M2L2 Transcriptional/translational control
RNA tumour retroviruses
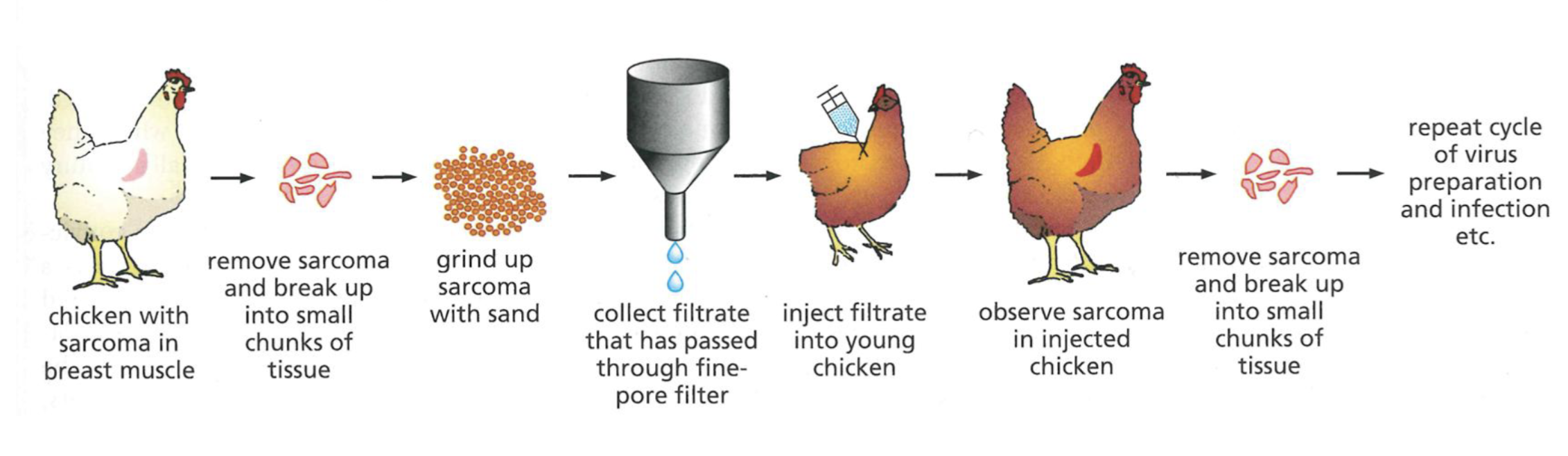
Cancer causing oncogenes like v-src introduced into genome of infected cells, reversing central dogma of biology
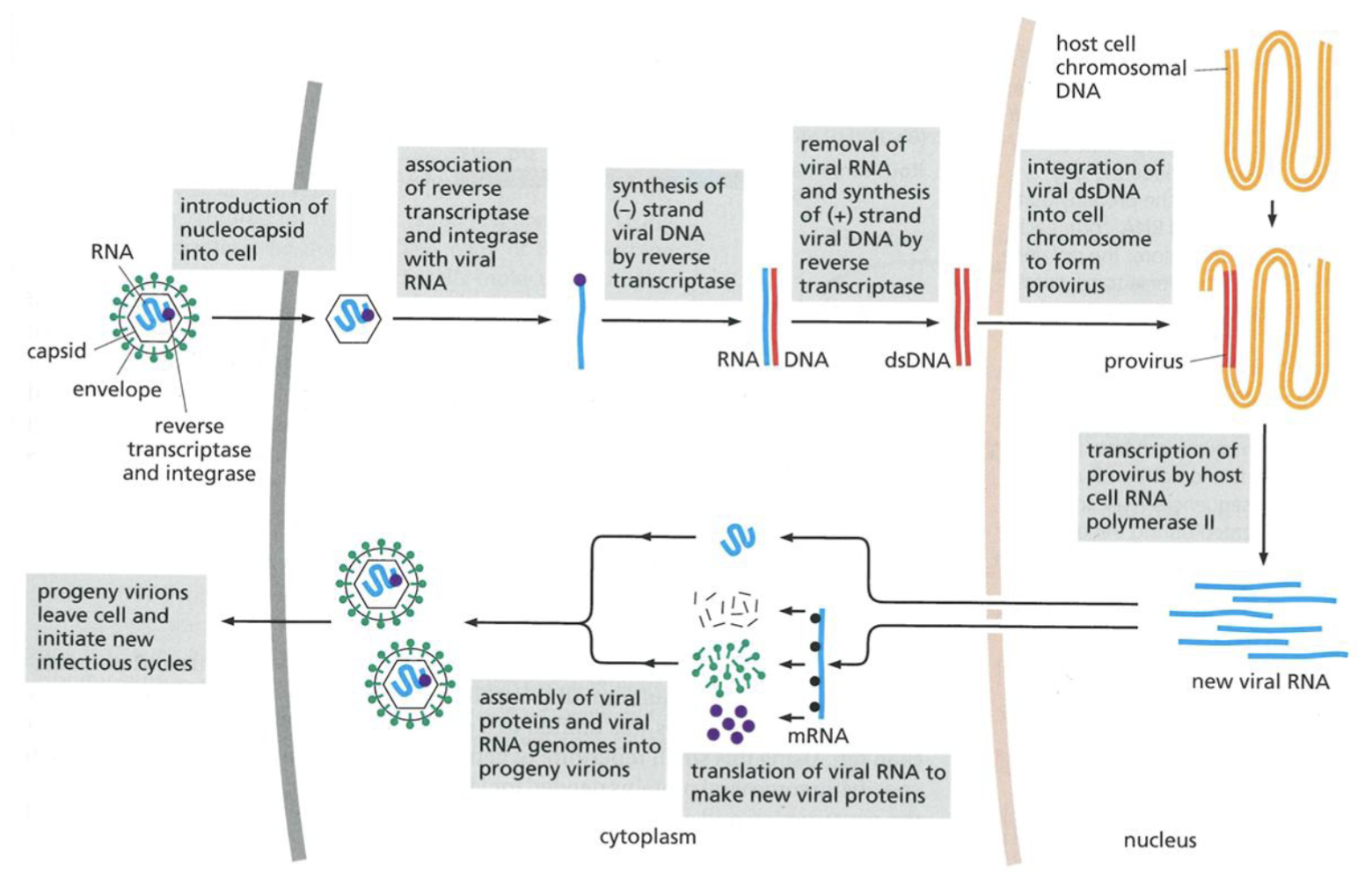
Src in …***
Transcription in the cancer cell
Activator protein 1 (AP-1)
TF that regulates genes related to proliferation, differentiation, apoptosis, inflammation
Target of MAPK cascade
Made of of products of two JUN and FOS family proteins
Both contain basic leucine zipper domains that facilitate their binding as dimers to response element in target genes
Activated in response to GFs, ROS and radiation, with the specific combination of dimers influencing the biological response
AP-1 oncogenes c-JUN and c-FOS cause aberrant expression and inappropriate increase in expression of AP-1 regulated genes
Associated with dysfunction and onset, development, invasion, and migration of cancer
Involved in drug/radiation resistance
Myc
Belongs to family of helix-loop-helix TFs that act as heterodimers to modulate the transcription of target genes with E-box sequences (5’-CACGTG-3’)
Myc-Max complexes act as enhancers also also promote elongation of nascent transcripts by releasing RNA pol II tcomplexes from pause sites immediately downstream of TSS, allowing it to proceed with transcription and synthesise full length pre-mRNA transcripts
As cells differentiate the Mad protein levels increase, displacing Myc and reducing the expression of Myc-Max
Mad-Max complex prevents RNA pol II release, losing its ability to stimulate transcription, perimitting cells to enter post-mitotic differentiated state
Protein function/stability modulated by phosphorylation
Under normal conditions Myc is ubiquitinated and degraded - short 15-20 min half life
Myc-Max heterodimeric TF complex —> expression of 1,000+ genes which can also affect the cell cycle, favouring proliferation over differentiation
In healthy cells the Myc proto-oncogene is regulated downstream of many signal transduction pathways including Wnt, Hedgehog, Notch, TGF-β and many RTKs
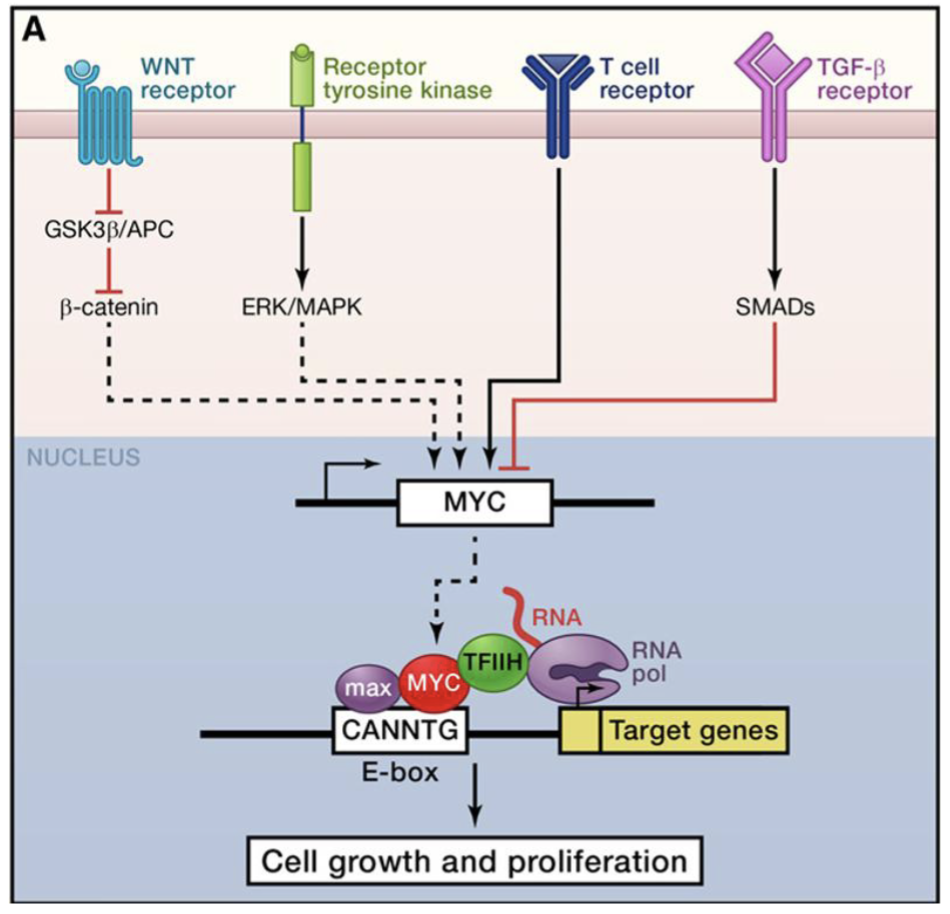
Myc activation in cancer can result from loss of this regulation, eg mutations in Wnt signalling APC, or direct alterations of Myc gene, eg. amplification or chromosomal transclocation
Acute sustained Myc expression —> inappropriate increase in transcription of Myc-regulated genes and E-box driven genes, unregulated cell cycle checkpoint progression, genomic instability due to Myc induction of ROS via mitochondrial biogenesis, and increased metabolism
Myc represses genes involved in cell-cell binding while cells undergo mitosis, linked to EMT and metastasis
Burkitt lymphoma - Myc gene translocates from Ch8 to Ch14, falling within the regulation of strong promoter of immunoglubulin genes
One of the most amplified among many different human cancers
Steroid hormone receptor superfamily
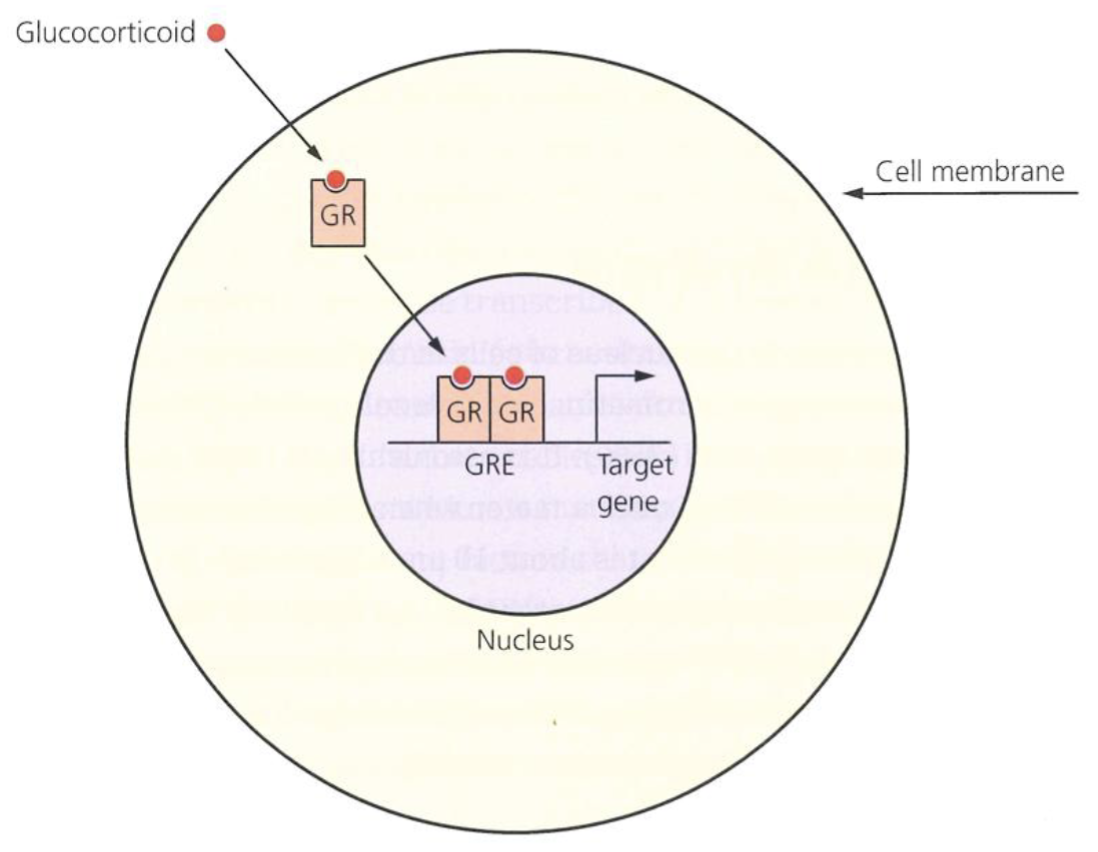
Steroid hormones pass through the cell memebrane and bind to their particular intracellular steroid receptors (aka nuclear receptors) in the cytoplasm
Acts as ligand dependent TF
Contain zinc finger type DNA binding domain, ligand binding domain for a specific steroid molecule, and dimerisation domain as they activate transcription as a dimer
Retinoic acid receptor (RAR) acts as RA-dependent transcriptional regulator - important during differentiation
RAR constitutively located in nucleus and acts as transcriptional repressor in the absence of RA
Binds to RA response element (RARE) in target genes as heterodimer with another member of the family called RXR
Aberrant forms of RARs, often due to chromosomal translocations, are characteristic of several leukaemias
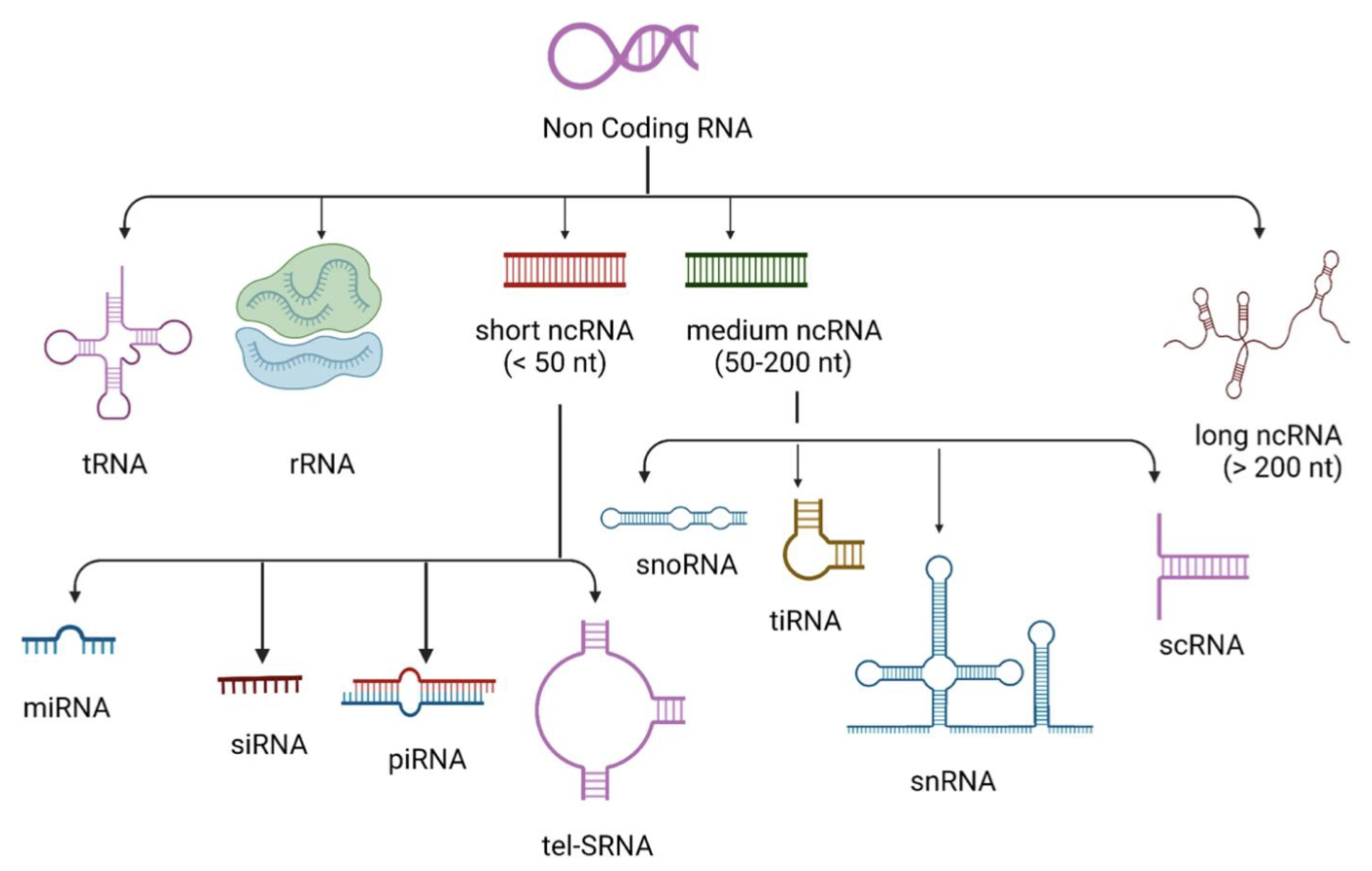
ncRNAs
Can be transcribed from either the coding or non-coding DNA strand, from intronic or intergenic (regions between genes) sequences
lncRNAs
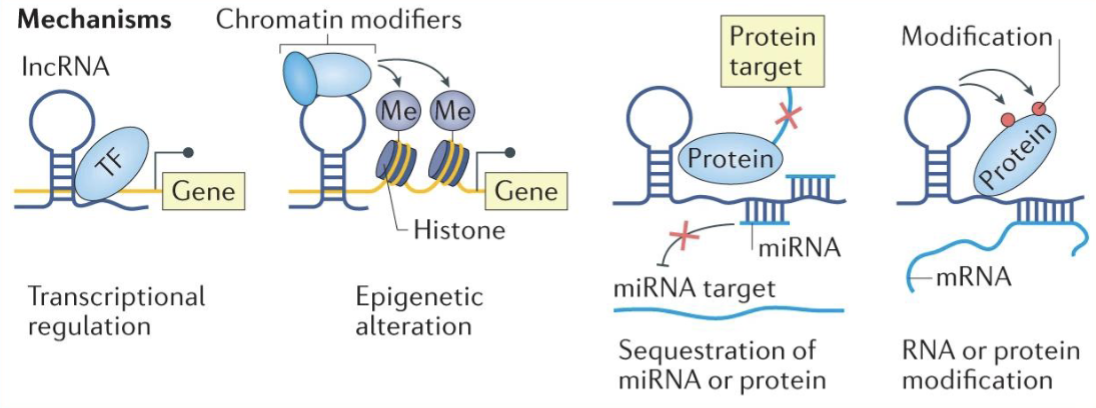
Endogenous polyadenylated RNAs which are >200nt and lack an open reading frame
Transcribed from gene enhancers and modulates gene activation and silencing, X chromosome inactivation, alternative splicing, and post translational regulation
Involved in epigenetic modifications and are themselves epigenetically regulated
Thousands ofn lncRNAs are differentially expressed in tumours vs normal tissue
The steroid receptor activator is a lncRNA
They can act as molecular scaffolds to guide chromatin modifying enzymes (eg. HOTAIR or DLX6AS24)
Can act as competing endogenous RNAs (ceRNAs) that sponge miRNAs or proteins
They may inhibit long range chromatin interactions
Can function through the act of transcription itself
Additional mechanisms emerging - eg. orchestration of nuclear archiecture, forming circular lncRNAs, destabilising interacting mRNAs
lncRNA-p21 involved in downstream effects of p53
In DDR, p53 binds to promoter of lncRNA-p21 and mediates repression of genes involved in cell cycle arrest and apoptosis
HOTAIR lncRNA is transcribed from HoxC gene cluster at Ch12, interacts with polycomb repressive complex 2 (PRC2) at 5’ end and lysine specific demethylase 1 (LSD1) complex which induce epigeneticc aleterations and cause gene silenceing/oncogenesis
HOTAIR dysregulation associated with cancer progression in 26 tumour types and predictive of metastasis in breast cancer
Can act as a sponge by binding/inhibiting specific repressive miRNAs
miRNA
Small ncRNA, 18-25nt, regulates mRNA expression
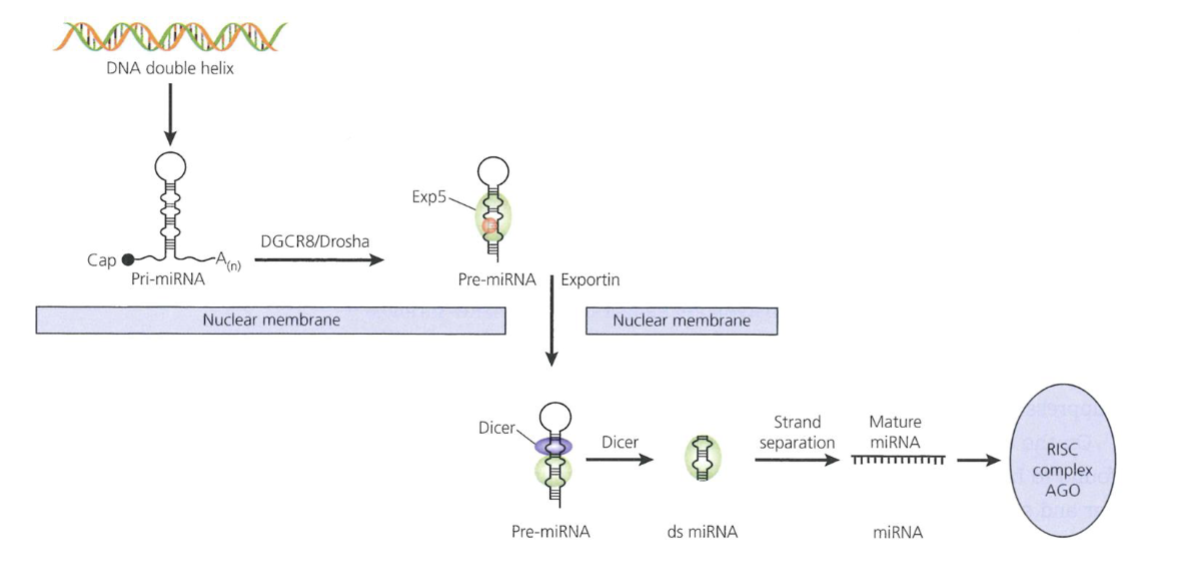
After being transcribed by RNA pol II from intergenic or intronic regions, primary transcript is processed by ribonucleases DGCR8 and Drosha in the nucleus, producing pre-miRNAs (hairpin shaped, 70-100nt intermediate)
Exportin-5 transports pre-miRNA into cytoplasm to by processed by the ribonuclease Dicer into double stranded miRNA
Strands separate and the mature single stranded molecule joins RNA-induced silencing complex (RISC) which contains an endonuclease component Ago which can degrade target mRNA
miRNA can hybridise perfectly to the 3’ UTR of the mRNA within RISC, causing mRNA cleavage/degradation, or it may bind to imperfect complementarity sites in the 3’ UTR in the RISC to block translation
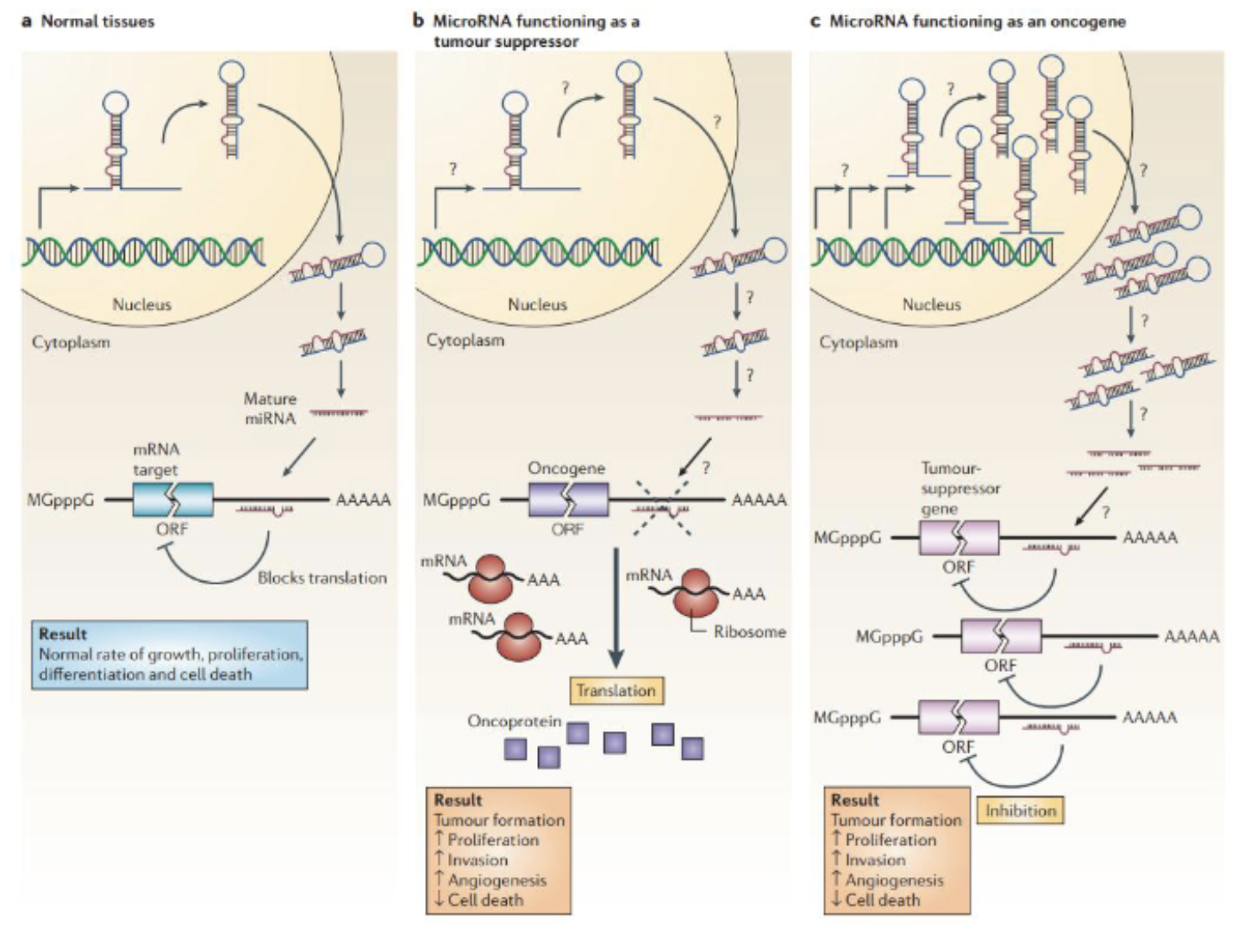
Reduction/deletion of tumour suppressing miRNAs promotes tumourigenesis, which could happen due to defects at any stage of miRNA biogenesis (amplification of miRNA gene, constitutively active promoter, increased processing or stability of miRNA)
Some miRNAs may act as oncogenic oncomirs (aka oncomiR) which function to suppress tumour suppressor mRNAs may be amplified in cancers
Let-7 family of miRNAs repress Ras oncogenes, refuced in lung, breast, urothelial, and cervical cancers
miR-21 inhibits apoptosis and is upregulated in glioblastoma and breast cancers
miR-155 cooperates with Myc to promote B cell malignancies, overexpressed in Burkitt lymphoma
Evidence that miRNA expression profiles may help discriminate different types of cancers, may be useful for diagnosis/prognosis where mRNA profiles fall short
Translational control
Most transcriptional activity in cells is directed for rRNA synthesis and synthesis of mRNA for ribosomal proteins
More cellular energy is directed towards translation than transcription and DNA replication
Tumour cells under physiological stress downregulate translation and become uncoupled from regulation, controlled by translational initiation
Translational initiation in eukaryotes:
Pre-initiation complex (43S)
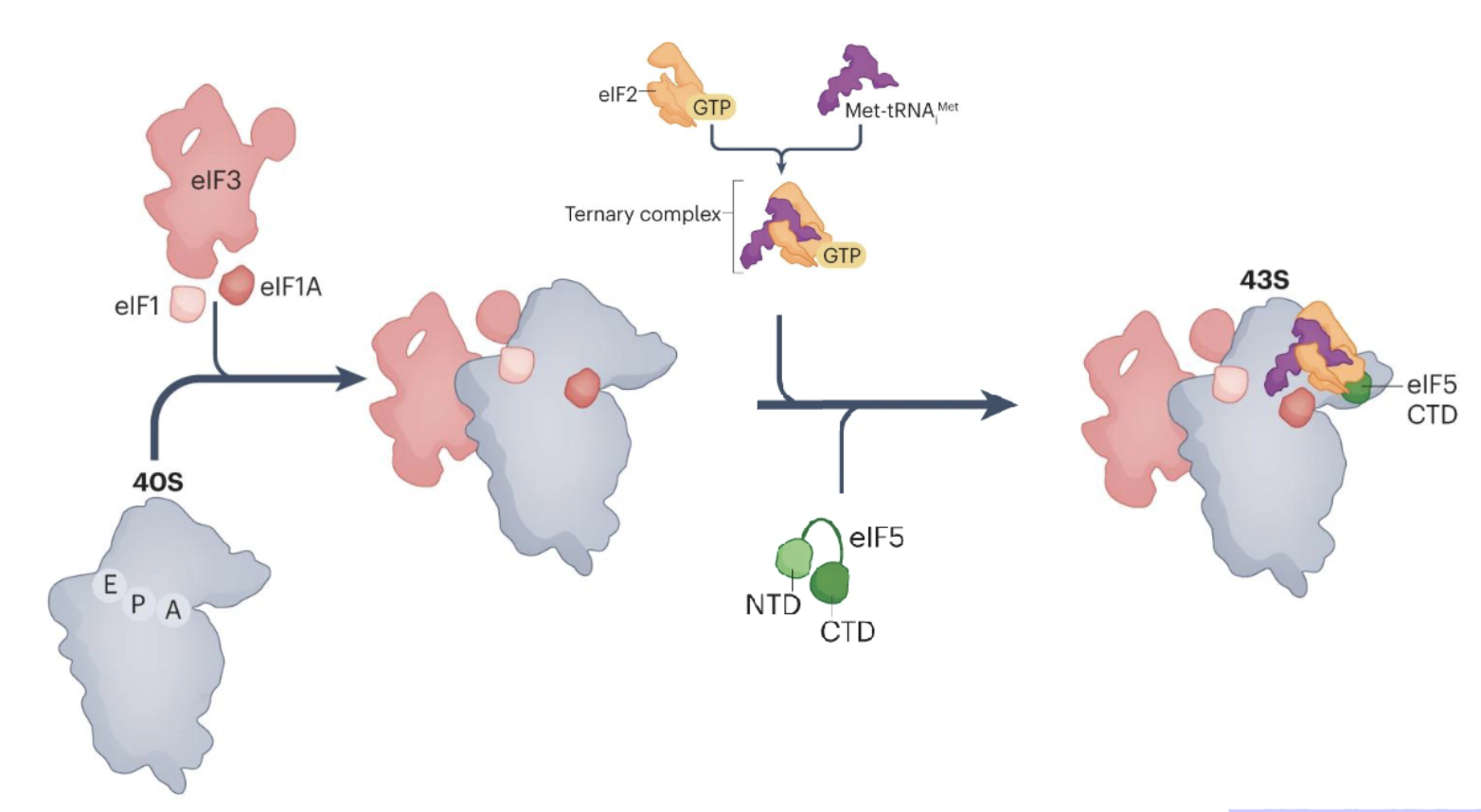
Binding of eukaryotic translation initiation factor 1 (eIF1), eIF1A, and eIF3 to the 40S subunit
eIF5 and ternary complex of eIF2, GTP and met-tRNA then bind to form a 43S translation PIC
mRNA recruitment
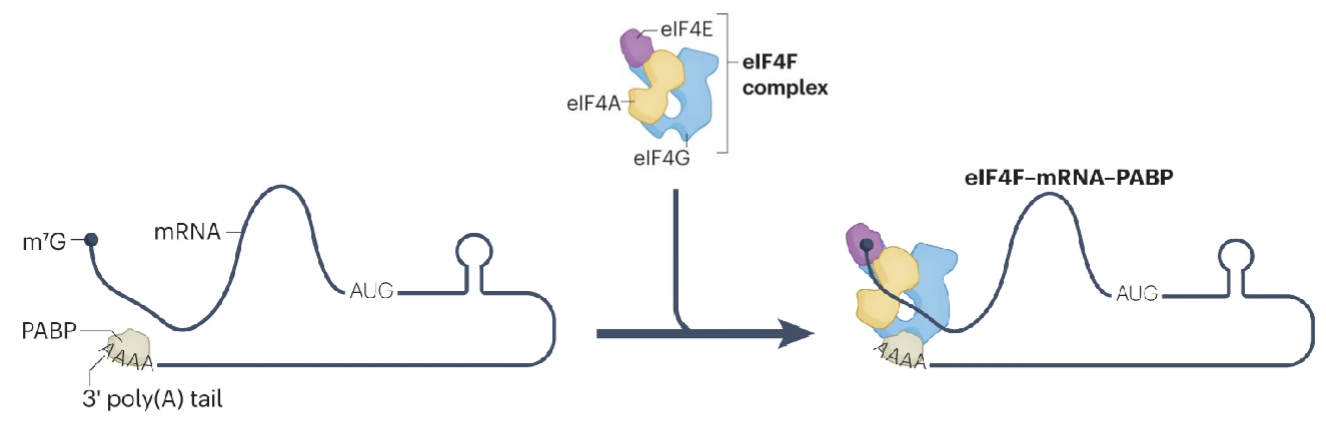
Activation of mRNA by cap binding complex eiF4F is crucial for recruitment
eIF4F and polyadenylate-binding protein (PABP) bind to the poly(A) tail, selecting mRNA for recruitment
43S recruitment to mRNA
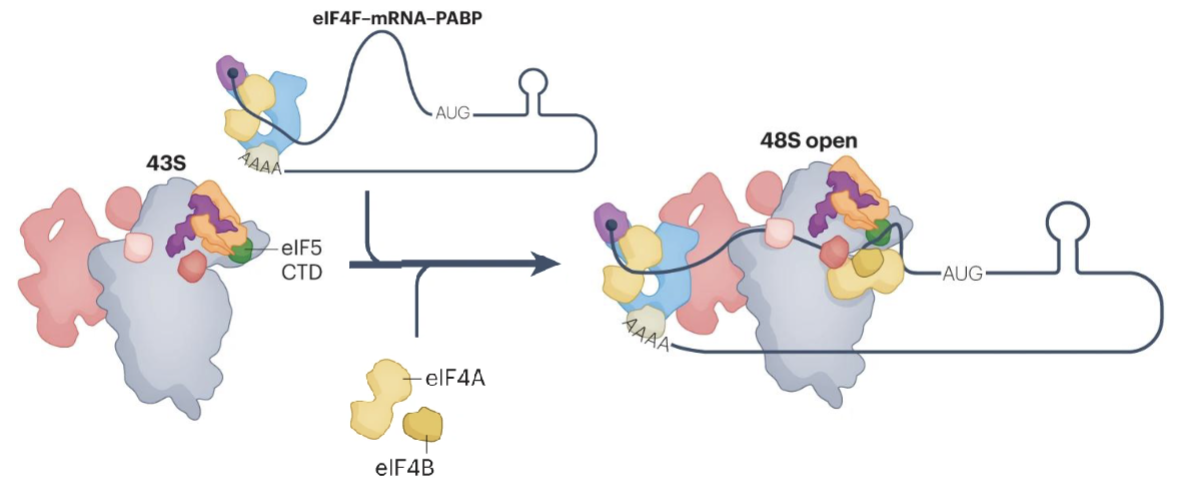
Assembled 43S is recruited to mRNA to form the scanning-competent 48S complex (48S open)
During scanning of 5’ UTR of mRNA, eIF5 interacts with eIF2 and accelerates eIF2-bound GTP hydrolysis
Scanning and AUG selection
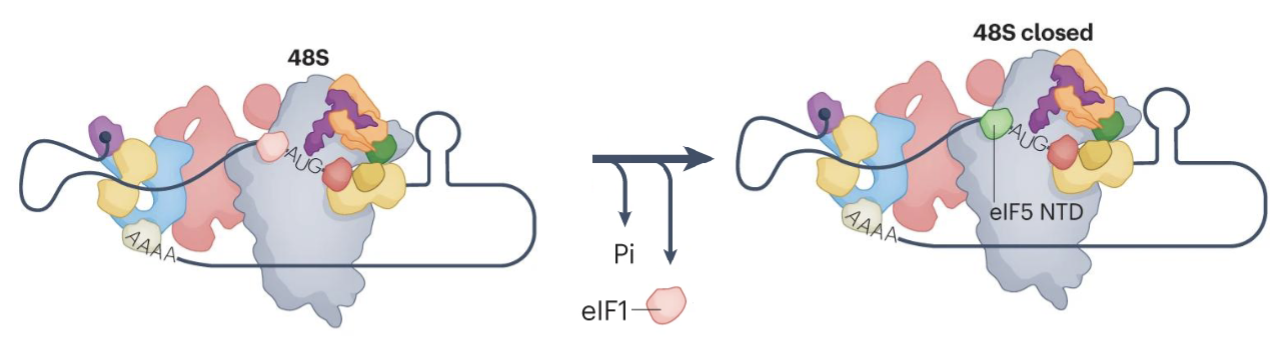
Start codon selection triggers release of eIF1 and Pi from the complex
Formation of 80S initiation complex
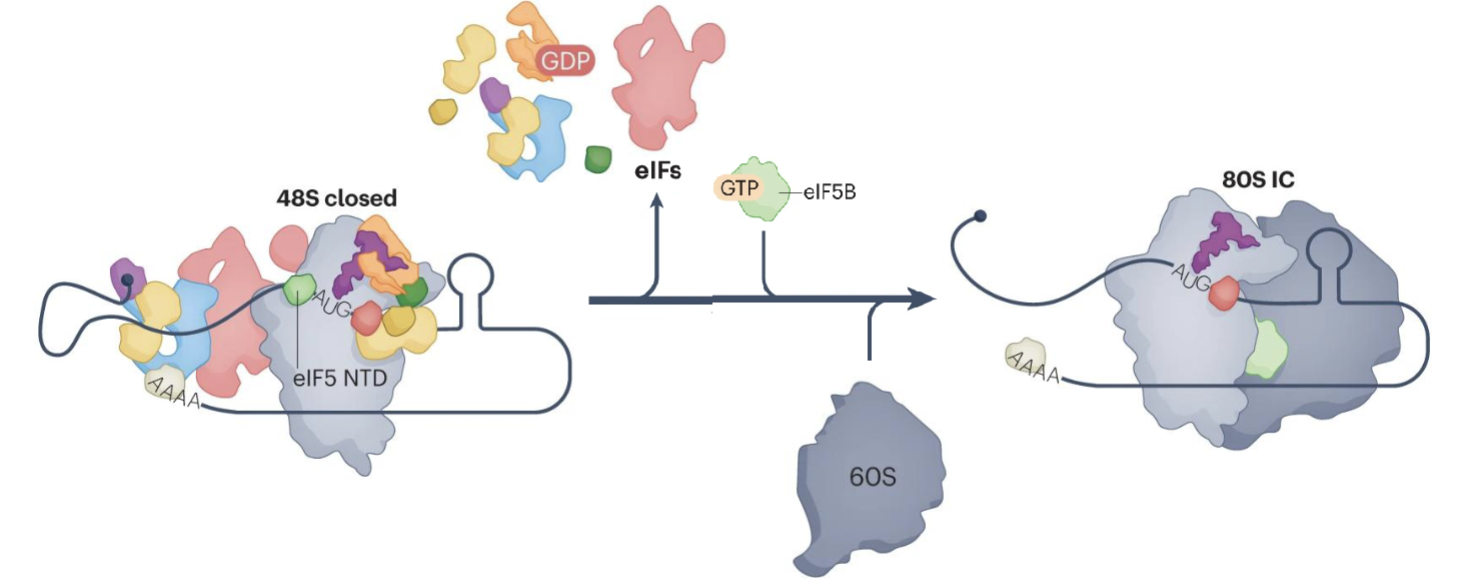
N terminal domain of eIF5 occipies the position vacated by eIF1 near the P site of the ribosome
eIF2-GDP has lower affinity for Met-tRNA so the release of Pi triggers the release of eIF2-GDP, eIF5, eIF3 and eIF4
eIF2-GDP release allows eIF5B to bind, promoting the joining of the 60S large subunit, forming the 80S initiation complex (80S IC)
Elongation and termination
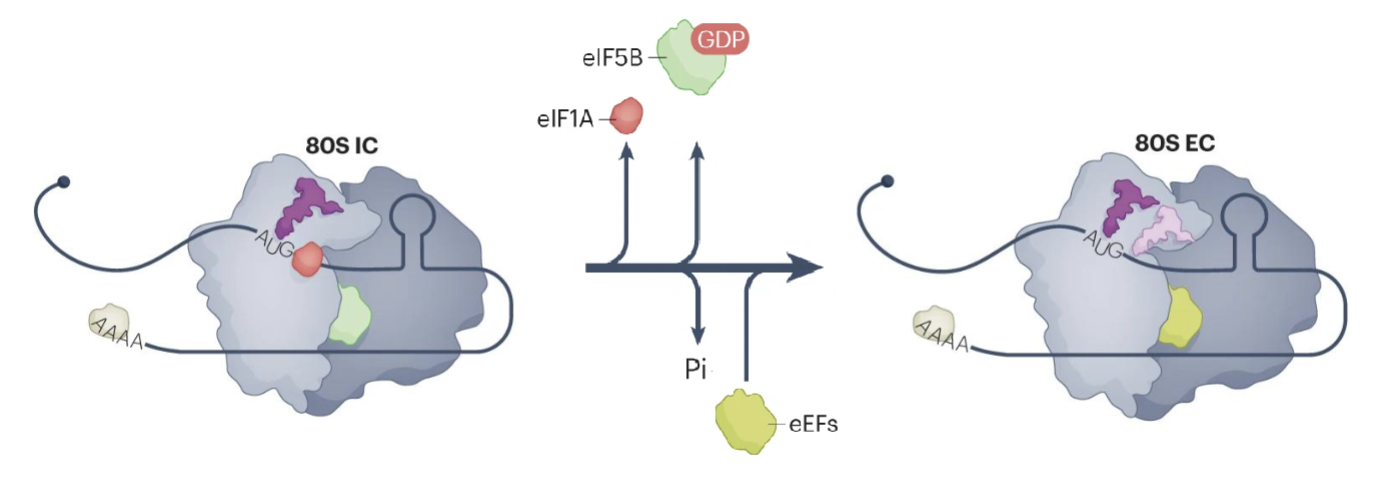
80S IC formation triggers hydrolysis of eIF5B-bound GTP and release of eIF1A
eIF5B undergoes conformational change that places the amino acylated end of Met-tRNA in the peptidyl transferase centre of the ribosome
eIF5B release marks the end of initiation and beginning of elongation (80S EC)
Eukaryotic elongation factor 1A (eEF1A)-GTP delivers amino-acylated tRNA to the A site of the ribosome
eEF2-GTP promotes tRNA translocation from A site to the P and E sites
Release of eEF2-GTP and deacylated tRNA from the E site allows a new elongation cycle to occur
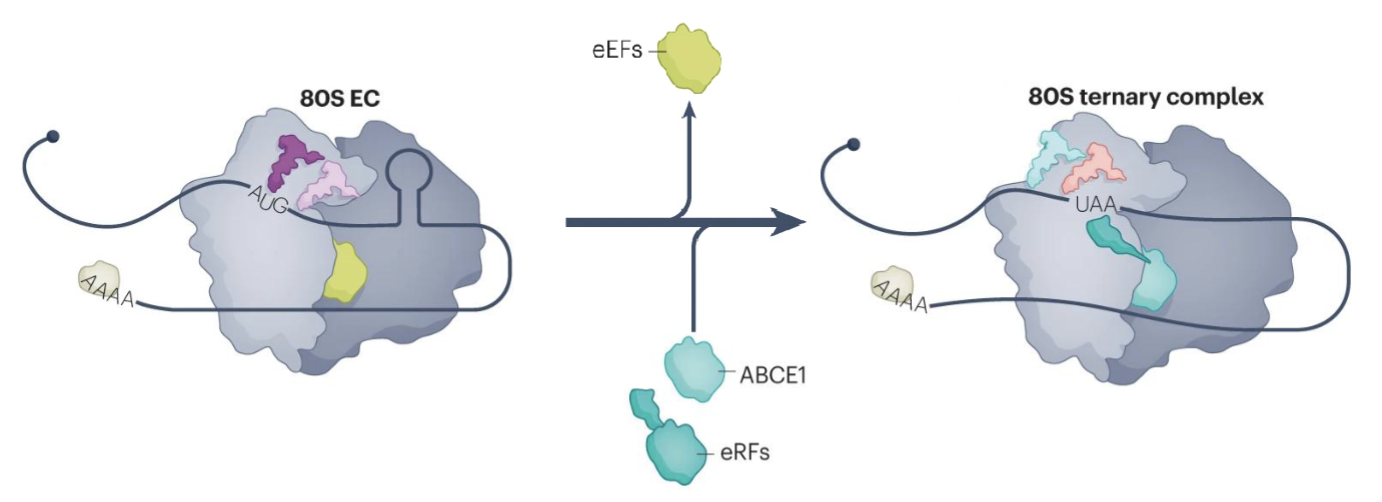
Translation termination by eukaryotic release factors (eRFs) occurs upon encountering a stop codon
ABCE1 binds to 80S termination coplex and stimulates peptidyl-tRNA hydrolysis by eRF1
Recycling
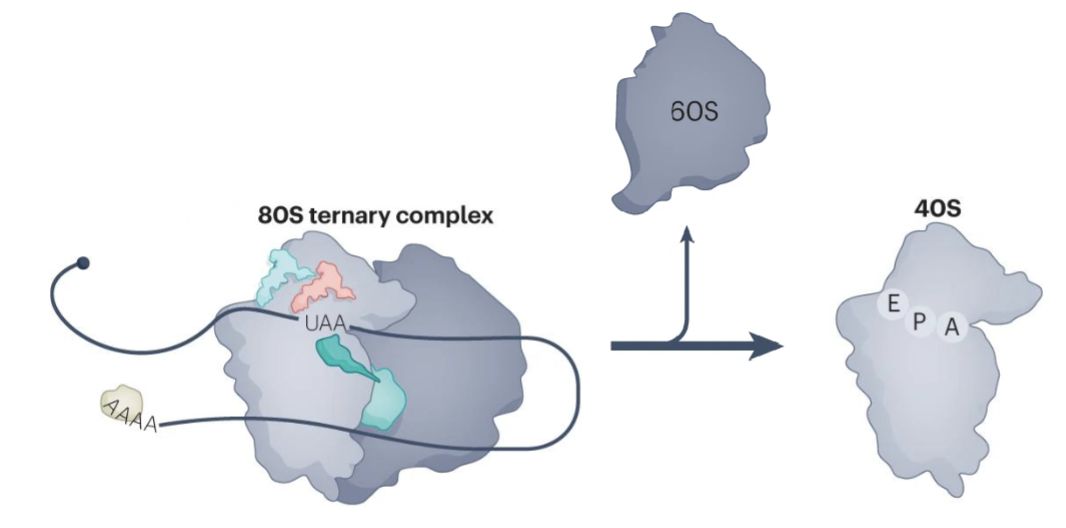
ABCE1 splites 80S into the 40S and 60S subunits
mRNA and tRNA are removed from the 40S by recycling factors, freeing the 40S subunit for a new round of translation
eIF4F complex formation

Ribosomes are recruited to the 5’ end of mRNA via the eIF4F complex, consisting of eiF4E, eIF4G, and eIF4A - these are all targets of Myc and can be dysregulated in cancer, amplified in human tumours
eIF4E and eIF4G are classical oncogenes
These components can also regulate translation initiation, eg. eIF4E phosphorylation by MNK1 and MNK2 can promote tumour development and dissemination
MNK-mediated phosphorylation of eIF4E is also involved in translational reprogramming driving tamoxifen resistance in ER+ breast cancer
eIF4A can be sequestered by tumour suppressor protein PDCD4, preventing formation of eIF4F complex
Loss of PDCD4 associated with cancer cell invasion and poor patient survival
eIF4E-binding proteins (4E-BPs) compete with eIF4G to bind to eIF4E, acting as tumour suppressors by inhibiting cap dependent translation
Loss of 4E-BP expression can also be lost or have impaired function due to inhibitory phosphorylation
4E-BP expression is increased in stage III non-metastatic oesophageal, breast, and prostate cancers, where is is proposed to oppose metastasis but lead to large, locally advanced tumours
Ternary complex (TC) formation
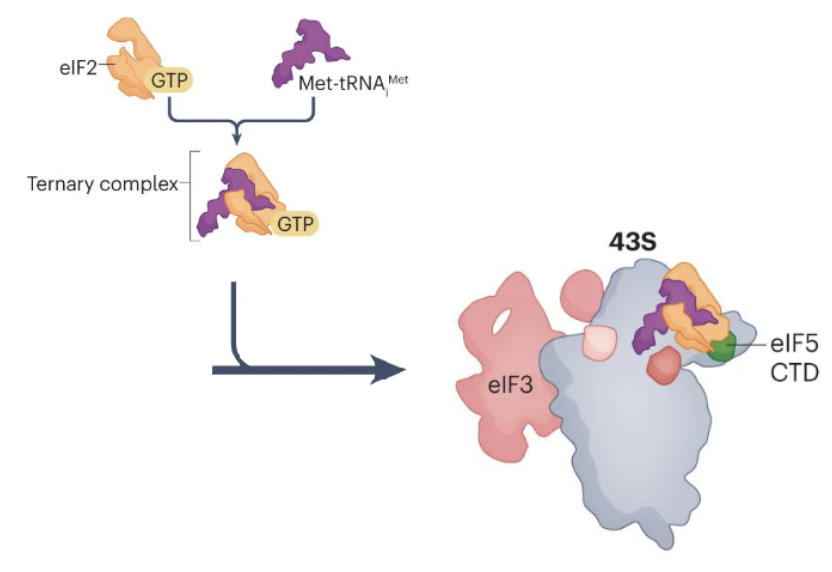
TC is composed of eIF2, GTP, and Met-tRNA
Deregulated TC formation in cancer has caused different findings relating to eIF2α phosphorylation
Generally thought that more eIF2α phosphorylation increases ability to respond to stress by promoting translation of upstream open reading frame (uORF) containing stress response mRNAs
Accordingly overexpression of eIF2α or its kinase PKR has been shown to promote transformation in some contexts, but the mechanism is unclear
In contrast long term eIF2α promotes apoptosis and prompted research on upregulating eIF2α activity or inhibiting eIF2α phosphatases for cancer therapy
Outcome of eIF2α phosphorylation is highly context specific and may be related to site of disease or underlying mutations
Additional ways to modulate TC activity could also include overexpressing eIF5 or its mimic proteins (MPs) 5MP1 and 5MP2 which can bind to and sequester eIF2 from the 40S ribosome when present ine xcess
eIF2 binding by eIF5 or 5MPs reduces global protein synthesis but enhances translation of uROF containing mRNAs which could be important for some cancer properties
eIF3 connecting eIF4F and PICs
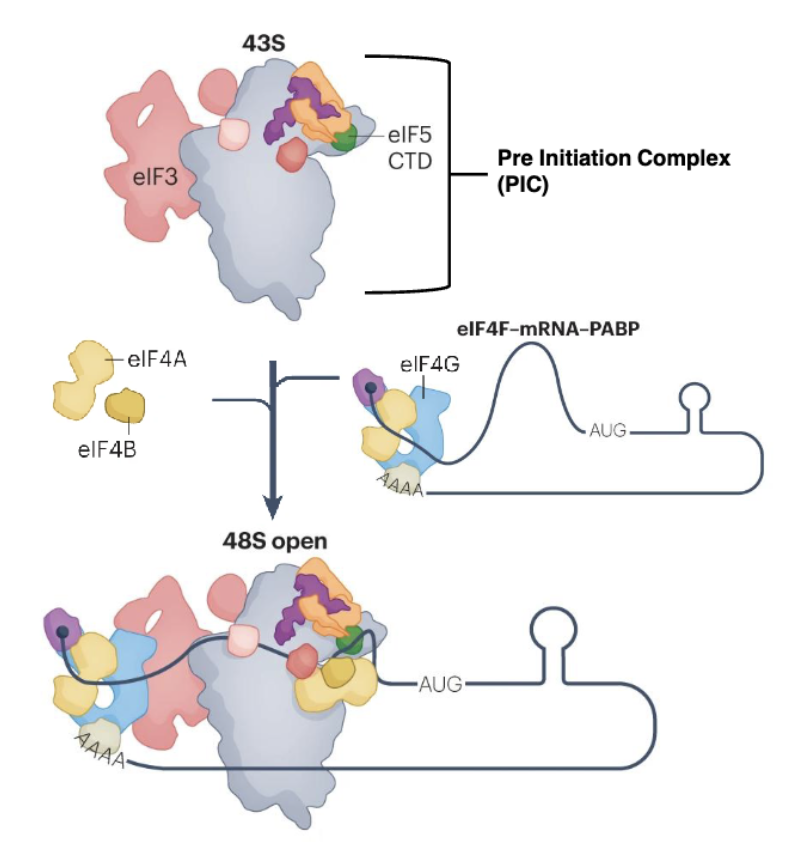
eIF3 is a multi-subunit complex that binds to eIF4G, bridging it to the PIC and thereby connecting mRNAs with the 40S subunit for scanning to occur
Increased eIF3 levels should therefore increase rate of translation initiation
Translation elongation and termination
Literature has focused on translation initiation but oncogenic changes in elongation and termination is also emerging, eg. dominant role for loss of inhibitory regulation of elongation by eEF2K for intestinal tumour formation
Increased availability of specific tRNA isoaccepting species in cancer cells has a role in tumourigenesis
Speed of amino acid incorporation during elongation is dependent on availability of corresponding charged tRNA
Several studies have reported translation programs in which proliferating undifferentiated cells and cancer cells express tRNAs optimised to correspond to codon usage of pro-proliferative mRNAs
Elongation can be deregulated in cancer via programmed -1 ribosomal frameshifting (-1 RPF), a process by which sequence elements force elongating ribosomes back by one base, leading to frameshift, premature stop codons, and nonsense-mediated mRNA decay (NMD)
May explain the oncogenic role of silent mutations inducing frameshifting in tumour suppressors
Termination at premature stop codons can be a cancer driver if it occurs as a result of somatic mutations in tumour suppressor genes, resulting in NMD of the corresponding transcripts
eIF6 initiation factor has confusing multiple roles in mRNA translation and associated with altered translational regulation in cancer
Ribosomal subunit anti-association factor that prevents aberrant interactions between the 40S and 60S ribosomal subinits
eIF6 must be displaced from the ribosome for the final step of 60S ribosome biosynthesis in the nucleolus and can promote 80S ribosome disassemby in cytosol by preventing the reassociation of post-termination 60S ribosomes
Aberrant expression observed in some cancers in which it accumulates in the nucleolus
Hallmarks of cancer
Sustained proliferative signalling
Reduced dependence on external growth stimulation signals
Involves alteration of growth signals, mutations to transcellular transducers or intracellular circuits
Evading growth suppressors
Acquired mutations or gene silencing interfere with response to growth suppressor signals
Enabling replicative immortality
Maintaining telomere length to bypass Hayflick limit
Activating
Proteins tethering cells to their surroundings are altered for cancer invasion
Modulating cell-cell adhesion proteins (notably Ig and calcium dependent cadherin families) which mediate cell-cell interactions, integrins which link cells to ECM
Inducing angiogenesis
Altering the balance between angiogenic inducers and inhibitors (angiogenic switch)
Supplying oxygen and nutrients, removing wastes
Evasion of cell death
Upregulating pro-apoptotic proteins and downregulating anti-apoptotic proteins
Includes other programs of cell death
Avoiding immune destruction
Avoids stimulating immune response to escape immunological killing
Deficiencies in the development or function of immune cells
Deregulating cellular energetics
Aerobic glycoloysis
Upregulating glucose transporters like GLUT1 to increase glucose import
Tumour-promoting inflammation
Tumour-associated inflammation has unanticipated, paradoxical effects on tumourigenesis and progression
Inflammation can supply bioactive molecules to the TME, including GFs, survival factors, proangiogenic factors, ECM modifying enzymes which facilitate angiogenesis, invasion, and metastasis
ROS production can contribute to mutagenesis and higher malignancy
Genome instability and mutation
Increased mutability due to breakdown in genomic maintenance machinery
Compromising surveillance systems that normally moniotor genomic integrity and force damaged cells into senescence/apoptosis
Non-mutational epigenetic reprogramming
Tumour specific pressures like hypoxia and nutrient deprivation in TME can reshape epigenetic lanscape to influence cell phenotypes
Promotes tumour evolution and therapeutic resistance
Can also corrupt stromal cells to support malignant progression
Polymorphic microbiomes
Microbial communities can influence tumour development and therapeutic response
Specific gut bacteria can induce DNA damage, stimulate proliferative signalling, and shape the immune response
Microbiota in the skin, lung, and vagina can modulate local cancer risk and behaviour
Intra-tumoural microbiomes can also have a role
Senescent cells
Traditionally viewed as tumour suppressive barrier
Senescence associated secretory phenotype (SASP) - releases cytokines, chemokines, proteases that enhance cancer properties eg. proliferation, angiogenesis, invasion, and immune escape
Senescent cancer cells can also escape senescence and resume growth with heightened malignancy
Not limited to cancer cells, also may affect stomal cells which can become senescent to remodel the TME to support growth/metastasis
Unlocking phenotypic plasticity
Can reverse or circuvent terminal differentiation
Dedifferentiation - mature cells revert to progenitor-like states that are more permissive for proliferation and tumourigenesis
Blocked differentation - progenitor cells prevented from completing maturation
Transdifferentiation - permits cancer cells to switch lineage and allow adaptation to new environments or therapeutic pressures
Often driven by altered developmental TFs and epigenetic reprogramming, frequently accompany malignant progression and therapeutic resistance