TEL - genetic variation and pedigree analysis
Dominant autosomal inheritance
Recessive autosomal inheritance
Y-Chromosome linked inheritance (there is only one Y chromosome that's why there is no need to determine is an allele on Y chromosome is dominant or recessive)
X-Chromosome linked dominance inheritance
X-Chromosome linked recessive inheritance
Humans are diploid organisms so they have two sets of each chromosomes. In total they have 23 pairs or 46 chromosomes. 22 pairs of chromosomes are known as autosomal chromosomes and 1 pair is a sex chromosome (male: XY; females: XX). Homologous chromosomes are the matching pairs e.g. the two chromosome 14 are homologous chromosomes.
Although majority of the cells in the human body are diploid cells (somatic cells), human's do have haploid cells. Haploid cells contain only one set of each chromosome rather than two. Gametes or germ cells are haploid cells (example: sperm and oocytes) containing only one set (or n) number of chromosomes and autosomal or somatic cells are diploid cells containing 2n number of chromosomes. The number of chromosomes (n) differs in different organisms. In humans a complete set (2n) comprises of 46 chromosomes
Sex-linked inheritance involves genes located on sex chromosomes, while autosomal inheritance involves genes on non-sex chromosomes.
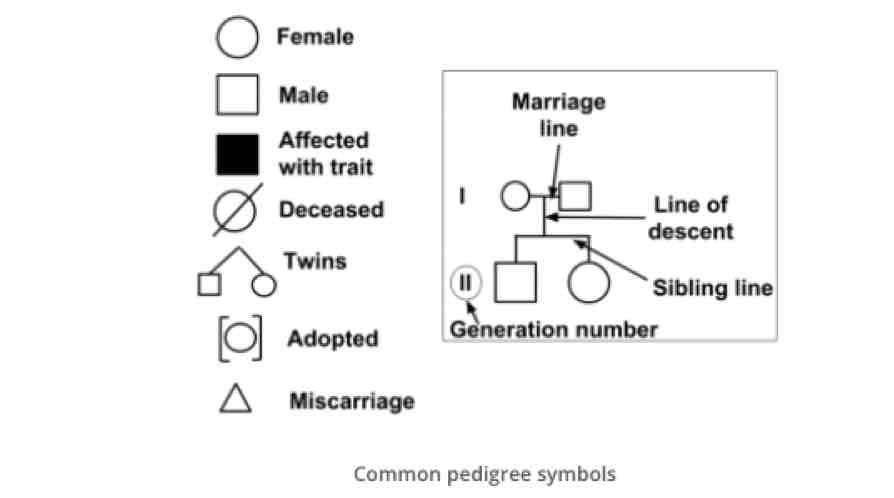 Pedigree analysis
Pedigree analysis
Pedigree analysis describes the process of interpretation of information displayed as a family tree. The family tree or pedigree is constructed using a standardised set of symbols and will include information about the disease status of each individual.
Analysing pedigrees can reveal
(1) whether a trait is dominant or recessive,
(2) the type of chromosome, autosomal or sex-linked to,
(3) genotypes of family members, and
(4) probabilities of phenotypes in future generations.
For families with a history of autosomal or sex-linked diseases, this information can be crucial to family planning.
What do the different symbols in a pedigree analysis mean?
Tips on How to Read a Pedigree:
1. Determine Trait Type:
• Dominant Traits: If a trait is dominant, at least one parent must have the trait. Dominant traits typically do not skip generations.
• Recessive Traits: For a recessive trait, neither parent is required to show the trait, as they can be heterozygous carriers.
2. Identify Inheritance Pattern:
• Autosomal Inheritance: In autosomal inheritance, both males and females are equally likely to be affected.
• Sex-Linked Inheritance: In X-linked recessive traits, males are more likely to be affected than females because males have only one X chromosome.
Autosomal dominant trait
Characteristics:
Affected children usually have affected parents
Heterozygotes (Ff) are affected
Two affected parents can have unaffected child
Two unaffected parents will not have affected child
Both males and females are affected with equally frequency
Mostly show a complete penetrance: all the individuals have the mutation show clinical symptoms
X-linked recessive trait
Characteristics:
Trait skips generation
Heterozygotes (XBXb) are unaffected
Parents affected can have unaffected child
Males are more often affected than females
Males always get X chromosome from their mothers
In more than 99 percent of people with Rett syndrome, there is no history of the disorder in their family. Many of these cases result from new mutations in the MECP2 gene.
Rett syndrome is a monogenic X-linked dominant pattern of inheritance. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. The inheritance is dominant if one copy of the altered gene in each cell is sufficient to cause the condition.
Males with mutations in the MECP2 gene often die in infancy. However, a small number of males with a genetic change involving MECP2 have developed signs and symptoms similar to those of Rett syndrome, including intellectual disability, seizures, and movement problems.
Genetic defects.
Rett syndrome and some variant forms of this condition are thought to result from defects in the MeCP2-associated machinery that recognises and 'reads' methylation marks. The genetic defects that are responsible for Rett syndrome affect the gene MECP2, which encodes methyl-CpG binding protein 2. Females with Rett syndrome are heterozygous for a de novo mutation in MECP2.
The first group of proteins that were discovered with the potential of binding to methylated DNA were the MBD (methyl-binding domain) protein family members. The mammalian MBD family consists of 5 nuclear proteins, MBD 1–4 and MeCP2 (Methyl CpG binding protein 2). MECP2 is an X-linked gene (Xq28), which has different functions in gene regulation and chromatin organisation. MeCP2 is widely expressed among various tissues, with higher expression in the brain. MeCp2 mutations have been detected in more than 90% of classical Rett syndrome patients.
The genomic locus of MECP2 spans approximately 76 kb and consists of four exons encoding two different isoforms (MeCP2E1 and MeCP2E2), due to alternate splicing of exon 2. MECP2E1 is more efficiently translated and show 10X more expression than MECP2E2 in brain.
The main functional domains of MeCP2 are the MBD, the TRD, and the C-Terminal Domain (CTD). The MBD facilitates binding to methylated CpG dinucleotides and the preference for adjacent A/T-rich motifs. It is also capable of binding to nonmethylated DNA sequences . However, the role of MeCP2 as a transcriptional repressor is mostly mediated through its TRD domain. The TRD interacts with corepressor complexes such as mSin3A, further recruiting HDAC1 and HDAC2, and thereby acting as a link between DNA methylation and chromatin remodelling
Although mutations in MECP2 are responsible for almost all known cases of classical Rett syndrome as consequence of abnormally expressed or structurally formed, they have been identified in just 47–50% of atypical cases. Interestingly, genetic defects that affect the X-linked gene cyclin dependent kinase-like 5 (CDKL5) have recently been found to be involved in atypical Rett syndrome; at least 12 point mutations and 2 translocations in this gene have been reported. However, scientist are still working to understand how these mutations cause the disorders. Indeed, scientists believe the remaining cases of Rett syndrome may be caused by partial gene deletions, mutations in other parts of the MECP2 gene, or additional genes that have not yet been identified, and they continue to look for other causes.
Diagnosis
Rett syndrome is confirmed with a genetic test to identify the MECP2 mutation on the X-chromosome. However, since the MECP2 mutation is also seen in other disorders, a Rett syndrome (RTT) diagnosis requires either the presence of the MECP2 mutation (genetic test) or fulfilment of the diagnostic criteria (clinical diagnosis including physical examination, brain imaging, blood test and lumbar puncture) or both.
Many people with Rett syndrome live well into their middle age and with the help of relatives, carers and a multidisciplinary care team can lead a fulfilling and happy life. The condition worsens over time so people will require more attention and care as they get older; they may lose the ability to move independently and their communication skills may deteriorate so they will require more help and support.
Uniparental disomy
Uniparental disomy is when a person receives two copies of a chromosome from the same parent instead of one from each parent. This can also involve parts of a chromosome rather than the whole chromosome.
Key Points:
1. Examples of Disorders: Conditions like Angelman syndrome and Prader-Willi syndrome can be caused by uniparental disomy.
2. How It Happens:
• Prezygotic Mechanism: This usually involves errors during the formation of eggs or sperm (meiosis).
• Postzygotic Mechanism: This involves errors that occur after fertilization as the cells divide (mitosis).
3. Imprinted Genes: Some genes are only active depending on whether they come from the mother or father (imprinting). If a child has UPD for an imprinted gene, they may only express the traits from the parent whose chromosome they received twice. For example, if both copies are from the father, the father’s genes will be expressed while the mother’s genes will be silenced.
Genomic imprinting is an epigenetic process result in silencing of one of the two alleles (maternal or paternal) based on the parent of origin.
Imprinting does not occur on every chromosome; only nine chromosomes are known to have regions of genes that are imprinted (~100 genes).
Imprinting occurs by a pattern of methylation, meaning the copy of the gene to be inactivated is coated with methyl groups. This takes place before fertilization, in the egg and sperm cells. The methylation prevents that gene from being expressed.
Genomic imprinting is a reversible form of gene inactivation and is not considered a mutation.
Angelman syndrome (AS) is a complex genetic disorder that primarily affects the nervous system. Characteristic features of this condition include delayed development, intellectual disability, severe speech impairment, and problems with movement and balance (ataxia). Children with Angelman syndrome typically have a happy, excitable demeanor with frequent smiling, laughter, and hand-flapping movements. With age, people with Angelman syndrome become less excitable, and the sleeping problems tend to improve. However, affected individuals continue to have intellectual disability, severe speech impairment, and seizures throughout their lives.
It is caused by a loss of function of a gene called UBE3A which is located on chromosome 15. The exact mechanism that causes this loss of function is complex (Genetic disease are complex!).
People normally inherit one copy of the UBE3A gene from each parent. Both copies of this gene are turned on (active) in many of the body's tissues. In certain areas of the brain, however, only the copy inherited from a person's mother is active while the copy inherited from the father is inactive. This parent-specific gene activation is caused as a result of imprinting. So the copy inherited from the father has been epigenetically silenced.
If the maternal copy of the UBE3A gene is lost because of a chromosomal change (e.g. deletion: 70% of cases) or a gene mutation (10% of the cases), a person will have no active copies of the gene in some parts of the brain.
What can cause deactivation or deletion of the active maternal copy?
Several different genetic mechanisms can inactivate or delete the maternal copy of the UBE3A gene.
Most cases of Angelman syndrome occur when a segment of the maternal chromosome 15 containing this gene is deleted.
In other cases, Angelman syndrome is caused by a mutation in the maternal copy of the UBE3A gene.
In a small percentage of cases, a person with Angelman syndrome inherits two copies of chromosome 15 from his or her father, instead of one copy from each parent (uniparental disomy). And because the parent allele is inactive this individual will have no active allele for the UBE3A gene.
Rarely, Angelman syndrome can also be caused by translocation chromosomal rearrangement, or by a mutation or other defect in the region of DNA that controls activation of the UBE3A gene.
The cause of Angelman syndrome is unknown in 10 to 15 percent of affected individuals. Changes involving other genes or chromosomes may be responsible for the condition in these individuals
These genetic changes can abnormally turn off (inactivate) UBE3A or other genes on the maternal copy of chromosome 15.
Deletion of section of chromosome containing UBE3A
The typical deletion region is indeed large and spans about 6 million base pairs of DNA. Most deletions extend from break point one (BP1) to either BP2 or BP3 and are termed class I or class II deletions. About 10% of the deletions extend further beyond BP3, for example, at site BP4. New methods of clinical testing can distinguish between class I and class II deletions. All the large deletions remove UBE3A from the maternally derived chromosome. The deletions also remove additional genes as pictured but UBE3A deletion causes essentially all the problems associated with AS.
Inheritance Pattern
Affected individuals typically have no history of the disorder in their family. Most cases of Angelman syndrome are not inherited, particularly those caused by a deletion or mutation in the maternal chromosome 15 (15q13) or by paternal uniparental disomy (UPD) or by a chromosomal rearrangement (translocation) involving imprinting defect (i.e monoallelically active). These genetic changes occur as random events during the formation of reproductive cells (eggs and sperm) or in early embryonic development.
Rarely, a genetic change responsible for Angelman syndrome can be inherited, but it is possible for a mutation in the UBE3A gene or in the nearby region of DNA that controls gene activation to be passed from one generation to the next
Inheritance of imprinting genes
A carrier father can pass on the genetic defect to his children without it causing any problems because the allele from inherited from the father is not active in the cells.
If female passes this same genetic defect on to her children, regardless of the sex of her child, that child will have AS. This is because the allele inherited from the mother should have been the active, functioning allele.
The UBE3A gene provides instructions for making a protein called ubiquitin protein ligase E3A. Ubiquitin protein ligases are enzymes that target other proteins to be broken down (degraded) within cells. These enzymes attach a small molecule called ubiquitin to proteins that should be degraded. Cellular structures called proteasomes recognize and digest these ubiquitin-tagged proteins.
Protein degradation is a normal process that removes damaged or unnecessary proteins and helps maintain the normal functions of cells. The UBE3A plays a critical role in the normal development and function of the nervous system helping to control (regulate) the balance of protein synthesis and degradation (proteostasis) at the junctions between nerve cells (synapses) where cell-to-cell communication takes place.
Proper gene dosage of UBE3A is crucial to normal brain development, as evidenced by the neurodevelopmental disorders associated with deletions, mutations, and copy number variations (CNVs) of UBE3A.
Diagnosis
A combination of genetic tests can reveal the chromosome defects related to Angelman syndrome:
•Parental DNA pattern. This test, known as a DNA methylation test, screens for three of the four known genetic abnormalities that cause Angelman syndrome.
•Missing chromosomes. A chromosomal microarray (CMA) can show if portions of chromosomes are missing.
•Gene mutation. Rarely, Angelman syndrome may occur when a person's maternal copy of the UBE3A gene is active, but mutated. If results from a DNA methylation test are normal, your child's doctor may order a UBE3A gene sequencing test to look for a maternal mutation.
Treatment
There's no cure for Angelman syndrome. Research is focusing on targeting specific genes for treatment. Current treatment focuses on managing the medical and developmental issues. Depending on the child's signs and symptoms, treatment for Angelman syndrome may involve:
Anti-seizure medication to control seizures
Physical therapy to help with walking and movement problems
Communication therapy, which may include sign language and picture communication
Behaviour therapy to help overcome hyperactivity and a short attention span and to aid in development
Single base-pair substitution
There are also known as single nucleotide polymorphisms (SNPs) and can be any nucleic acid substitution:
Transition
interchange of the purine (Adenine/Guanine)
or pyrimidine (Cytosine/Thymine) nucleic acids
Transversion
interchange of a purine and pyrimidine nucleic acid
DNA substitution mutations are of two types. Transitions are interchanges of two-ring purines (A
G), or of one-ring pyrimidines (C
T): they therefore involve bases of similar shape. Transversions are interchanges of purine for pyrimidine bases, which therefore involve exchange of one-ring & two-ring structures.
Fill the gaps the some common genetic disorders. Use the letter P-Point mutation, or any insertion/deletion entirely inside one gene), D-Deletion of a gene or genes and C-whole chromosome extra, missing, or both.
Disorder | Chromosome or gene | Type of inheritance | Variation |
Phenylketonuria (PKU) | PAH | Autosomal recessive | P |
Cystic fibrosis | CFTR | Autosomal recessive | P |
Sickle cell anemia | HBB | Autosomal recessive | P |
Angelman syndrome | UBE3A | Autosomal recessive | DCP |
Down syndrome | 21 | ----------------------------- | C |
Hemophilia A | FVIII | X-linked recessive | P |
Polycystic kidney disease | PDK1 or PDK2 | Autosomal Dominant | P |
Turner syndrome | X | Monosomy | C |
Effects of the Genetic variation
1) PCR (Polymerase Chain Reaction)
This technique is used to amplify specific regions of a DNA strand millions of times. A region may be a number of loci, a single gen, a part of a gen, or a non-coding sequence. This technique produces a useful quantity of DNA for analysis, be it medical, forensic or some other form or analysis. Amplification of DNA form as little as a single cell is possible.
A basic PCR involves a series of repeating cycles involving three main steps:
1. Denaturation of the double stranded DNA.
2. Annealing of specific oligonucleotide primers.
3. Extension of the primers to amplify the region of DNA of interest.
2) Hybridisation techniques (Fluorescent In Situ Hybridisation, FISH)
Hybridization (of nucleic acids) is a technique in which single-stranded nuclei acids are allowed to interact to form complexes, or hybrids with sufficiently similar complementary sequences. This technique allows the detection of specific sequences. FISH uses fluorescence probes that bind to only those parts of a nuclei acid sequence with a high degree of sequence complementarity to detect and localize the presence or absence of specific DNA sequence or chromosomes. Fluorescence microscopy can be used to find out where the fluorescent probe is bound to the chromosomes. FISH is often used for finding specific features in DNA for use in genetic therapy, medicine, and species identification.
3) DNA microarray technology (for detection of for example SNPs)
Microarrays are a technology in which millions of nucleic acids are bound to a surface and are used to measure the relative concentration of nucleic acid sequences in a mixture via hybridization of nucleic acids and subsequent detection of the hybridization events through fluorescence.
SNP array is a type of DNA microarray which is used to detect polymorphisms within a population. A Single Nucleotide Polymorphism (SNP), a variation at a single site in DNA, is the most frequent type of variation in the genome.
The basic principles of SNP array are the same as the DNA microarray: DNA hybridization, fluorescence microscopy and solid surface DNA capture. The three mandatory components of the SNP arrays are: An array containing immobilized allele-specific oligonucleotide (ASO) probes. Fragmented nucleic acid sequences of target, labelled with fluorescent dyes. A detection system that records and interprets the hybridization signal.
4) Genomic Sequencing
A genomic sequencing allow to get the complete list of the 3 billion nucleotides (A, C, G, and T for DNA genomes) that compose the human genome. Within a species, the vast majority of nucleotides are identical between individuals, but sequencing multiple individuals is necessary to understand the genetic diversity.
Ataxia-telangiectasia disease
Description
Is a rare inherited disorder that affects the nervous system, immune system, and other body systems. This disorder is characterised by progressive difficulty with coordinating movements (ataxia) beginning in early childhood, usually before age 5. People with ataxia-telangiectasia often have a weakened immune system, and many develop chronic lung infections. They also have an increased risk of developing cancer, particularly cancer of blood-forming cells (leukaemia) and cancer of immune system cells (lymphoma).
Frequency
Ataxia-telangiectasia occurs in 1 in 40,000 to 100,000 people worldwide.
Inheritance pattern
This disorder is inherited in an autosomal recessive pattern, which means both copies of the ATM gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but do not show signs and symptoms of the condition.
Causes
Multiples mutations in the ATM gene (chromosome 11q) cause ataxia-telangiectasia. The ATM protein assists cells in recognizing damaged or broken DNA strands and coordinates DNA repair by activating enzymes that fix the broken strands through the DNA double-strand break repair pathway (DBS). Efficient repair of damaged DNA strands helps maintain the stability of the cell's genetic information. Mutations in the ATM gene reduce or eliminate the function of the ATM protein. Without this protein, cells become unstable accumulating DNA damage and leading to the formation of the cancerous tumour. Therefore, the ATM gene provides instructions for making a protein that helps control cell division and is involved in DNA repair. This protein plays an important role in the normal development and activity of several body systems, including the nervous system and immune system.