Chapter 5 Notes
Structure & Shape of Organic Molecules
4.1
Functional Groups
Alkane (alkyl): single C–C bond
 Alkene (alkenyl): double C=C bond
Alkene (alkenyl): double C=C bond
Alkyne (alkynyl): triple C≡C bond
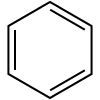
Aromatic compound (aryl): benzene ring
Alkyl halide (halo) R – X
Alcohol R – OH
Ether R – O – R
Amine R – NH3 (primary, secondary R – NH – R, tertiary R – N – R2 )
Thiol R – SH (smell bad)
Sulfide R – S – R
Nitro compound R – NO2
Aldehyde (doubled bonded O at the end of a chain)
Ketone (double bonded O in the middle of the chain)
Carboxylic Acid (double bonded O and 1 H bond)
Ester (double bonded O and ether (O link) group)
Amide (double bonded O and an N group)
4.2
Infrared Spectroscopy
IR light excites nuclear vibrations in a molecule
Different functional groups, and structures of the molecules absorb IR light at different frequencies
Energy Absorption
Due to quantum theory, energy levels in atoms are quantized, therefore a molecule can only exist in certain vibrational states
The difference in energy between the ground & excited states is equal to the energy absorbed
Wavenumber (cm-1) (ν), used because they have value in a more manageable range
When a molecule absorbs IR light at the proper wavelength(/wavenumber) the bonds are excited, not the electrons (excited e- cause UV/visible light)
Energy in the 5-50 kJ mol-1 range
Types of Vibration
Stretching Vibrations:
- Symmetric Stretching: both arms stretch out
- Asymmetric Stretching: one arm stretches while one condenses, vvBending Vibrations:
- Scissoring: arms move inward (like scissors)
- Rocking: side-to-side
- Wagging: back & forth
- Twisting: arms twist
Fingerprint Region: 1500-500cm-1, very complex pattern but unique to compounds
Functional Group Region: 4000-1500cm-1, useful for identifying functional groups
Harmonic Oscillator
Molecular bonds can be thought of balls and spring that vibrate
Bond stretches & compresses more but average bond length remains the same
, where v= frequency, k= force constant, μ= reduced mass (kg)
Stronger bonds vibrate at higher frequencies (wavenumbers)
Triple bonds > double bonds > single bonds
Hybridization: sp > sp2 > sp3
Mass difference: bonds of higher masses vibrate at lower frequencies
Vibrational frequency of an A – X bond decreases with increasing atomic mass of X (as m2 ↑, μ ↑)
Alcohols have a wide, deep peak
Carbonyls have an overtone ~3500cm-1, echo of the C=O stretch (close to fingerprint)
4.3
Alkanes
Saturated hydrocarbons, only single bonds
CnH2n+2
Homologous series: structurally related
The bigger the molecules, the higher the boiling point (dispersion forces are stronger)
C1-C4= gases, C5-C17=liquids, C>17 = solids or waxes @ room temp.
 Constitutional Isomers: same molecular formula, different bonding sequence (connectivity)
Constitutional Isomers: same molecular formula, different bonding sequence (connectivity)
3D Structures
Dot-line-wedge: dotted line goes into the page; wedge comes out of the page
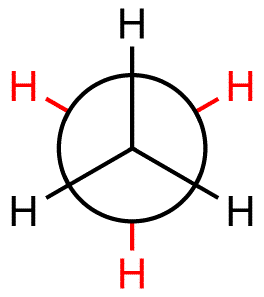
Newman projections: looks down the C – C axis
Eclipsed conformation: front atoms cover back atoms (less stable)
Staggered conformation: rotated 60º
Conformations: non-permanent spatial arrangements of atoms by rotation about single bonds
Relative Energies of Conformations:
Dihedral angle: angle between methyl groups
Lower energy if the dihedral angle is larger because charges repel
Gauche is to the left (or right), right next the atom
Most stable is staggered anti (methyl groups are as far as possible)
“steric strain/hinderance” replusive forces between molecules
Cycloalkanes: CnH2n because 2 H atoms were removed to close the ring
Cyclopropane (60º) are highly strained because all C – H bonds have a eclipse conformation
Cyclobutane (~90º) & Cyclopentane (~109.5º) are puckered (reduces repulsion of adjacent eclipsed C – H bonds & lowers strain)
Cyclohexane (109.5º, sp3 hybrids)
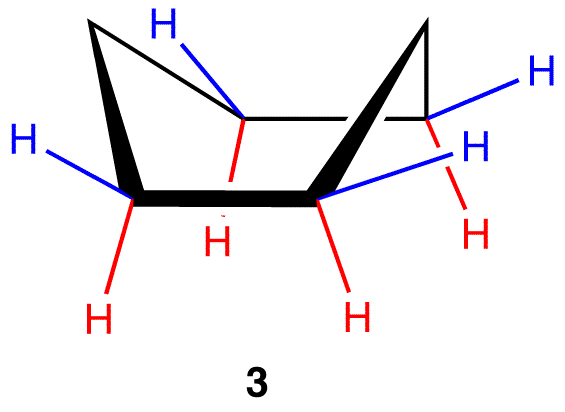
Boat Conformation: base of the base Cs are eclipsed, uncommon
due to flagpole interactions (peaks interact)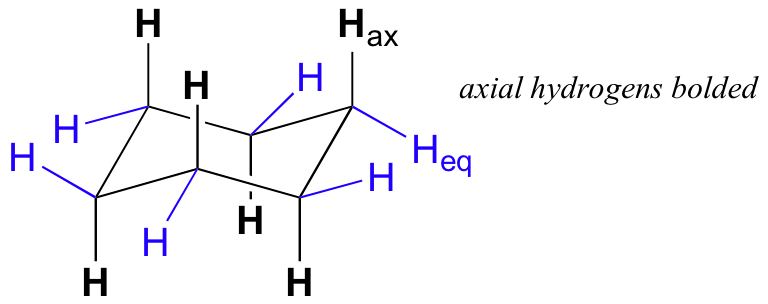
 Chair Conformation: all C – H bonds are staggered & no flagpole
Chair Conformation: all C – H bonds are staggered & no flagpole
interactions, therefore lower energy
Axial Substituents: point straight up or down
Equatorial Substituents: point laterally (outward)
If you change which C is the top peak in a chair conformation, all the axial substituents become equatorial, & eq 🡪 axial
Change if there are axial substituents repelling each other (steric repulsion), equatorial makes them further away, more energetically stable
Change rapidly interconvert 🡪 not isomers
Constitutional Isomerism in Cycloalkanes
Cis/trans isomers: non-hydrogen substituents on different Cs occupy different positions relative to the plane of the ring
Cis: both on the same side
Trans: opposite sides of the ring
Affects properties of the molecule
Stereoisomers: same molecular formula, same bonding sequence (connectivity), different non-interconverting 3D structures
Oriented differently in space, cannot be interconverted by rotations around single bonds
Can’t have cis/trans isomers if there is ambiguity (don’t know which substituent to compare to)
Alkenes & Alkynes
Unsaturated, 1/more double bonds
Alkene C=C (CnH2n)
Alkynes C≡C (CnH2n-2)
For every 2 H atoms removed, 1 “unit of unsaturation”, each unit of unsaturation could be a π bond of a ring
Each C involved in C=C bond is bonded to 2 atoms & is sp2 (trigonal planar 120º)
Each C involved in C≡C bond is bonded to 2 atoms & is sp (linear 180º)
Stereoisomerism in Alkenes
No rotation about the π bond, therefore cis/trans may occur
Rings with 7 members/less, a double bond within the ring can have only the cis geometry because the trans would be too strained
Possible number of cis/trans isomers= 2n, where n is the # of bonds with 2 different substituents on each C
Vitamin A
In the body, as retinal (Vitamin A aldehyde), deficiency causes blindness
Retinal contains chain of 4 C=C bonds in trans configuration
An enzyme breaks down the C=C bond & then restores the C=C in cis conf.
Cis-retinal allows vision, when light hits cells in reconverts & cycle continues
E,Z Nomenclature for Alkene Stereoisomers
Uses priorities to identity which substituents are cis/trans
E is opposite, Z is same side
RULES:
Higher priority to higher atomic number
If there’s a tie… continue down the chain to determine the highest out of those 2 sets
Multiple bonds are given single bond equivalencies (double bond is treated as 2 single bonds)
4.4
Chirality
Chiral objects: not superimposable, mirror images, no plane of symmetry
Achiral objects: superimposable, same object
Enantiomers
2 molecules that have the same molecular formula & bonding sequence but not superimposable (mirror images)
type of stereoisomer
exist in pairs
Stereocenters (Chiral Centers)
an atom bonded to 4 different groups in a tetrahedral geometry
most common is a chiral carbon
 * if you switch any 2 groups @ a chiral center you get the mirror image,
* if you switch any 2 groups @ a chiral center you get the mirror image,
if there are 2 chiral centers in the molecule you have to switch both
Fisher Projections
3D structure of molecules
“hug me but don’t kiss me”
tetrahedral shape from a different perspective
useful for chiral carbons
R & S Designation of Enantiomer Configuration
assign priority using same rules as E,Z
orient molecule so that lowest priority group is behind the plane
direction of rotation can be R or S
R is clockwise, S is counterclockwise
Chirality in Biomolecules
Many physiologically active molecules exist as enantiomers
Enantiomers behave very differently (“life or death”)
Thalidomide
Anti-morning sickness drug in 1960s
The R form had the desire anti-nausea effect
The S form caused birth defects
Number of Stereosiomers
The maximum # of stereoisomers in a molecules with n stereocenters is 2n
Molecules can exhibit cis/trans and R,S stereoisomerism simultaneously
Plane-Polarized Light & Optical Activity
When all waves oscillate in the same plane, light is said to be plane-polarized
Achiral molecules have no effect
Chiral molecules rotate the plane of polarization (are “optically active”)
Clockwise (right) + is dextrorotation (d–)
Counterclockwise (left) – is laevorotation (l–)
D/L is based on how fisher projections are drawn
Same Formula
Same Connectivity Different Connectivity
∴ same molecule ∴ constitutional isomers
Different non-interconverting same non-interconverting
spatial arrangement spatial arrangement
∴ stereoisomers ∴ not stereoisomers