Unit 1
Jan 8 - Types of Molecular Interactions
ionic - interaction of 2 charged atoms based on coulombs law
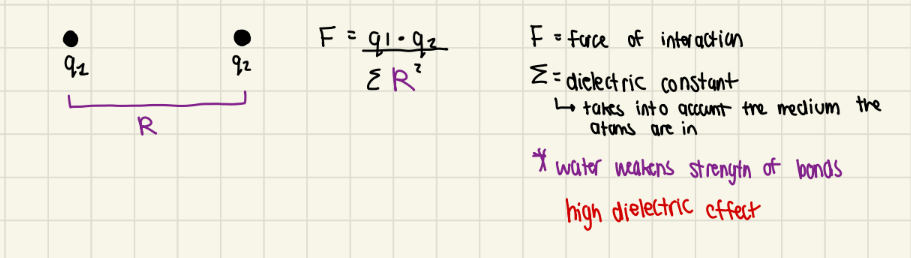
hydrogen bonds - unequal sharing of a hydrogen atom; a hydrogen atom that is partly shared by 2 electronegative atoms
the hydrogen donor is electronegative and tends to pull electrons away from the hydrogen, the result is:
hydrogen donor: delta -
H is covalently bonded to
H: delta +
hydrogen acceptor: delta -
both are usually O2 or N2, sometimes Suflur
is also electronegative and develops a delta -
needs a lone pair of electrons
how a hydrogen bond is formed: the delta + charged H is attracted to the delta negative acceptor and hydrogen bond is formed
H-bonds are weak 4-13 kJ/mole and longer 1.5-2.6 A than covalent bonds
Van Der Waals Interactions - temporary dipoles; attraction of any 2 molecules
at any given time, the charge distribution around an atom is not symmetric
result: this asymmetry causes complimentary asymmetric on other atoms, causing them to be attracted to each other
has smallest energies of 2-4 kJ/mole
attraction inc until the electron clouds start to overlap and repel
contact distance is the distance maximal attraction of 2 atoms
Hydrophobic Interaction - almost all biochemical reactions occur in water
water has a big impact on these reactions and interactions
structure: has a bent shape, making the molecule polar and capable of forming multiple H-bonds
result: water is very cohesive (can form hydrogen bonds with each other); a hydrogen-bonding machine
water is an excellent solvent for polar or charged molecules
hydrophilic: soluble in water
hydrophobic: insoluble in water
amphipathic: hydrophilic + hydrophobic groups
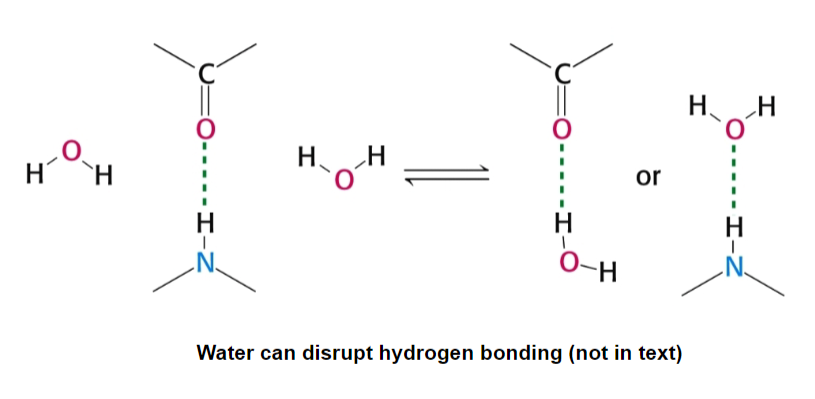
water can weaken electrostatic interactions by competing for their charge (or partial charge)
water reduces electrostatic interactions by 80x (high dielectric constant)
result: serious consequences for biological systems, water often needs to be excluded/manipulated to allow various electrostatic reactions to occur
water is a double-edged sword: we need it to dissolve, but it interferes with electrostatic interactions
Jan 10 - Laws of Thermodynamics
All biological events are governed by a series of physical laws of thermodynamics
I. The total energy of a system (matter in a defines space) and its surroundings is constant; cannot be created or destroyed, can only change form
ex. burning wood or dropping a ball off a roof
Enthalpy (H): the heat content of our system
II. The total energy of a system and its surroundings always increases for a spontaneous process
entropy (S) = randomness
system or universe increase in entropy for a reaction to go forward
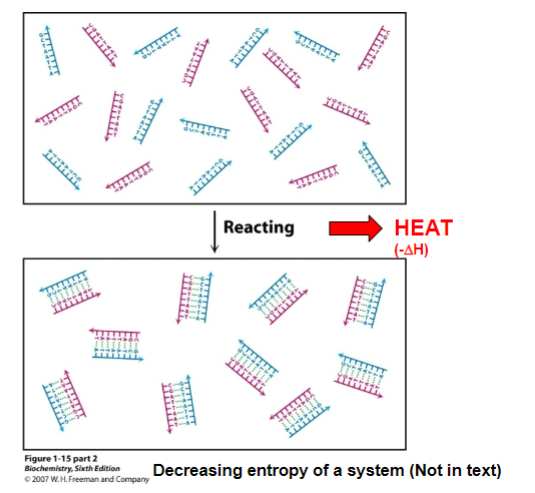
ex. goes from ordered system to a more disordered one
Things can become more ordered
entropy can be decreased locally in the formation of ordered structures ONLY if the entropy of the universe is increased by an equal or greater amount
entropy decreases in system (2 molecules going to 1 molecule) but heat is released causing the entropy around the system (ex. the universe) to increase
TLDR: reaction releases heat, causing entropy of the universe to increase, which is greater than the decrease of entropy in the system
we can exist only if we make the universe more disordered
How to Measure Spontaneity
ΔG = ΔHsys - TΔSsys
T = temp in K
ΔG is gibbs free energy measured in KJ/mole
Spontaneity = how likely a reaction or process will occur
doesn’t define how fast a reaction is, only if the reaction will occur
ΔG < 0 = the reaction is spontaneous
ΔG > 0 + the reaction is non-spontaneous
Enthalpy/Entropy
Affect on ΔG
ΔH < 0 (ex. releasing heat)
ΔG becomes more negative and the reaction becomes more spontaneous
ΔS > 0 (ex. reaction becomes more disordered)
ΔG becomes more negative and the reaction becomes more spontaneous
Why you want to know ΔG → reactions needs to be spontaneous to occur in the cell (ΔG = no reaction)
Jan 10 - Entropy & The Hydrophobic Effect
Hydrophobic molecules cluster together since the entropy is more favorable
hydrophobic molecule entropy decreases
water entropy increases - more hydrogen bonds with water, more ordered
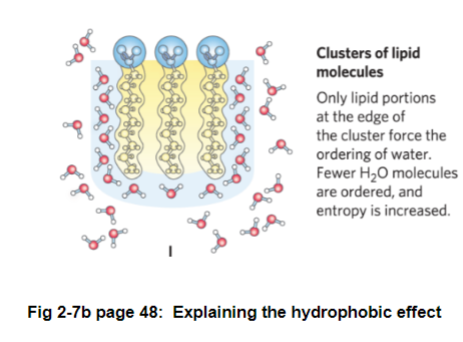
Fig 2-7b page 48: when a non-polar molecule is added to water, the water molecules are forced into a cage around the hydrophobic molecule
result: lowers entropy since water is becoming ordered
when 2 non-polar molecules come together, fewer water molecules are needed to form the cage around the non-polar molecules
result: entropy increases; favors non-polar molecules to cluster together (not a direct force pulling the 2 molecules together)
Jan 10 - Ph and Buffers
The concentration of hydrogen ions within biological systems is critical
Why? Most biomolecules (not all) act as weak acids or bases (ex. their functional groups can be ionized)
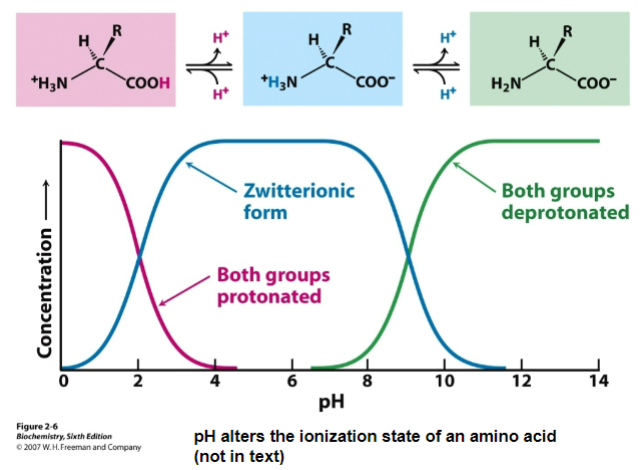
Figure 2-6: amino acid and nucleotide charge changes with plot
changing pH may change the way a molecule behaves since it can affect its ionization state
molecule behavior depends on ionization state
[H] is measured as pH
pH = -log[H]
scale of 0-14
0 = strongly acidic
14 = strongly basic
How do we maintain a pH of a cell (7.2-7.4)? Weak acids can act as buffer
weak acid = an acid that is not completely ionized in solution
acid - gives up protons
base - proton acceptor
HA ←→ A^- + H^+
pKA = -logKa

we can calculate the pH of any solution of a weak acid if we know the molar ratio of a conjugate base to acid and pKa → Henderson Hasselbalch Equation
pH = pKa + log[A]/[HA]
if we titrate a weak acid (ex. acetic acid) with a strong base (NaOH) we can see the following:
pH = pKA → [HA] = [A]
pH < pKa → [HA] > [A] → more acid than conjugate base
pH > pKa → [HA] < [A] → conjugate base is more dominant form
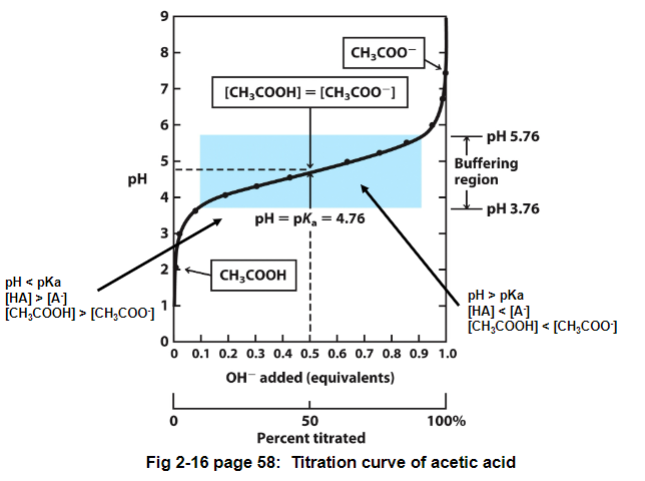
Fig 2-16: there is a region where the pH doesn’t change very much even though a large amount of acid (or base) are being added
buffering region: a mixture of a weak acid and a conjugate base in a 1:1 ratio
function: resists changes in pH because both forms are present and can either soak up or release a proton
pH = pKa here
the buffering region is usually +/- pH unit from pKa unit
ex. if pKa is 4.76, pH is 3.76
buffers fail when there is only the acid or conjugate base present
you want the buffer to have a pKa = 7
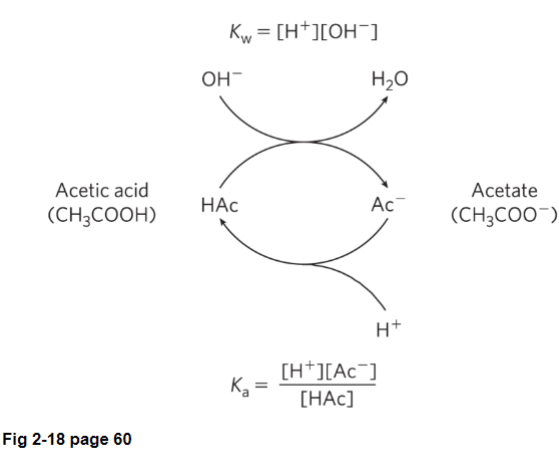
Fig 2-18: buffer gives away proton, turning into the conjugate base (acetate)
if HCI is added, the pH will drop (increasing [H])
pH will go down slowly if you add both acetic acid and acetate since acetate will soak up the proton, turning it into acetic acid
Types of Buffers
Bicarbonate (more complicated)
CO2 (dissolved) + H2O ←→ H2CO3 (carbonic acid) ←→ H + HCO3 (bicarbonate)
approximate pKa of 6.1, would buffer from 5-7
Phosphate
H2PO4^- ←→ H + HPO4^-2
pKa of 7.1, buffering rage 6.2 - 8.2
Histidine and Cysteine
amino acids
Histidine pKa = 6; Cysteine pKa = 8
Jan 13 - Protein and Amino Acid Structure
protein: linear polymer built of amino acids
structure: final shape depends on its sequence of a.a
a.a contain a variety of functional groups, allowing for massive diversity
proteins can interact with each other or molecules to form complexes
proteins can be flexible or rigid
amino acid: the “ultimate lego set”
Structure:
alpha carbon
carboxylic acid/ carboxyl group (COOH/COO-)
the form that exists depends on pH
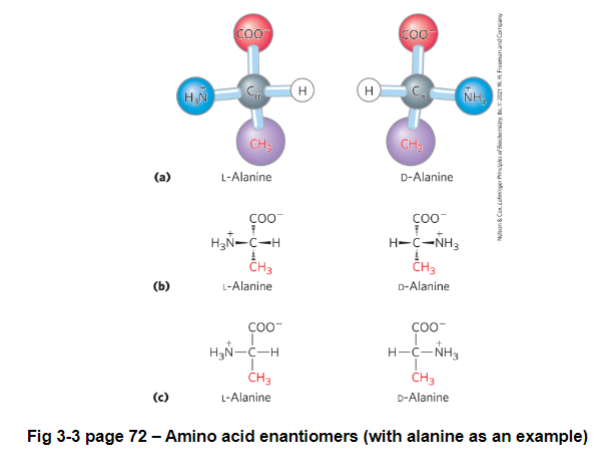
the carbon is chiral: an atom that has 4 different functional groups; not superimposable on its mirror image
thus there are 2 enantiomers of each amino acid, except for glycine (R group is hydrogen)
enantiomer: a pair of molecules each with one or more chiral center that are mirror images of each other
hydrogen
unique R side-chain
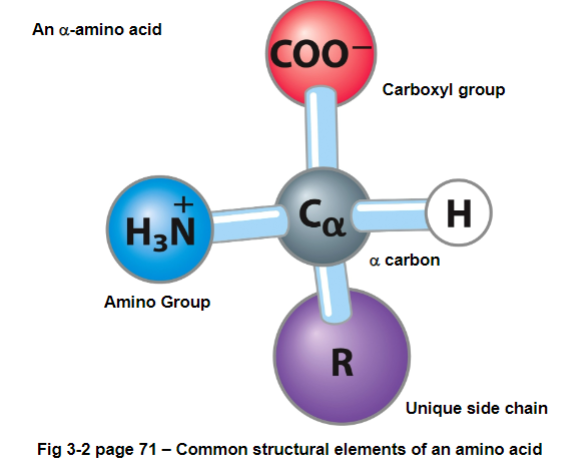
FIG 3-2: amino refers to NH3 group, acid refers to carboxyl group - all attached to an alpha carbon
Determining L or D
carboxyl group is above central carbon, and R group is below
L amino acid = if amino group (NH3) is on left to alpha carbon
D amino acid = if amino group (NH3) is on right to alpha carbon
In biological systems only the L-amino acids exist (with very few exceptions) in proteins and living systems
furthermore almost all L-amino acids are in the S-configuration
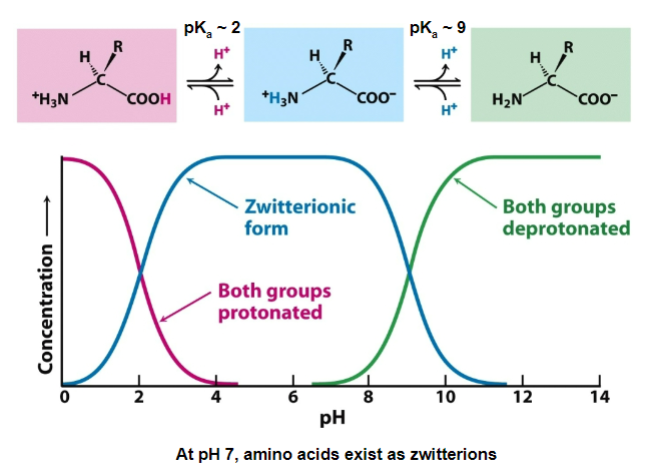
Ionization of amino acids
all amino acids have 2 ionizing group (carboxyl and amino group), often 3 (R-group)
carboxyl pKa = 2
pH 0-2 - COOH dominates
pH 2 - 14 - COO- dominates
amino group pKa = 9
0 - 9 - NH3+ dominates
Hence, at pH 7 amino acids exist as zwitterions: ions with both positive and negative charges
Types of Amino Acids
basic and acidic amino acids are often found on the surface of proteins (interact with water and away from the hydrophobic amino acids)
you can vary the amino acid by changing the side-chain (R-side chain)
there are 20 key amino acids and these (or slight modifications are used in all living things
you have to know all 20 amino acids (draw them), their structures, names, and abbreviations
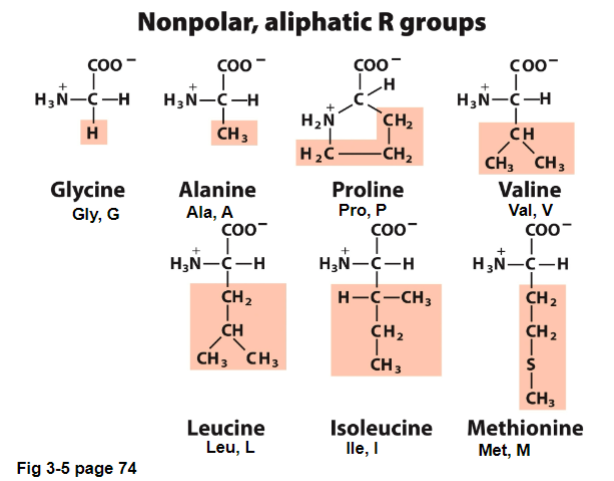
Jan 15 - I. Nonpolar, Aliphatic R groups
Non-polar, aliphatic: all are hydrophobic and will tend to cluster together
different sized and shaped R-groups allow for close packing
usually found in the center of a protein, away from the water
usually not reactive
aliphatic: compounds with an open chain structure (alkane)
Types of Polar, uncharged R groups | Characteristics | How to remember |
Glycine (Gly,G) |
| |
Alanine (Ala, A) |
|
|
Valine (Val, V) |
|
|
Leucine (Leu, L) |
|
|
Isoleucine (Ile, I) |
|
|
Methionine (Met, M) |
|
|
Proline (Pro, P) |
|
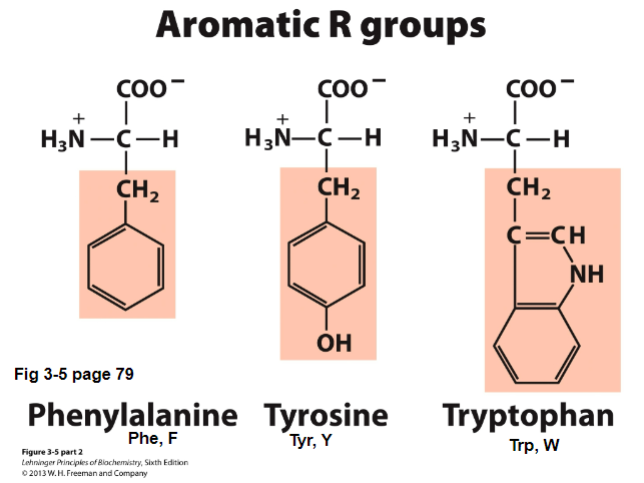
II. Aromatic R-groups
Aromatic Amino Acids: contain aromatic rings (ex. phenyl rings)
participate in hydrophobic interactions
usually found in the core of proteins
Types of Aromatic R-groups | Characteristics | How to remember |
Phenylalanine (Phe, F) |
|
|
Tyrosine (Tyr, Y) |
|
|
Tryptophan (Trp, W) |
|
|
III. Basic Amino Acids - Positively Charged R Groups
Basic Amino Acids (positively charged, high pKas)
lysine and arginine are both positively charged at pH 7 (pKas > 10)
Mea culpa: my grievous fault; the imidazole of His is aromatic, but we don’t classify it as aromatic
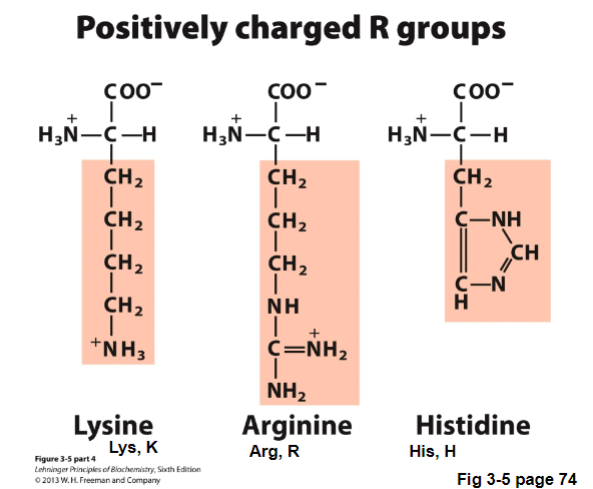
Types of Basic Amino Acids - Positively Charged R Groups | Characteristics | How to remember |
Lysine (Lys, K) |
| |
Arginine (Arg, R) |
|
|
Histidine (His, H) |
|
|
Jan 17 - IV. Acidic Amino Acids - Negatively Charged
Acidic Amino Acids - negatively charged
contain carboxyl groups as R groups
negatively charged at pH 7 (pKas < 4)
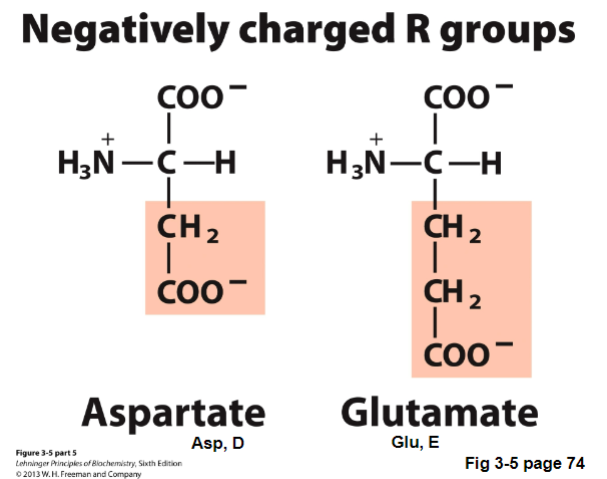
Types of Acidic Amino Acids - Negatively Charged | Characteristics | How to remember |
aspartate (Asp, D) |
|
|
glutamate (Glu, E) |
|
|
V. Polar, Uncharged R groups
not charged at pH 7
can H-bond, more hydrophilic
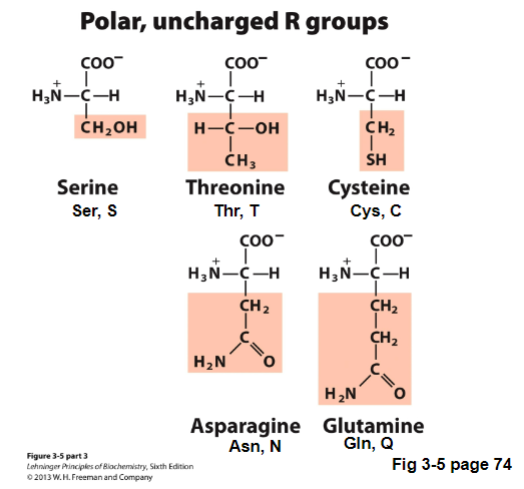
Types of Polar, uncharged R groups | Characteristics | How to remember |
serine (Ser, S) |
| |
threonin (Thr, T) |
| |
cysteine (Cys, C) |
| |
Asparagine (Asn, N) |
|
|
Glutamine (Gln, Q) |
|
|
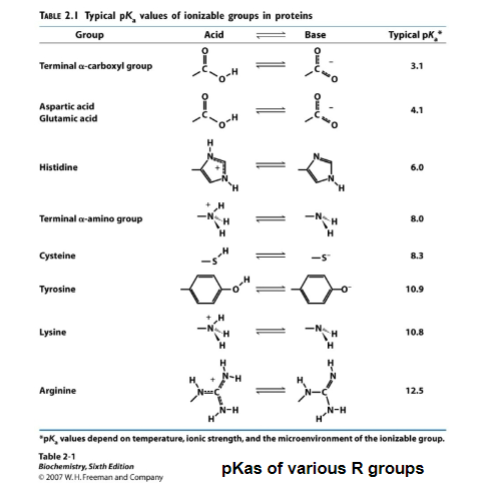
note: the pKa for an R group of an amino acid can change depending on the environment (see page 2 of problem set 2)
dont have to remember pKa, just now the conjugate base and acid form
Protein Structure
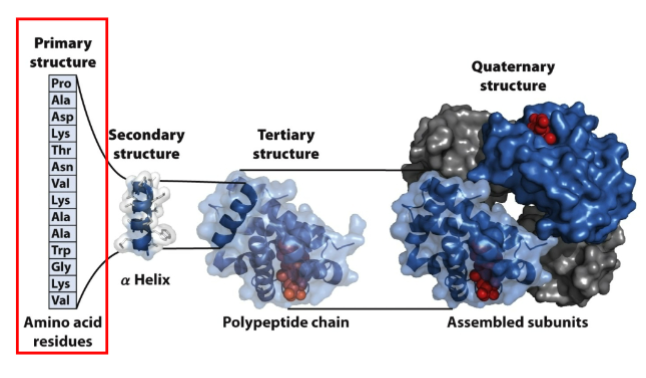
Jan 20 - I. Primary Structure
I. Primary Structure (1 degree) - linear sequence of amino acids linked by peptide bonds to form a polypeptide
peptide bond - linkage of alpha-carboxyl linked to the other alpha-amino group of another amino acid with the loss of water
not energetically favorable, but once formed stable
delta G to break the peptide bond is more favorable (rather be single amino acids) - the activation energy to break a peptide bond is very high
polypeptide - a series of amino acid residues linked by peptide bond
structure
have polarity
not referring to charge, the ends are different (the end that has a free amino - NH3 group)
free amino group should always be on left, free carboxyl group is on right
residue - an amino acid unit in a polypeptide
contain a backbone which repeats (NCC)(NCC) with variable side chains
the backbone is hydrophilic
carbonyl and amino group can H-bond, except proline which has limited H-bonding ability
molecular weights of proteins are expressed in Daltons or more commonly Kilodaltons (kDa)
1 Da = mass of hydrogen atom = 1g/mole
each protein has a unique primary amino acid sequence
primary sequence is encoded in the DNA
knowing the primary amino acid sequence allows us to determine the
determine shape - 3D shape of a protein depends on primary sequence
determine function
understand disease
understand evolutionary history
polypeptides are flexible but conformationally restrained
what does this mean? the peptide bond has double bond characteristics due to resonance between the peptide bond and the carbonyl
result:
the peptide bond is planar. This in turn locks a series of atoms into a plane
no rotation about the peptide bond
since peptide bond acts like a double bond, the peptide bond could be in cis or trans orientation
cis = same direction
trans = pointing in diff directions
due to steric hindrance, all peptide bonds are in trans (except for x-pro peptide bond where both cis and trans occur)
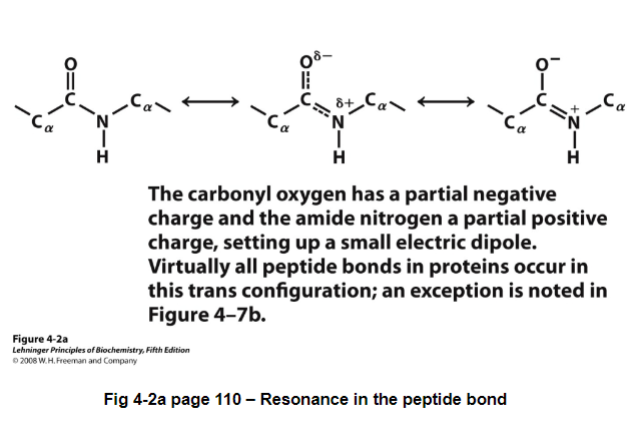
fig 4-2: are all in one plane; can’t rotate around the peptide bond
this partly explains the conformationally restrained part, but what about flexibility?
the bond between N-Calpha and C=O-C-alpha are free to rotate
result: provides flexibility, allowing the backbone to fold in many ways
we measure the amount of rotation about the bond as the dihedral angle (torsion angle)
ranges from - 180 to + 180
N - Calpha dihedral angle = phi = φ
C=O-C-alpha dihedral angle = psi = Ψ
not all combinations of phi and si are allowed due to steric hinderance, limiting the number of structures a protein can adopt
the combination of phi and si that are possible are shown in a Ramachandran plot
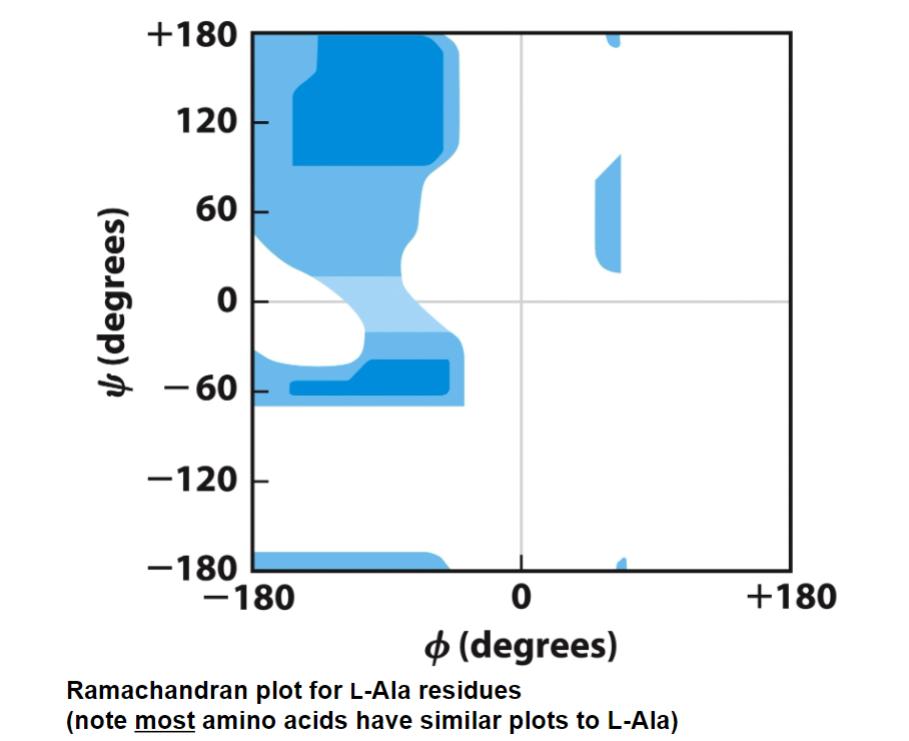
ex. lysine, has H as an R-group, less clashing so more blue
ex. proline, has an R-group ring and covalently bonded to nitrogen - phi are the ones restrained since ring will block it from doing so
Ramachandran plot
darker shade = very favorable
lighter shade = less favorable, but possible
white shade = not permitted
most amino acids have similar plots to L-Ala
note: ¾ of all dihedral angles are not permitted
bringing this all together
large molecules that can freely rotate among many bonds will assume random coils (no defined final structure; a mix of different structures)
because proteins have a series of limitations on what orientation they adopt
1st limitation: double bond characteristic, planar peptide bonds
2cnd limitation: the dihedral angles, phi and si
thus, they can often spontaneously fold into a single structure
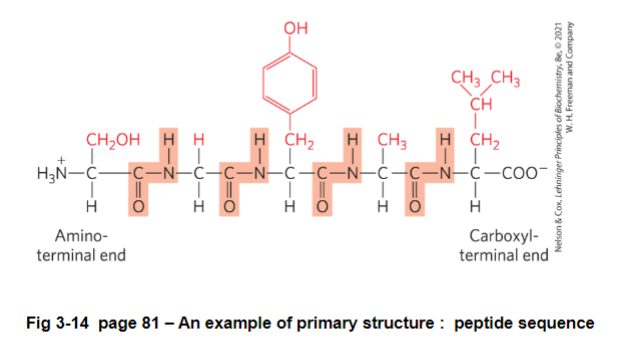
FIG 3-14: should be written as H3N - S - G - Y - A - L - Coo^-
Point Mutations
K 614 P
K = original amino acid
614 = location in primary sequence from the N terminal end
P = new amino acid
ex. describe E6V mutation in the beta-glonin protein
E = glutamine - acid/negatively charged charge
V = valine - r-group is non-polar aliphatic
went from negatively charged to a hydrophobic group
each hemoglobin protein consists of 2 alpha-globin and 2 beta-globin subunits
creates a new hydrophobic patch on the surface of T-state hemoglobin and allows to self-polymerize
rods are inflexible, causing sickle cell anemia
sickle cells go through capillary beds, doesn’t twist and flex and hurts our capillaries (don’t need to know, just know the effect of a singe point mutation)
Jan 22 - II. Secondary Structure - Alpha Helix
II. Secondary Structure - spatial arrangement for amino acid residues in a polypeptide that are relatively close to each other in linear sequence
alpha helices and beta strands/sheets - the common types of secondary structures
alpha helix - a polypeptide backbone forms the inner part of a right handed helix, with the side chains sticking outwards
structure
the helix is stabilized by intrachain hydrogen bonds between the NH and C=O groups of the backbone
intrachain = hydrogen bonds are within the backbone
R-groups are almost perpendicular to the axis of the helix
has ideal dihedral angles of phi - 60 and psi -45
the C=O of residue i forms an H-bond with the N-H or residue i + 4 (ex. 4 residues further towards C-terminus) - see diagram below
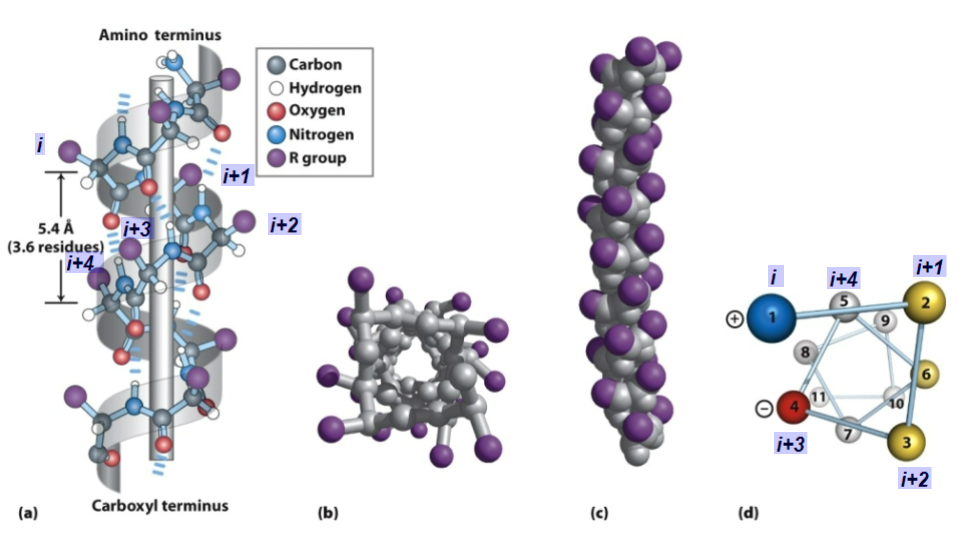
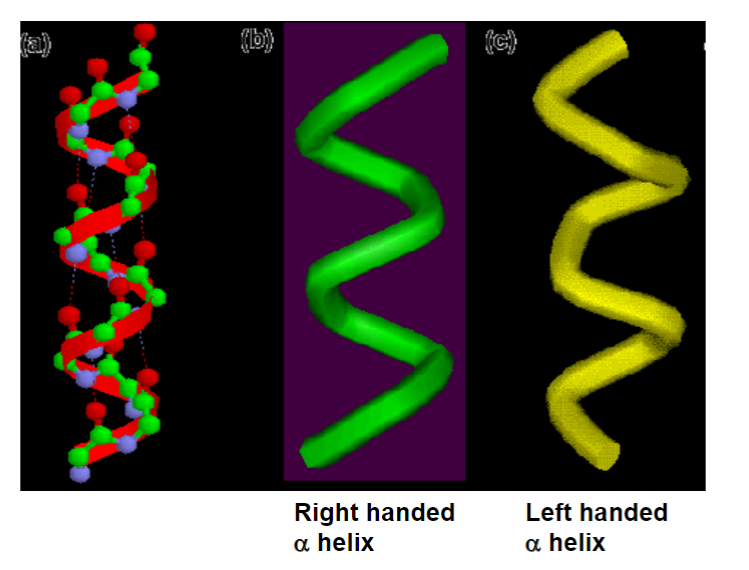
the helix is almost always right handed
left handed and right handed alpha helix are not the same (not superimposable)
all the NH and C=O in the backbone are H-bonded, except the ends
each amino acid residue in the helix increases the helix length by 1.5 angstroms
helix rises by 1.5/# of amino acids
r-groups of i, i +1 and i+2 point in different directions
r-groups of i, i +3 and i + 4 point in similar directions
important because it aids in an ampithatic molecule
left handed helices are permitted but rare (not as stable)
alpha helices are show as twisted ribbons or rods
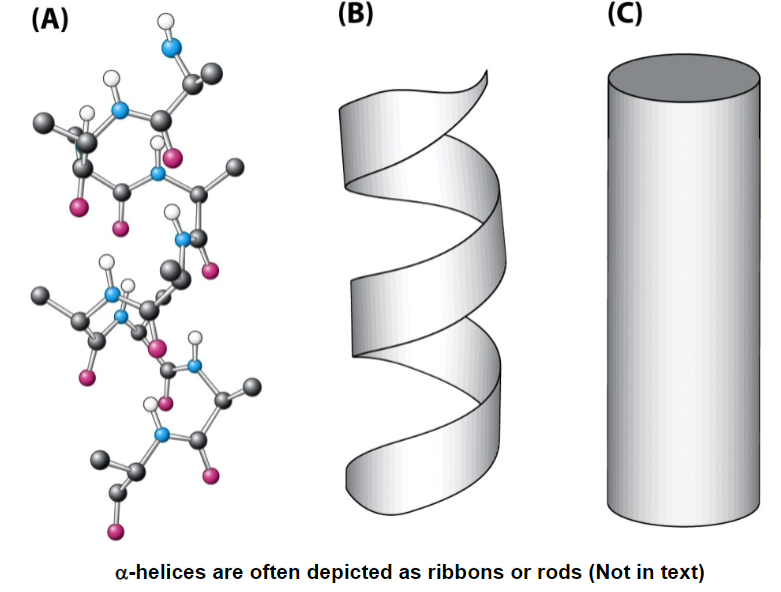
alpha helical content of a protein can vary
some proteins have no alpha helices
some proteins are just alpha helices
usually alpha helices are less than 45 angstroms
2 alpha helices can intertwine into coiled coils (ex. our hair)
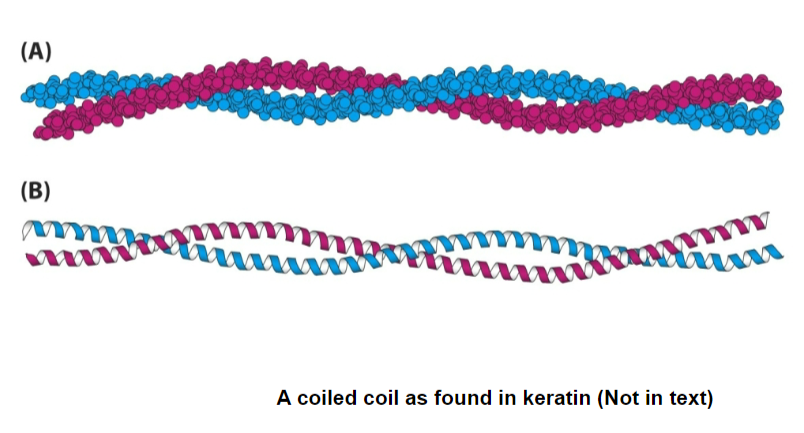
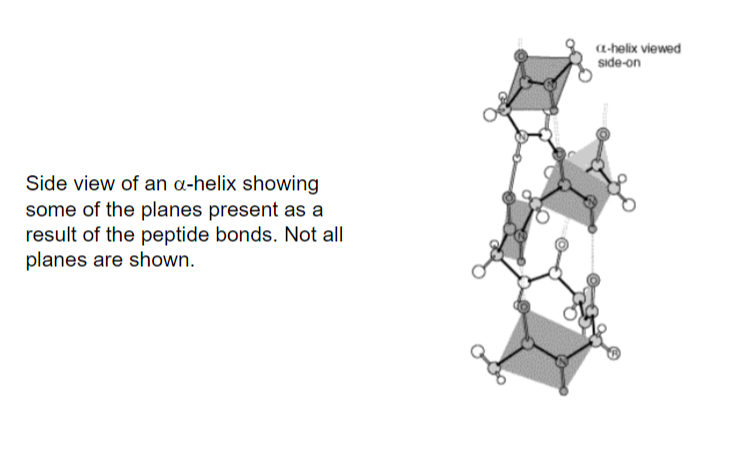
alpha helices are a series of planes that are coiling due to double bond characteristics of peptide bonds
beta-pleated sheets - two or more beta-strands (usually a polypeptide strands from the same molecule) associated as stacks on chains in an extended zigzag form
stabilized by interstrand hydrogen bonds
beta strands are the components that make up a beta sheet
has little zigzags or pleats (each pleat is the plane of a peptide bond)

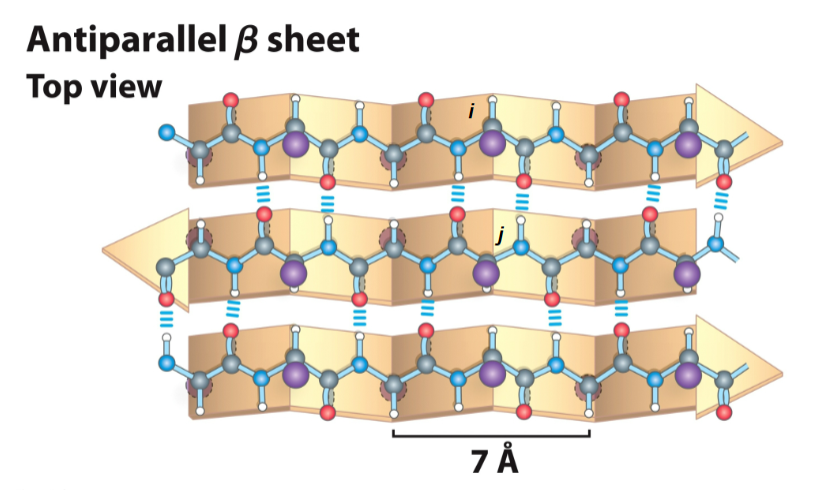
for an antiparallel beta-sheet
each amino acid extends the beta strand by 3.5 A (more spread out than an alpha-helix)
r-groups are adjacent of adjacent residues point in opposite directions perpendicular to the plane of the strand or sheet
the strands are organized into beta-sheets
strands run in opposite directions, the N-H and C=O of a single residue i on one beta-strand h-bonds to the single residue j on the other beta-stran opposite to it
has ideal dihedral angles of phi - 139 and psi + 135
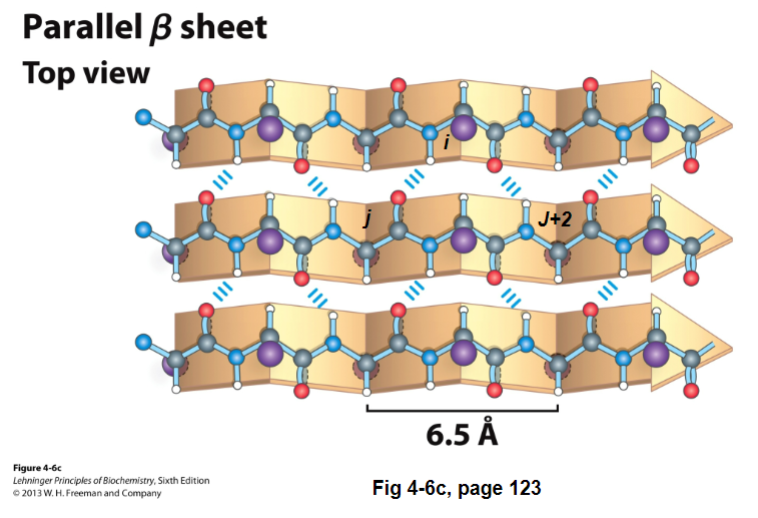
B-sheets can also be parallel. It’s like an antiparallel B-sheet, except:
instead of doing straight down H-bond, it does 2 reaches (shortens length of beta strand + causes dihedral angle to be twisted)
each residue in the beta strand only extends the B-strand by 3.25 A
has ideal dihedral angles of -119 phi and +113 psi
the N-H of residue i on one B-strand H-bonds to residues j C=O in the other strand
C=O of residue in i in the first strand H-bonds to j + 2 N-H in the second strand
Mixed B-sheets (both parallel and antiparallel)
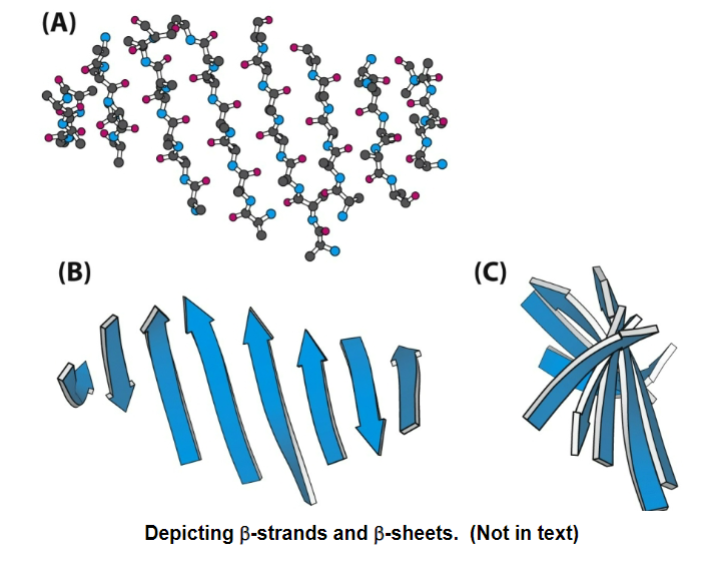
B-strands are depicted as broad arrows pointing towards the C-terminal end
the distance between B-strands in primary amino acid sequence can be small or large
beta-strands can be short or long
beta-sheets can be flat or twisted
it can even twist back on itself to form a beta-barrel
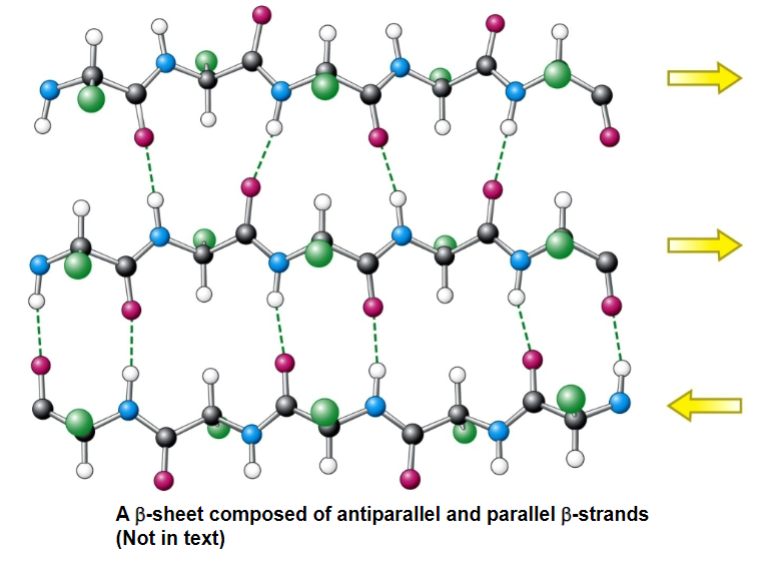
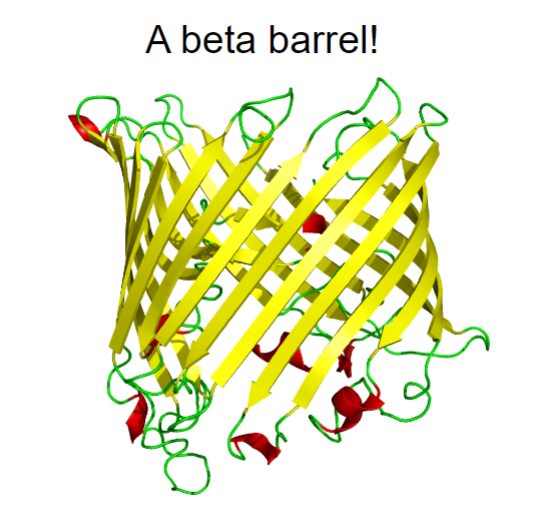
when drawing out a polypeptide backbone, draw the backbone as a zigzag
forces us to draw it as trans
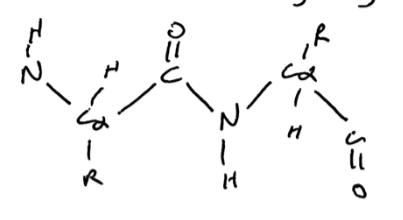
III. Tertiary Structure
Tertiary Structure - the spatial arrangement of amino acid residues that are far apart from each other in linear sequence as well as the pattern of disulfide bonds
BIG MONEY = the 3D structure
each protein tertiary structure is different
tertiary structure is limited to a single polypeptide that folded into a structure
ex. myoglobin
O2 storage protein in mammalian muscles
single polypeptide chain of 153 amino acids
contains a heme group (iron in a protophyrin ring) where the O2 binds to
70% of the chain is in alpha helices (8 helices)
most of the rest are in loops and turns
the core of the protein is almost exclusively composed of hydrophobic residues (except for a his which is needed by the heme)
the surface is composed of more polar/charged residues (and some non-polar)
quick note about myoglobin: binds O2 with high affinity and only releases if when [O2] is really low
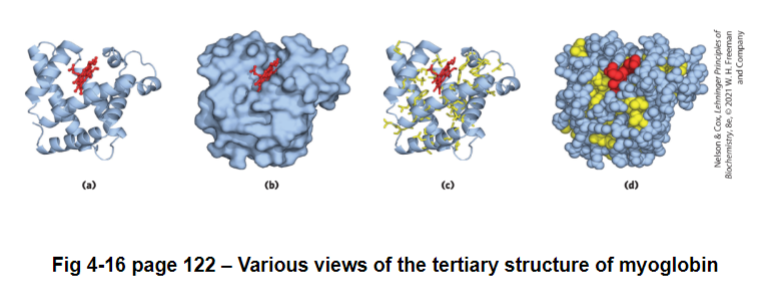
why would you wanna know the structure of myoglobin? structure helps us understand function
helps us identify possible places for drug binding
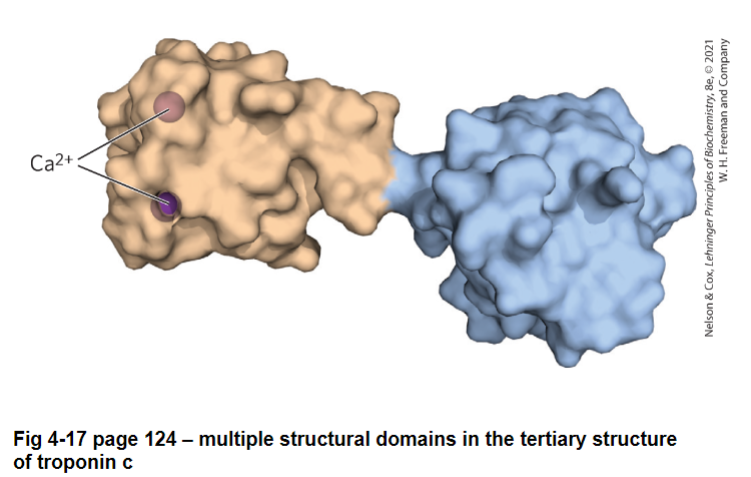
some proteins have multiple compact structures called structural domains linked by flexible sections of the polypeptide (often with no defined structure)
no defined structure = structure is moving around
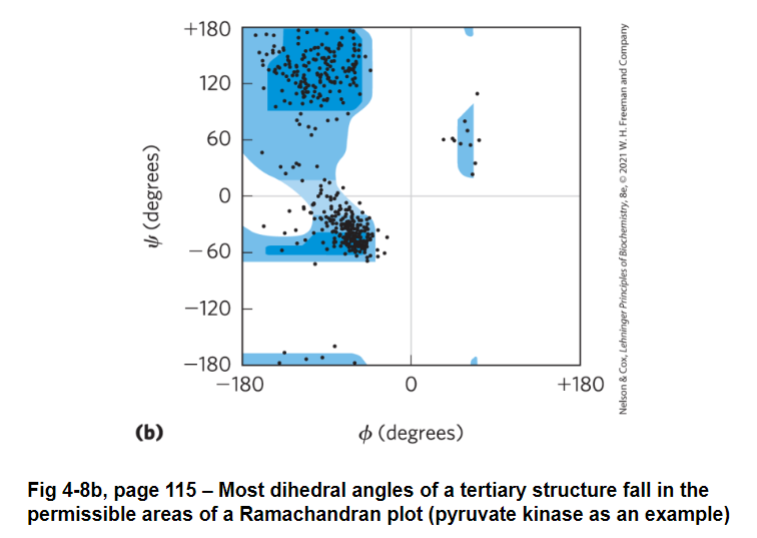
in most tertiary structures, the dihedral angles for each residues fall into permissible areas of a Ramachandran plot
jan 27 lecture
IV. Quaternary Structure
Quaternary structure: the spatial arrangement of multiple folded subunits (folded polypeptides) and the nature of their interactions along with the disulfide bonds between subunits
some proteins are composed of more than one folded polypeptide chains (subunits)
homomers - the subunits can be identical
heteromers - when the subunits are different
some proteins must be multimers in order to function
protomer - the base unit in a quaternary structure
usually repetitive
usually (not always) monomeric in nature but the structure is not the same as the monomer
protomer is what it looks like in the final structure
fig 2-22 pg 128
ex. hemoglobin
the O2 transporter in mammals
composed of 4 subunits
2 alpha subunits (alpha-globins)
2 beta subunits (beta-globins)
will cover in much more detail later, but it is the interactions between the subunits that are critical for function
hemoglobin cannot function unless it is a tetramer
the protomer for hemoglobin is an alpha-beta dimer
ex. minute virus of mice
9 UPI 2 subunits of 51 UP2 subunits to form an isohedral capsid with just enough room to fit the viral DNA
Protein Folding
even with limitations on the backbone, there are trillions of possible structures a polypeptide could adopt and it would take forever for a protein to try every single structure
however, most proteins spontaneously fold into just one structure in seconds
up until a few years ago, we couldn’t predict a protein’s tertiary structure based off of primary sequence, but now AI programs such as Alphafold2 or rosettafold can!
we do know that folding is driven by thermodynamics
ex. finding the most stable complex (most negative delta G)
note: the difference in free energy between folded and unfolded is small (20-60 kJ per mole)
this is mostly (but not all) driven by entropy (ex. the hydrophobic residues are excluded from water in the core, while hydrophillic residues are on the surface)
backbone of a polypeptide is polar, when it folds, some parts of the backbone is in the core (not favorable).
in order to bury the polar backbone of a polypeptide in the core, it needs to H-bond
many alpha-helices and beta-sheets are amphipathic (one side is hydrophobic, the other is hydrophilic)
unpaired charged or polar group in the hydrophobic core tend to destabilize the structure
Q: can a portion of a primary sequence define a secondary structure?
A: yes and no
certain amino acid residues are more likely or less likely to be found in a stabilized (or destabilized) alpha-helices and beta-strands/sheets
table 4-2 - proline and glycine are alpha-helix wreckers, and to a lesser extend beta strand wreckers
proline has the ring, dihedral angle is limited and can’t twist into the optimal angle for an alpha helix
proline has limited h-bond capabilities since it doesn’t have N-H in its backbone (has N-R instead)
glycine has a hydrogen in its r-group
however, experiments have shown that the exact same portion of sequence in 2 different proteins can adopt 2 different secondary structures
thus we cant always determine the secondary structure by looking at a portion of primary sequence
the overall teritiary structure influences secondary structure
what else do we know?
folding tends to be an all or non process
usually you’re folded or you’re not
it is cooperative
ie. as one portion of the protein folds (ex. beta sheet) it will influence how another portion folds
thus, we don’t have to sample every structure FIG 4-26 pg 131
however, there is usually many possible pathways, so we often depict folding as a free-energy funnel FIG 2-27 pg 132
in a proteins unfolded state, there are many possible structures with high free energy, but as those species fold the free energy decreases until you reach the folded state
the farther you go into folding, there are more limitations to the types of structures you can get\
there are multiple folding routes to get you to the final structure
jan 29
not all proteins have a single 3D structure
some proteins only fold into a single structure when they bind something
some proteins are in equilibrium between 2 structures
determining structure of intact proteins (big deal)
x-ray crystallography
use x-ray to measure e- density
how the structure of myoglobin; hemoglobin were determined
nuclear magnetic resonance (NMR)
measures the location of nuclei
cryo-electron microscopy
uses a beam of e- to visualize a frozen protein
post-translational modifications (after protein synthesis)
phosphorylation - attachment of phosphate group, usually to the OH of an R-group
commonly phosphorylated residues: Ser, Thr, Tyr
activates or inactivates a protein by adding a negative charge
glycosylation - attachment of a sugar or carbohydrate to an amino acid residue (usually, Asn, Thr, Ser)
surface labelling
most proteins on the cell surface are glycosylated
hydroxylation - adds an OH (usually proline)
result: becomes more polar
fibre stabilization
carboxylation - adds a carboxyl group (usually to glu)
important in clotting
acetylation - addition of an acetyl group to an NH3 (amino) - (ex. lysine)
(insert pic of the acetyl group)
most proteins are cleaved or trimmed after synthesis
this could activate or inactivate them
ex. fibrinogen → cut → fibrin (forms clot)
multiple proteins can be formed from a single long polypeptide
result: multiple mature protein
ex. viruses like to do this
Enzyme Thermodynamics & Kinetics
enzyme - biological macromolecule (usually protein) that acts as a catalyst for biochemical reactions
“work horses of the cell”
catalyst - chemical that increases the rate of a reaction without being consumed
Thus, enzymes speed up reactions
enzymes are very specific, they will only catalyze on specific set of reactions
papein: enzymes that cleaves peptide bonds in a polypeptide
trypsin: only cleaves peptide bonds on the carboxyl side of lys and arg
thrombin: only cleaves peptide bonds only between and arginine and glycine
specificity is based on a series of weak interactions between the substrate and the enzyme, specifically in the active site
dont want too many interactions
the shape of the enzyme determines specificity and function
active site: region of the enzyme that binds the substrate
it contains the residues that directly participate in the reaction
characteristics of active site:
they are clefts (aka dimples) in the enzyme made up of residues from all over the primary sequence
take up a small volume of the enzyme
water is usually excluded or manipulated from the active site
the substrate is bound to the active site by a series of weak interactions
thus, the substrate and the enzyme must be partly complimentary, otherwise the substrate can’t bind and catalysis cannot occur
can occur by
lock and key
active site is a complimentary match to the substrate (rare)
induced fit
binding of the substrate causes the active site to assume a matching shape
jan 31
the hyperbolic curve = equilibrium
enzymes do not alter the final equilibrium of products to reactants (see slide)
enzymes do not alter delta G of reaction, they obey the laws of thermodynamic
ex. they can’t change the spontaneity of a reaction if delta g reaction is positive, it is non-spontaneous and adding enzymes will not change that (fig 1-29 pg 26)
enzymes do speed up the rate of a reaction
how do enzymes do this?
enzymes accelerate reactions by decreasing the activation energy (delta g double dagger) by facilitating the formation of the transition state (X double dagger)
imagine a substrate being converted to product (S → P)
in order to form products, the substrate goes through a transition state (x double dagger), the transition state has the highest G in the reaction and the lowest concentration
the energy needed to get x double dagger is known as the activation energy (delta g double dagger)
usually the activation energy (g double dagger S → P =) (insert picture of formula
activation energy from S → P
activation energy P → S
the activation energy controls the reaction rate
only a fraction of S will have enough energy to form the transition state (x double dagger)
note: the activation energy is not part of the overall delta g of reaction calculations because the energy put in is returned when the transition state is converted to products
fig 6-3 page 181
for S+ E ←→ES ←→ EP←→ E + P
enzymes interact with the transition state such that the activation energy is lowered
therefore, the reaction speeds up as as greater fraction of S has the energy to reach the transition state (X double dagger)
where does the energy come from to lower the activation energy?
comes from the enzyme binding and stabilizing the transition state (x double dagger)
the active site of an enzyme is perfectly complimentary to the transition state (x double dagger)
we call the energy stabilizing the transition state in an enzyme the binding energy (delta Gb)
delta Gb = delta delta G (change in activation energy) = delta g ++ - delta g ++ catalyzed
the energy is derived from the non-covalent interactions between the enzyme and the transition state
END OF MATERIAL FOR MIDTERM