lecture 2: structural biology of the GroEL molecular machine
introduction to GroEL
GroEL enables bacteria to help assist when our proteins are failing to fold properly which may lead to aggregation. It is a Group1 HSP60 chaperonin family of proteins and takes part in protein folding especially under conditions of cell stress. It forms double ring structure with 7 subunits of 60 kDa in each ring. Each subunit can take the energy of ATP and through hydrolysis undergo a conformational change to work. It works in tandem with GroES
A combination of biochemical and structural techniques has helped unravel the GroEL structure.
how does it work
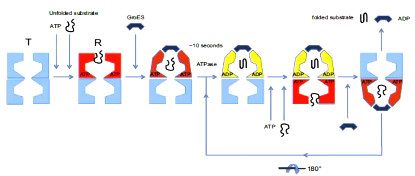
ATP hydrolysis in one ring is required to enable subsequent ATP binding to the opposite ring but hydrolysis is not required for the folding to proceed within the chamber. The conformational change that means that the new folding chamber that forms eventually releases the protein when ATP is bound to the opposite chamber.
early cryo-en of GroEL complexes showed low contrast images however some features were still visible such as the ring structure with 7 subunits. Side views showed rings stacked on top of each other. these images allowed the roll of GroES to be discovered where it was viewed that it formed a cap on top and bottom.
Later, they produced cryo-em maps of GroEL in the presence of different nucleotides such as ADP, ADP analogues and ATP. these showed the effect of different nucleotides on the conformation of the subunits such as opening of rings and the rotation of the domains.
crystal structures of GrowEL gave atomic resolution information of the precise structure it adopted. These did have the disadvantage of not having the same shape as the low resolution images obtained with nucleotides. This means that crystallization suppressed conformational changes. No crystal structure of GroEL is available with ATP as ATP would be hydrolysed as the enzyme is still active in the solution while the crystal forms. A non hydrolysable analogue of ATP did not induce conformational changes caused by ATP.
Although generally at lower resolution, cryo-em maps represent the whole range of structures adopted by a protein in solution rather than what crystallizes. This is solved by merging the data of high resolution crystallography with the low resolution images with cryo-em
effect of ATP binding on the conformation of GroEL
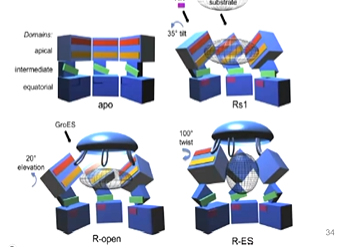
an advantage of cro-em is trapping intermediates. It was discovered that GroEL with nothing added had an apical domain at the to then the intermediate domain and a larger equatorial domain. The equatorial domain has the ATP binding domain. The second ring is a mirror image of this:

They were able to get the crystal structure with nothing added and fitted this to the cryo-em image. They then worked to try to trap the ATP bound state of gro-el. Initially, it didn’t work as they tried adding ATP but the structures were not high resolution as adding ATP changed the conformation adopted by groel which did not freeze quick enough. A mutation (D398A) was able to hydrolyse ATP at a slower rate and so the bound state of ATP was captured. They were able to see that there was positive cooperativity across one of the rings - asymmetric ring stricture. ATP was bound to the top ring and this changed the shape but was not bound to the bottom ring.
They were able to look at the fitted structure and found that in the ring without ATP, there is a salt bridge between E386 and R197, however this is not found in the bound form and instead there is a salt bridge formed between K80 ans E386.
effect of ATP binding on inter-subunit interactions
apical domains undergo an anticlockwise twist which buries hydrophobic protein binding sites, reducing GroEL’s affinity for its substrate protein. This forces it into pocket with GroES binding. ATP binding results in formation of a new salt bridge between adjacent subunits (E386 to helix C, residues 66 to 84). This means that ATP binding in one subunit is communicated to the ATP binding site of the next subunit around the ring.
At the interface between the two equatorial domains, the connections are weakened by ATP ro allow it to have more cooperativity
what happens when ATP is hydrolysed
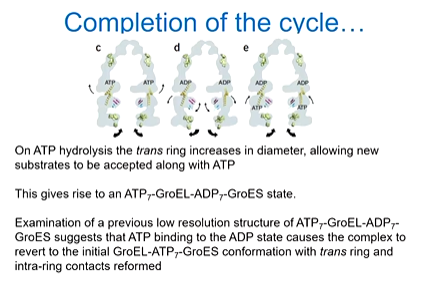
Two high resolution maps of GrowEL with ATP and GroES present and added one with ADP with GroEL and GroES and looked for the transition in the folding complex. It was found that there is a shared beta sheet between two subunits and when ATP is hydrolysed, this is separated as the strands are pulled apart. This is because of a small rotation in the equatorial domains. As you move from the ATP bound to the ADP bound, there is a shift to the apical domains so the opposite ring open out.
ATP hydrolysis changes the shape of the complex beyond where it actually binds - hydrolysis in the cis ring causes radial expansion of the trans ring. Relay helix D is the one that is offset and the Trans equatorial domains pivot around the Glu461 inter-ring contact disrupting the beta strands intra ring contact. The radial expansion is propagated throughout the trans ring, resulting in an opening of the substrate binding cavity
In the folding chamber, the protein is ready to be released and so the new substrate can bind on the opposite ring to accept the new ATP and substrate.
Sorting of conformations combined with a study of the slow ATPase GroEL mutant has lead to some more results. Clare et al in 2012 collected 60,000 images of GroEL and originally needed 6000 due to the lack of side views due to preferred orientation. They went through a computational sorting process. They got 7 maps with slightly different structures through this:
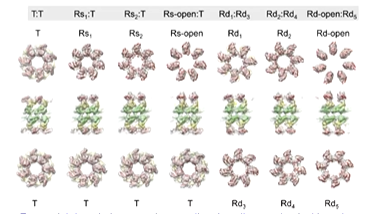
The new structures gave a better picture of key intersubunit salt bridge movement:
substrate binding to GroEL
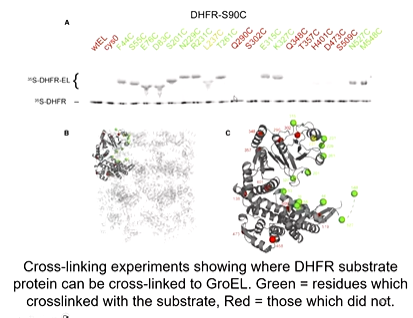
A development in cryo-em was required to obtain a high resolution structure of the sample with the substrate and GroEL. This is because it is a moving target and so there are different conformations in the solution. The development was so that multiple conformations could be analysed. A large data set had to be collected and they reveled 5 distinct complexes. They found out that the substrate found to 3 subunits at the same time on the apical domain. Cross linking experiments showed where DHFR substrate protein was crossed linked to GroEL. green represents residues which crosslinked with the substrate and so bound and red are those which did not. they found binding in any part of the cavity so there is heterogeneity
Another group looked at substrate binding to GroEL in the presence with GroES and visualised binding across the cavity. The substrate binds in several places at inlet or inside the cavity making contact with 3 or 4 apical domains. Apical domains were shown to cluster together around those binding sites and biochemical evidence showed that substrate protein could bind virtually anywhere inside the GroEL cavity. The structure of a folding intermediate encapsulated in the GroEL/GroES chamber has been determined.
It was also found that there were samples of GroEL that were single rings that could still function properly. Cryo-EM structures of ringle rings and class averages revealed multiple conformations that were significant. There was one conformation that was expanded that created 80% more volume in the folding cavity. This allowed a 86 kDa complex of mitochondrial branched chain a-ketoacid dehydrogenase to be enclosed and this is the largest substrate ever to be observed in the cis activity.
There were 4 structure one of which was expected but the other 3 were bigger as was suggested.
The single ring phenomenon shows that 2 rings are not essential to the function. Higher resolution single stricture ring structures may show how the mechanism adapts to compensate for the lack of a trans ring