W1 L3: Sequencing genes and genomes
Sequencing of Biomolecules:
DNA:
- Work out the order of the four bases (A, C, G, and T) in fragments of DNA, usually amplified by PCR or DNA cloning
- Possible since 1977 - Increasingly sophisticated and increasingly affordable
RNA:
- In principle and in practice, RNA is sequenced indirectly through the sequencing of cDNA
Protein:
- Possible since 1950 (insulin)
Classical protein sequencing is time consuming and requires relatively high amounts →Increasingly replaced by modern methods
DNA sequencing
Single-read sequencing
Maxam-Gilbert method
- Based on chemical degradation
- NOW OBSOLETE
Sanger method
- Based on primer extension chain termination
- Dominating method until NGS – still used
Massive parallel sequencing = Next-generation sequencing (NGS)
- Based on primer extension
- Fully automated
- A revolution in progress
Sanger Sequencing
- DNA sequencing with chain-terminating inhibitors
- dideoxy sequencing (ddNPTs)
- Used for sequencing single genes and fragments of DNA
- Understanding the structure of the fragment
- Mutation screening in specific genes
- Validations of findings from NGS
- Highly accurate
Pre-sequencing
Need to produce multiple copies of the DNA fragment to be sequenced
- Clone the DNA fragment into a plasmid and grow in E Coli or
- Amplify the DNA fragment by PCR
Denature the sequence by heating (or by adding NaOH) to produce single-stranded DNA
Prepare a DNA polymerase, primer, dNTPs and ddNPTs
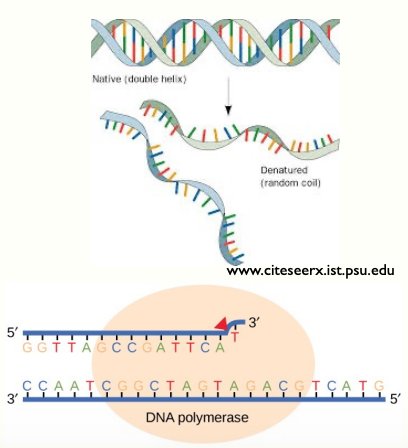
Primer Extension
DNA synthesis does not start from scratch
Primer sequence needed to anneal to template strand
Synthesis of new strand - by adding bases to the primer that are complementary to the template→ extending the primer
DNA polymerase synthesises complementary strand
- Starting from the primer
- Forms a complementary copy to the template strand
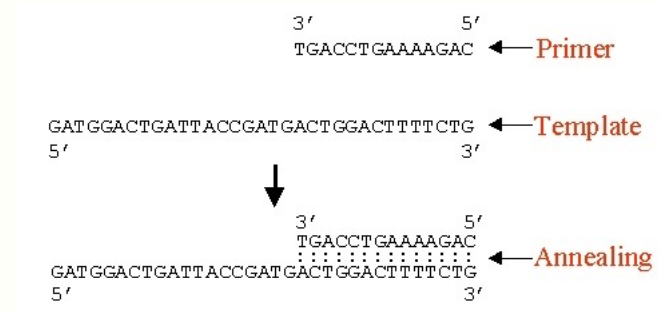
Termination of DNA synthesis
Dideoxyribonucleic triphoshates (ddNTPs) — terminator nucleotides
Modified version of the normal DNA building blocks (dNTPs)
Uses the same bases (A/C/G/T), but the sugar is modified
Wherever the ddNTP has been incorporated, DNA synthesis can proceed no further
The lack of a 3’ OH group in the dideoxynucleotide prevents the formation of phosphodiester bond
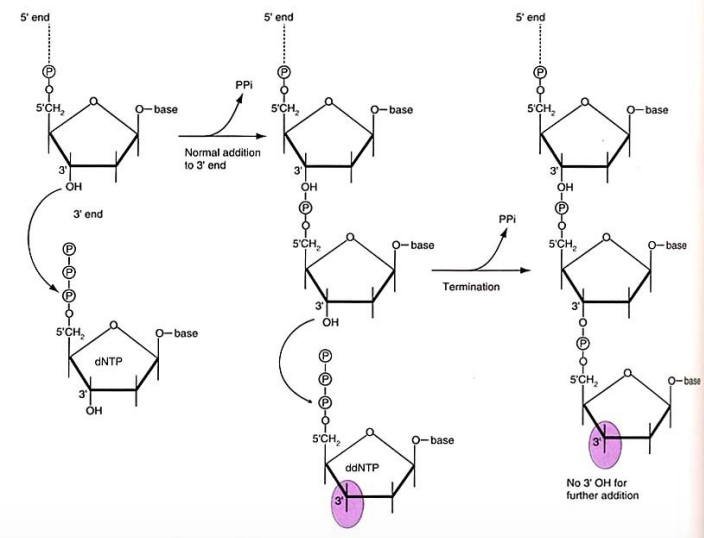
Multiple strands due to DNA amplification
Excess of normal dNTPs against the amount of ddNTPs (100:1), which compete against each other in DNA synthesis
Termination happens at different places at different strands→ result is a set of DNA sequences of varying length, each ending to a ddNTP
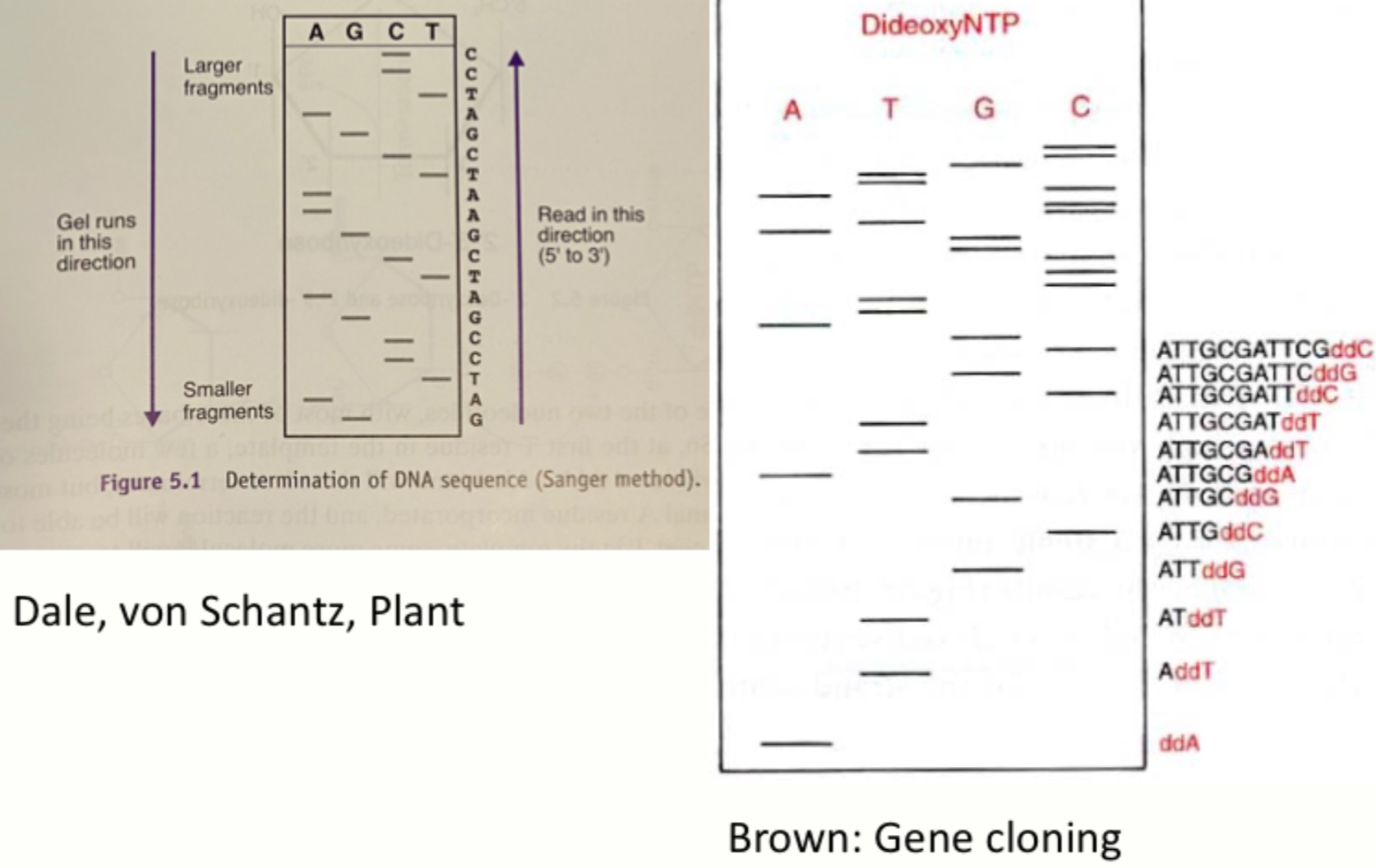
Sanger sequence
- Primer, DNA polymerase & mix of normal (unlabelled) dNTPs & labeled ddNTPs
- Labeled used to be radioactive - now fluorescent dyes used
- Correct order reached through gel electrophoresis & fluorescence detection
- In genotyping, where only single SNP is genotyped, process is similar, but no normal (unlabelled) dNTPs are added
Separating nucleic acids according to size: Slab gel electrophoresis
Nucleid acids cary many -ve charged phosphate gps
Migrate towards +ve electrode when placed in electric field
Porous gel acts as sieve → small mols. pass more easily than larger ones
Advances in Sanger sequencing technology
1977-1985
- Radioactive labelling, requiring 4 separate reaction tubes for each ddNTP - separated individually on large (30cm x 50cm) slab electrophoresis gels→ dry & X-ray
- X-ray film, heavy X-ray film cassettes, darkroom film manipulation
- Manual reading of autoradiogram & feeding into computer (re-reading often required)
- 1.5 days from set up to results
1986 onwards
- Fluorescent dye labelling - using mixed reaction tube containing all 4 ddNTPs
- Automated, optical detection system using scanning laser
- Direct automated entry of DNA base sequence data into computer
- Time-consuming due to handling of gel plates
Capillary gel electrophoresis (1995 onwards)
- Slab gel electrophoresis is manual (slow, prone to human error) - capillary gel electrophoresis is automated
- Fluorescently-labelled DNA samples migrate through long, v. thin tubes containing polyacrylamide gel (instead of running gel for a finite time)
- Machine uses laser to detect fluorescence a fixed point just before end of gel
- Allows longer reads (up to 1000 bases)
- separation not stopped at specific point
- each fragment is allowed to proceed to bottom of gel where resolution is highest
- Faster & cheaper
Pros & cons of Sanger sequencing
Pros
- Highly accurate sequences ~99.95%
- Several 100 bases long (800-1000bp)
Cons
- Gel eletrophoresis not suitable for handling large no. of samples at a time - not fully automated
- not suitable to genome sequencing
1st sequenced human genomes
- Human Genome Project 2001 → daft genome compromising of DNA from several volunteers
- BAC - based sequencing (bacterial artificial chromosome)
- Celera company 2001 → genome of J. Graig Venter
- Expressed Sequence Tags (ETS)
- Highly time-consuming & extremely expensive
- HGP→ took 10 yrs & cost $2.7 billion (In 2022, costs ~$1000)
Next-generation sequencing (NGS)
- Also known as massively-parallel sequencing - sequencing millions of DNA fragments simultaneously
- From 2005 onwards a tech revolution
- Vast ↑ in amt. of sequencing data per run (seq. throughput) → dramatic ↓in cost
- Moving from sequencing single gene & exons to:
- Whole-genome sequences (WGS)
- Whole-exome sequences (targeted DNA sequencing) (WES)
- Whole-transcriptomes (RNAseq) & targeted transcriptomes
- Methyl-Seq, ChIP-seq
- Ribo-seq
Key Terminology:
- Throughput→ amt. of seq. data (in Mb) that are processed in 1 run
- Read length→ length of DNA fragments -measured in nucleotides
- short read lengths can be processed w/ high throughput
- Read depth→ seq. coverage i.e. how many times each seq. is represented
- 30x to 50x for WGS
- 100x for WES
- important for small read lengths for genome assembly
- Genome assembly→ aligning & merging sequenced pieces to make sense of seq. genome
- de novo - 1st time sequencing a species or,
- alignment against a previously obtained reference sequence
Methods based on amplified DNA templates
- 2nd-gen DNA seq.
- From short (35 nucleotides) to medium-length seq. (up to 800 nucleotides)
- High to v. high seq. throughput
- Rel. high R of seq. errors in individual reads (overcome by ↑ coverage)
Commonly used 2nd-gen sequencing platforms
- Roche/454 pyrosequencing
- 1st NGS tech in market (2005)- discontinued support in 2016
- Rel. long reads & speedy but low throughput
- Emulsion PCR
- ABI SOLiD technique
- 2007
- checks each base independently twice → low error rate
- “Wildfire” method for preparing seq. templates (v. similar to Bridge PCR by Illumina)
- Illumina/Solexa sequencing
- 2008 - now market leader
- Bridge PCR
- Ion Torrent systems
- 2010
- Emulsion PCR
- Rel. long reads & speedy but low throughput (similar to Roche/454)
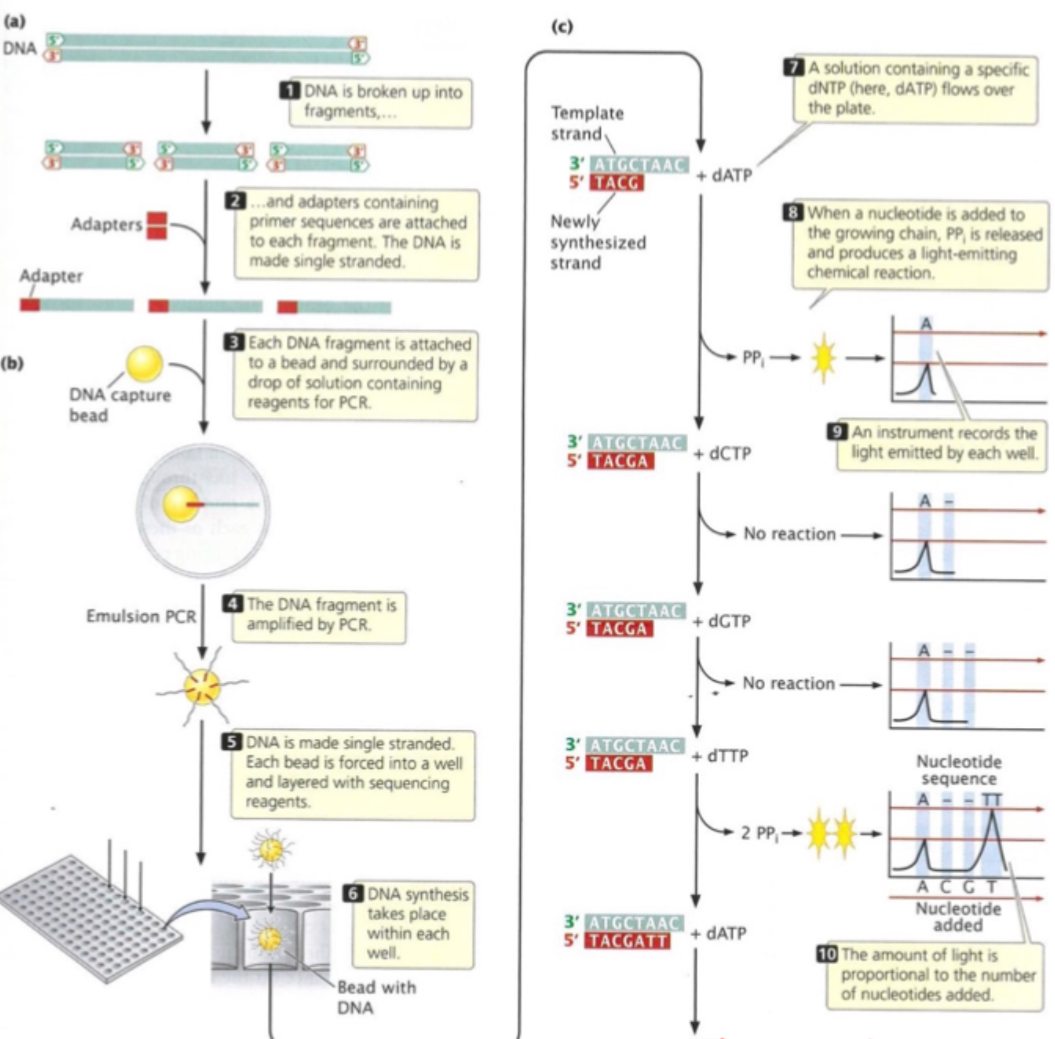
Sequencing w/ emulsion PCR
Uses bead surfaces, H2O & oil
Simultaneous amplification of each seq. w/o risk of contamination
each bead act as microreactor for PCR - each containing 1 strand of DNA
Terminators are reversible - after chem deprotection, synthesis is possible
Sequencing w/ bridge amplification
Whole Process
Genomic DNA→ Cut DNA→ Add linkers
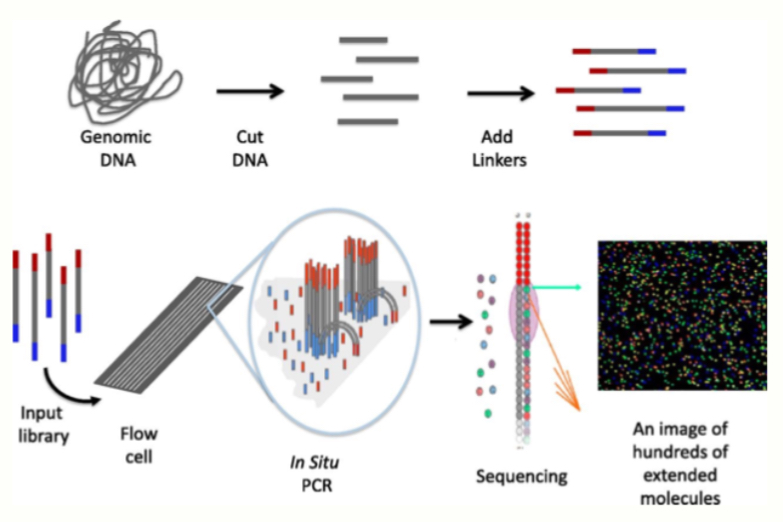 Bridge amplification (𝘪𝘯 𝘴𝘪𝘵𝘶 PCR in figure above)
Bridge amplification (𝘪𝘯 𝘴𝘪𝘵𝘶 PCR in figure above)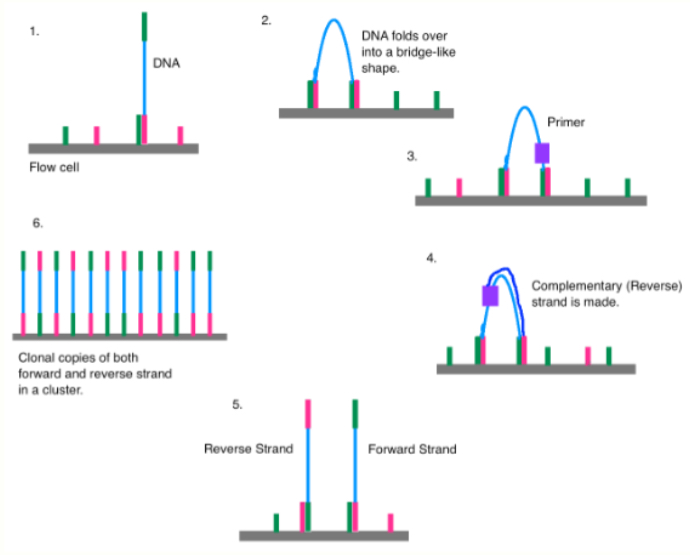
Methods based on unamplified DNA
3rd-gen DNA sequencing - ‘single-molecule sequencing’ (SMS) → long-read sequencing
Long seq. (1000s of nucleotides) -modest throughput
- important for assembly of genomes from newly seq. species & for distinguishing large-scale variations e.g. copy number variations in known ones
Release of protons (instead of fluorophore or phosphate) - recorded as electric current
Avoids problems related to DNA amplification (underrepresentation or overrepresentation of DNA after amplification)
Simple & cheap tech (small portable machines)
Higher error rates

GTEx portal
- Genotype-Tissue Expression (GTEx) project - build a comprehensive public resource to study tissue-specific gene expression & regulation
- 54 non-diseased tissue sites across 1000 individual - for molecular assays inc. WGS, WES & RNA-Seq
- Remaining samples available from GTEx Biobank
- GTEx Portal provides open access to data inc. gene expression, QTLs & histology images
Single-cell sequencing
- Possible to perform sequencing of genome, transcriptome & epigenome in a single cell
- Traditional sequencing works on cell pop.→ resulting data are aggregate values
- For understanding of cell-to-cell variation & identifying new cell types
- Catalog of human cell types
- stable cell properties
- transient cell features
- cell positions
- lineage relationships (identification of novel stem cells)
- Widely employed in cancer research
Analysis of NGS data
- Output files consist of millions of short(~100bp) reads (2nd gen) or longer reads (3rd gen)
- Reads are mapped to reference genome (if species known - as opposed to 𝘥𝘦 𝘯𝘰𝘷𝘰 sequencing)
- Variants are identified
- Heavily computational project requiring several software tools & computational skills
Sanger sequence vs. NGS
- Sanger sequence
- cheap, fast, simple
- highly accurate
- gives an answer about a single, specific question
- NGS
- getting cheaper w/ more tech
- more error-prone
- highly versatile (WGS, WES, RNA-Seq, methyl-seq…)
- computationally laborious