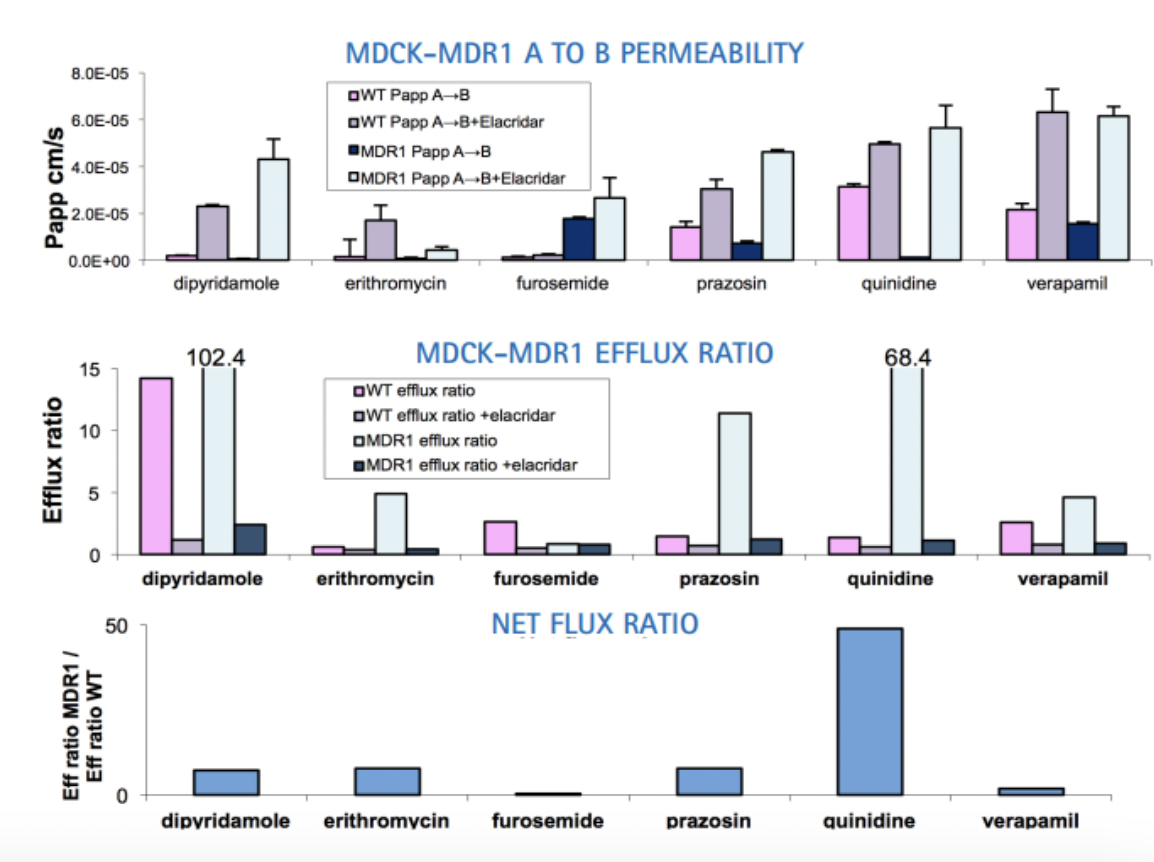
transfect cells with transporter of interest and see if that changes cell behaviour. important to have control. net flux ratio of greater than 2 indicate that transported thats been transfected into cells is responsible for efflux of drug.
inhibitor of transporter of interest. to use model inhibitors. add appropriate inhibitors with appropriate controls to see if anything changes.
distribution: reversible transfer
factors that influence drug distribution
pH of compartment
blood flow. drug will distribute into organs or tissues that normally receive rich blood supply. if poorly perfused, drugs will be distributed slowly or not at all.
tissue binding. drugs can potentially accumulate in certain tissues/organs based on characteristics of either drug or organ/tissue. very lipophilic drugs can accumulate in fat, so therefore obesity can complicate drug distribution
TETRACYLINES: high affinity for calcium, accumulates in bones and teeth. classed as category D drug by TGA, not used when pregnant or in young children. can affect growth of bones or lead to birth defects in pregnancy.
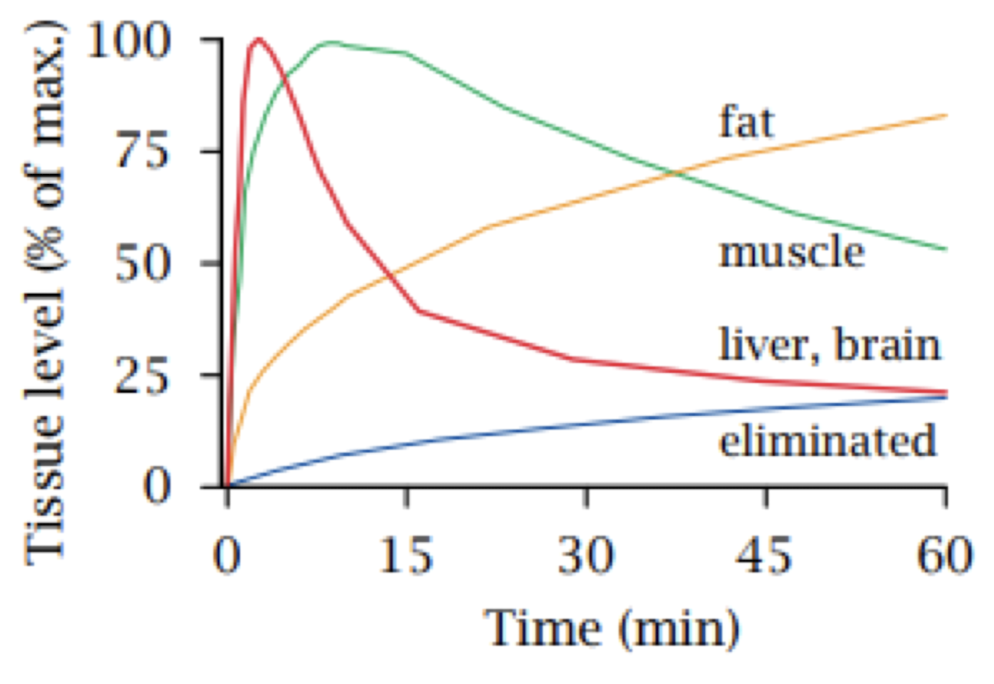
THIOPENTAL: v lipophilic. used IV as anaesthetic to induce anaesthesia, not to maintain anaesthesia. in graph below, liver and brain are well perfused, so drug is first distributed into these. slow into fat and muscle. to induce anaesthesia, action should be fast into brain. if it was continuously infused, then drug will slowly accumulate in body fat, drug distribution is reversible so after drug is no longer being administered, it will slowly return to systemic circulation which will makes plasma concentration decline slowly. could go back into the brain, ends up with hangover effects after anaesthesia is required.
solubility of a drug. hydrophilic drugs stay in extracellular compartment. lipophilic drugs can cross cell membranes and penetrate tissues. very lipophilic drugs distribute into tissues and accumulate in fat.
ETHANOL: lipid soluble enough to cross the blood-brain barrier. distribution is dependent on the relative water content of tissues. difference in males and females. females less DME’s and lower % of water content. distributed into TBW
plasma protein binding. plasma protein act as carriers for endogenous compounds and xenobiotics. binding is reversible, and lipophilic drugs are prone to plasma protein binding. only free (unbound) drugs will be distributed or metabolised or excreted. if bound to plasma protein, there are unable to undergo these processes.
ALPHA1 ACID GLYCOPROTEIN: binds basic drugs.
HUMAN SERUM ALBUMIN (HSA): binds many acidic drugs. each albumin contains 2 drug binding sites
permeability of blood-tissue barrier. identify why drugs are normally difficult to penetrate brain. what are characteristics of BBB that make it difficult to penetrate.
BLOOD-TISSUE BARRIER: endothelial cell layer. has fenestrations that allow exchange of molecules between intravascular space and interstitial fluid. has basal membrane (matrix of proteins with spaces filled with water)
BLOOD-BRAIN BARRIER (BBB): endothelial cell layer. no fenestrations, but tight junctions. has basal membrane. has 1 more feature - glial cells joined by tight junctions. for drug to get into brain, there is 1 more hurdle. glial cells perform protective role. efflux transporters are expressed. protects brain from harmful drugs.
expression of transporters. how transporters can influence how a drug is distributed to various organs.
LIVER: once drug is administered orally, first taken to liver (first point of contact). makes liver vulnerable to toxicity. if drugs are toxic, or undergoes metabolism to form toxic metabolites it can be harmed. rich in DME, detoxifies. in presence of inhibitor, reduced uptake of drugs into organs/tissues.
SINGLE NUCLEOTIDE POLYMORPHISM: not every single individual will respond in the same way to each drug, due to genetics. variation at single position in a DNA sequence. enzyme or protein may have gain or loss of function, can be either homozygous or heterozygous. drug could bind to other targets which can produce adverse effects.
TRANSPORTERS AND BRAIN PENETRATION: why drugs are difficult to gain access to brain. efflux transporters - MDR1 and BCRP. brain barrier expresses these efflux transporters so drugs can be returned to systemic circulation without reaching brain. if brain tumour, tumour overexpresses same efflux transporters, so for drug to get to the tumour cells, it is challenging due to the over expression.
VOLUME OF DISTRIBUTION: the apparent volume of body fluid into which the drug is distributed. when plasma concentration is high, volume of distribution (size of human body) is low as the plasma has less volume to be distributed into. TBW is ~60% of body weight. highly protein-bound drugs stay in plasma, high [plasma] and small volume of distribution. v lipophilic drugs are distributed into fat and have a low [plasma] and large volume of distribution.
Q: if the Vd of a drug is 400L for an average male of 70kg, the drug is distributed into what body compartment(s)?
a. the plasma
b. the extracellular fluid
c. TBW
d. TBW + tissue reservoirs -
METABOLISM: the chemical modifcation of a drug within the body. protects the body against xenobiotics. liver (primary); other organs/tissues (eg. brain, kidneys, lungs, plasma).
FIRST PASS METABOLISM: gut wall and liver. FPM decreases the bioavailibiltiy of orally administered drugs. extensive FPM → decreased bioavailability
DMEs: hepatic → membranes of endoplasmic reticulums of hepatocytes (eg. Cytochrome p450), also in cytosol. drug transporters are expressed on liver and intestine
PHASE 1 METABOLISM: parent drug → metabolite(s). oxidation, hydrolysis, dealkylation.
OXIDATION: add oxygen atom to a parent drug OR remove hydrogen from a parent drug. ETHANOL can be oxidised by having hydrogen removed.
HYDROLYSIS: ester and (less readily) amide bonds are removed from molecule. prodrug (drug with little or no pharmacological activity) converted in vivo to active drugs. eg. TENOFOVIR DISOPROXIL can be hydrolysed by ESTERASE to TENOFOVIR.
DEALKYLATION: demethylation, removal of a methyl group. eg. CODEINE is metabolised by CYP2D6 into MORPHINE. CODEINE is prodrug.
PHASE 2 METABOLISM: metabolite(s) → conjugated metabolite(s) OR parent drug → conjugated metabolite(s). glucuronidation, sulfation and glutathione conjugation.
CONJUGATION: attach a polar endogenous molecule to a FG in a drug, increases hydrophilicity to facilitate excretion
GLUCURONIDATION: UGT transfers glucuronic acid from UDP-glucuronic acid to a drug or drug metabolite. MORPHINE to two conjugated metabolties. M3G (major, inactive) and M6G (minor, active)
SULFATION: SULT (Sulfotransferase) transfers sulfonate group from PAPS to a drug or drug metabolite
GLUTATHIONE-CONJUGATION: GST (glutathione-S-transferase) attach glutathione to a drug or drug metabolite
! some drugs just undergo phase 2, some undergo phase 1 and 2.
OUTCOMES OF DRUG METABOLISM:
increase molecular size and hydrophilicity. reduces tendency to accumulate and facilitates renal excretion. if drug is lipophilic, will undergo hepatic metabolism to form hepatic metabolites, then either undergoes hepatobiliary excretion to be excreted in bile OR undergoes renal excretion to be excreted via urine. hydrophilic drugs undergo renal excretion and are excreted via urine. lipophilic undergo metabolism, hydrophilic drugs don’t undergo metabolism, excreted unchanged.
decreases half life of drug: time taken for drug in the body to drop by half. if drug undergoes metabolism, the half life decreases.
alters pharmacological activity or induces activity. drug metabolism can form one or more metabolism.
PARENT DRUG | METABOLITE |
|---|---|
pharmacologically activeMorphine → | pharmacologically INactive (most common)morphine-3-glucuronide (M3G) |
pharmacologically activeMorphine → | pharmacologically activemorphine-6-glucuronide (M6G) |
pharmacologically activeparacetamol → | toxic and reactiveNAPQI |
prodrug (little or no pharmacological activity)codeine → | pharmacologically activemorphine |
Q: regarding drug metabolism, which of the following statements is accurate?
a. drug metabolism increases half life of a drug
b. drug metabolism increases lipophilicity of a drug
c. drug metabolism reduces the molecular size of a drug
d. drug metabolism facilitates the renal excretion of a drug -
CYP450: most important is ==CYP3A4 ==→ most important, broad substrate specificity. CYPs are heme-containing proteins, use NADPH (cofactor) and O2 to metabolise xenobiotics/drugs. O2 → one atom goes to parent drug and one goes to water molecule. Isolation of Subcellular fractions. limitations of using microsomes for CYP→ can’t tell which CYP it is. in vitro study.
PARACETAMOL METABOLISM: CPY2E1-mediated formation of NAPQI
CYP-mediated drug-drug interactions: different CYPs have distinct but often overlapping substrate specificity
CYP induction. certain xenobiotics can increase CYP expression. when expression of enzyme increase, metabolism of relevant drug will change (can lead to problems). outcomes: reduced or lack of drug effects due to enhanced hepatic metabolism.
CYP inhibition: when drugs compete for the active site of an enzyme when co-administered. competitive inhibition: may still undergo metabolism, but to a lesser extent. other drug may block the drug from being metabolised. reversible, if other drug is removed, plasma conc can go back to normal. mechanism-based inhibition: irreversible inhibition due to formation of a complex via covalent bonding. outcomes: a. exaggerated drug effects or toxicity due to reduced hepatic metabolism b. lack of drug effects due to impaired metabolic activation of prodrug
ALCOHOL AND PARACETAMOL: in acute alcohol consumption, CYP2E1 plays a minor role in metabolism of alcohol as alcohol dehydrogenase is the major role. paracetamol and alcohol are competing for CYP2E1, so may reduce risk of paracetamol overdose due to less NAPQI being formed. however in chronic drinker, CYP2E1 is induced which leads to increased formation of NAPQI
RITONAVIR: potent CYP3A4 inhibitor, HIV protease inhibitor. inhibit metabolism of other drugs. PK ENHANCER
COBICISTAT: CYP3A4 inhibitor. inhibit metabolism of elvitegravir. PK ENHANCER
Ultrarapid metaboliser: contains 2 or more copy of the CYP2D6 genes
extensive: 2 copies
intermediate: 1 cpoy of the functional gene and 1 copy of loss of function gene
poor: 2 copies of loss of function gene
which metaboliser is most likely to experience adverse effects when given normal doses of a parent drug (most cases- pro drug)?
which of the following explains the clinical use of ritonavir as a pharmacokinetic enhancer?
a. CYP2D6 induction
b. CYP2D6 inhibition
c. CYP3A4 induction
d. CYP3A4 inhibition -
ion channel | G protein-coupled | enzyme linked | nuclear | |
|---|---|---|---|---|
mechanism of signal transduction | ||||
receptor location | membrane | membrane | membrane | intracellular |
effector protein(s) | ion channel | channel or enzyme | enzyme - protein kinases | gene transcription |
time scale of action | milliseconds | seconds | hours | hours |
agonists | ||||
example | nicotonic acetylcholine receptor | muscarinic acetylcholine receptor | cytokine receptors | oestrogen receptor |
reverse transcriptase inhibitors | protease inhibitors | integrase inhibitors | |
|---|---|---|---|
basic mechanism | phosphorylated to form triphosphate | cleaves Gag-Pol fusion polyprotein | bind to integrase and prevent DNA strand transfer |
drugs | tenofovir disoproxil fumarate/tenofovir alafenamide | -navirritonavir (CYP3A4 inhib) | -tegravirelvitegravir |
side effects | GI disturbance, CNS effects | insulin resistance, hyperlipidemia, CVD | GI disturbance, CNS effects |
xtra things | non-nucleoside reverse transcriptase inhibitors (NNRTIs) |
TDF WORSE: converted to tenofovir in lasma