Chemistry 12HL Reactivity 1-3
Reactivity 1
1.1: Measuring enthalpy changes
Heat transfers
energy is a measure of the ability to do work
to move an object against an opposing force
can be transferred through heat, light, sound, electricity, etc.
heat - form of energy transfer that occurs as a result of a temperature difference
when heat is transferred to a system, the average KE of molecules and the temperature are increased → KE is more dispersed among the particles
System and Surroundings
system → area of interest
open system → energy/matter can be exchanged with surroundings
closed system → energy can be exchanged but matter cannot
isolated system → energy and matter cannot be exchanged with surroundings
surroundings → everything else in the universe
Enthalpy of a system
enthalpy → chemical potential energy of a system
system acts as a reservoir of chemical potential energy/enthalpy
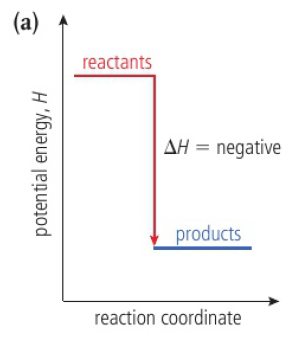
when heat is added to a system from its surroundings, its enthalpy increases (+ΔH)
when heat is given out by a system, its enthalpy decreases (-ΔH)
ΔH = change in enthalpy
endothermic:
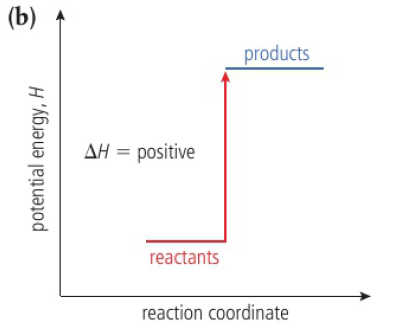
exothermic:
Direction of a reaction
there is a natural direction for change
to lower stored/potential energy (exothermic)
chemicals change in a way that reduces their chemical potential energy
products of an exothermic reaction are more stable than the reactants
stability is a relative term
most combustion reactions are exothermic
the energy needed to break the bonds is less than the energy produced as bonds form
activation energy → minimum KE to react
some bonds must be broken before new bonds are formed

Measuring enthalpy changes
standard enthalpy change (ΔHꝊ)
pressure → 100kPA
concentration of 1mol/dm³ for all solutions
all substances in their standard states
usually 298K as temperature
thermochemical equations
ex: CH4(aq) + 2O2(g) → CO2(g) + 2H2O(l) ΔHreaction = -890kJ/mol
1 mole of CH4 reacts with 2 moles of O2 to produce 1 mole of CO2 and 2 moles of H2O and releases 890kJ of heat energy
Calculating enthalpy changes
absolute temperature (K) is a measure of the average KE of the particles
more/less particles with same heat energy added will result in a different temperature
q=mcΔT
q: heat added (J), m: mass (g), c: specific heat capacity (J/gK), ΔT: temperature change (K)
specific heat capacity → heat needed to increase the temperature of a unit mass of a substance by 1K
depends on the number of particles present
Combustion: enthalpy changes
enthalpy change of combustion (ΔHc) can be determined using this apparatus:
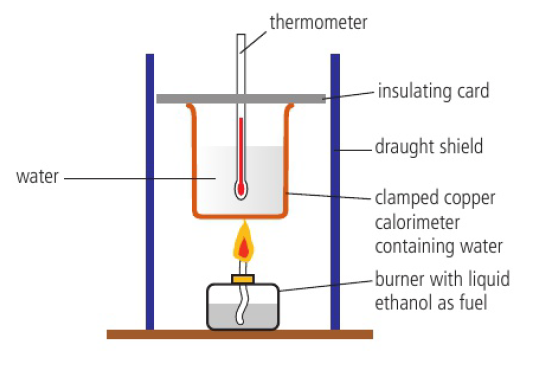
heat absorbed by the water can be calculated from the temperature change and mass
heat absorbed by calorimeter can also be calculated using c
temperature of the water increases due to heat released from the combustion reaction
heat released as ethanol and oxygen turn into carbon dioxide and water
there is a decrease in enthalpy in this reaction
Reaction in the solution: enthalpy changes
calculated by carrying out the reaction in an isolated system
heat released/absorbed by reaction can be measured from the temperature change in the water (solvent)
calorimeter made from an insulator to maximize heat transferred to water by reaction
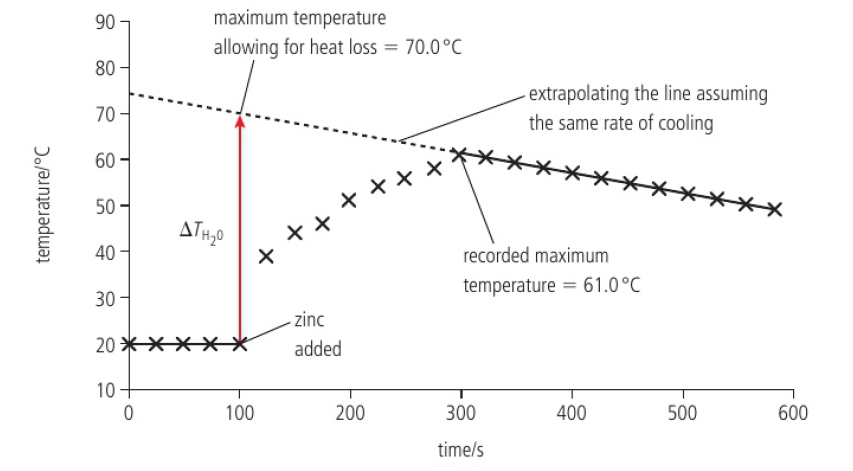
error → all heat produced in the reaction is absorbed by the water
heat is lost from the system as soon as its temperature rises above the temperature of its surroundings
by extrapolating the cooling section to the time when the reaction started, it now creates some allowance for heat loss, so now we can assume:
no heat loss from system
all heat goes from reaction to water
solution is dilute
water has density of 1g/cm³
1.2: Energy Cycles in Reactions
energy changes occur when bonds are broken or new bonds are formed
energy is required to separate particles and energy is released when particles come together
net enthalpy change is the difference between these two energy contributions
law of conservation of energy → energy cannot be created or destroyed, only transferred
Bond enthalpy
bond enthalpy → energy needed to break one mole of bonds in gaseous molecules in standard conditions
ex: Cl2(g) → 2Cl(g) ΔH=+242kJ/mol
breaking bonds is an endothermic process (positive enthalpy change)
bond enthalpies differ, it may be harder to break depending on environment
to compare bond enthalpies which occur in different environments, average bond enthalpies will be used
all bond enthalpies refer to reactions in the gaseous state
any enthalpy changes resulting from the formation/breaking of intermolecular forces are not included
multiple bonds (involves more bonding electrons) generally have higher bond enthalpies and shorter bond lengths
the more polar the bond, the stronger it will be
making bonds is an exothermic process (negative enthalpy change)
the same amount of energy is absorbed when a bond is broken as is released when a bond is formed
Energy changes in reactions
ex: complete combustion of methane
CH4 + 2O2 → CO2 + 2H2O
energy is taken in to break the C-H and O=O bonds in the reactants
energy is given out when the C=O and O-H bonds are formed in the products
reaction is exothermic overall as the bonds formed are stronger than those broken
opposite would be true for endothermic reactions
ΔH = ΣEbonds broken - ΣEbonds formed
some reactants need to be given an initial energy (activation energy) before they will react
some bonds in the reactants must break before new bonds can form
rate of some reactions can be explained by the relative bond enthalpy of the bonds broken
Hess’s Law
the enthalpy change for any chemical reaction is independent of the route, provided the same starting conditions and final conditions, and reactants and the products, are the same
ΔH3=ΔH1+ΔH2
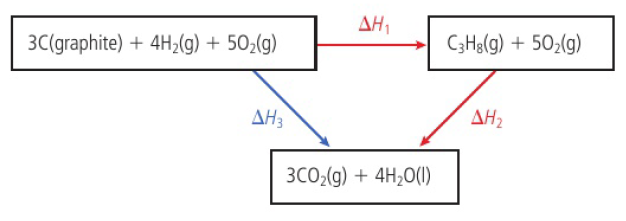
due to the conservation of energy
can be used to find enthalpy change of reactions that cannot be measured directly
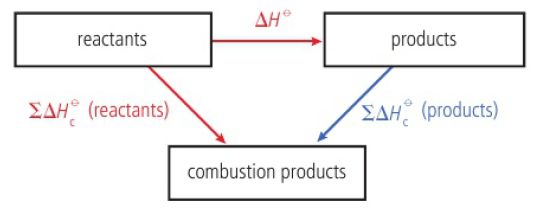
Calculating enthalpy changes
standard enthalpy change of combustion (ΔHcꝊ)
enthalpy change that occurs when one mole of the substance burns completely under standard conditions
can be measured by calculating temperature change of water heated by the combustion
reactants - products
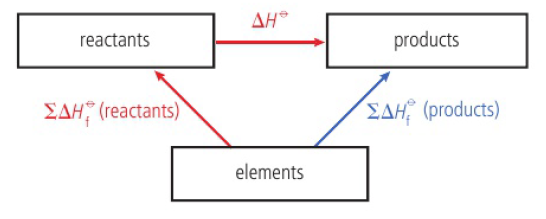
standard enthalpy of formation (ΔHfꝊ)
enthalpy change that occurs when one mole of the substance is formed from its elements in their standard states
standard measurements taken at a specific temperature (usually 298K) and pressure of 1×105 Pa
standard state of an element is its most stable form under these conditions
products - reactants
Lattice enthalpies
first ionization energy (ΔHiꝊ) → energy needed to form the positive ion of a gaseous atom
endothermic process (pulling electron away from electrostatic force)
first electron affinity (ΔHeꝊ) → enthalpy change when one mole of gaseous atoms attracts one mole of electrons
exothermic process (electron is attracted to positively charged nucleus)
lattice enthalpy (ΔHlatꝊ) → formation of gaseous ions from one mole of a solid crystal breaking into gaseous ions
ex: NaCl(s) → Na+(g) + Cl-(g) ΔHlatꝊ=+790kJ/mol
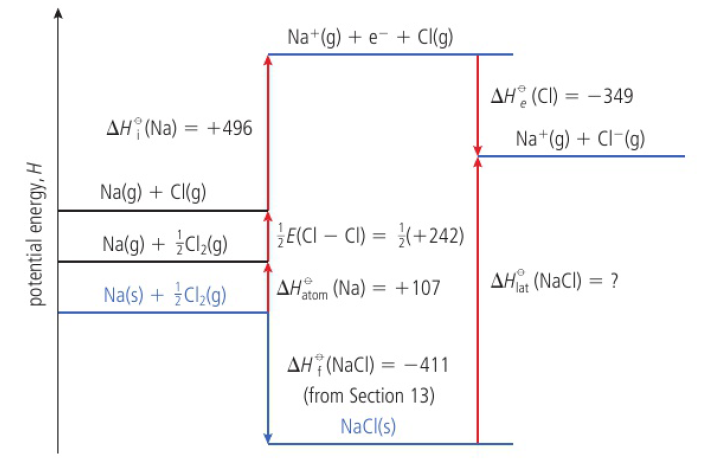
Born-Haber cycles
formation of an ionic compound from its elements is supposed to take place in a number of steps, including the formation of the solid lattice from its constituent gaseous ions
from Hess’s Law, the enthalpy change for the overall formation of the solid must equal the sum of the enthalpy changes accompanying the individual steps
ex: Na(s)+1/2 Cl2(g) → NaCl(s) ΔHf(NaCl)=-411kJ/mol
Na(s)→Na(g) sodium is atomized ΔHatom(Na)=+107kJ/mol
½ Cl2(g)→Cl(g) 1/2E(Cl-Cl)=1/2(+242KJ/mol) E=bond enthalpy
Na(g)→Na+(g)+e- ΔHi(Na)=+496kJ/mol
Cl(g)+e-→Cl-(g) ΔHe(Cl)=-349kJ/mol
Na+(g)+Cl-(g)→NaCl(s) -ΔHlat=+786kJ/mol → sum
1.3: Energy from fuels
Combustion reactions
many substances undergo combustion reactions when heated in oxygen
s-block metals form ionic oxides (basic)
p-block non-metals generally form covalent oxides (acidic)
many hydrocarbons/alcohols are used as fuels as their combustion reactions release energy at a reasonable rate to be useful
high activation energy → do not spontaneously combust, safe transport and storage
kinetically stable
complete combustion of organic compounds break the carbon chain → results in CO2 + H2O and a release in (a lot of) heat energy
complete combustion → products are fully oxidized
when oxygen supply is limited, incomplete combustion occurs
incomplete combustion of organic compounds
if the air supply is limited/compound has high carbon content, incomplete combustion occurs
results in carbon monoxide / carbon (soot)
releases less heat than complete combustion
Fossil fuels
an ideal fuel releases significant amounts of energy at a reasonable rate and produces minimal pollution
fossil fuels are non-renewable → used at a rate faster than they are replaced
liquid fuels have significantly greater energy densities than gases (per unit volume)
fossil fuels were formed by the reduction of biological compounds
oxygen is lost from biological molecules which generally result in hydrocarbons
coal → most abundant, 80-90% carbon (by mass)
crude oil → mixture of straight-chain and branched-chain saturated alkanes, cycloalkanes, and aromatic compounds, used as fuel for transportation and electricity
natural gas → primarily methane, cleanest fossil fuel → low carbon content
coal
advantages
cheap, abundant
longest lifespan (compared to other fossil fuels)
can be converted into synthetic liquid fuels and gases
safer than nuclear power
ash produced can be used to make roads
disadvantages
contributes to global warming (CO2 emissions)
contributes to acid rain (SO2)
produces particulats (electrostatic preceptors can remove most of these)
difficult to transport
waste can lead to visual + chemical pollution
mining is dangerous
petroleum
advantages
easily transported in pipelines/tankers
convenient fuel for use in cars → volatile, burns easily
high enthalpy density
sulfur impurities can be easily removed
disadvantages
limited lifespan and uneven world distribution
contributes to acid rain and global warming
transport can lead to pollution
carbon monoxide is produced through incomplete combustion (pollutant)
photochemical smog is produced
natural gas
advantages
higher specific energy
clean and easily transported
does not contribute to acid rain
disadvantages
limited supplies
contributes to global warming
risk of explosion (leaks)
Combustion of alkanes
increase in %carbon content down the homologous series suggests that incomplete combustion increases with length of the carbon chain
mass of CO2 produced per unit mass of fuel increases with %carbon content
the higher %carbon content (and lower %hydrogen), the lower the specific energy
The greenhouse effect
it is estimated that CO2 contributes to about 50% of global warming
greenhouse gases allow shortwave radiation from the Sun to pass through the atmosphere but absorb the longer wave infrared radiation that was re-radiated from Earth’s surface
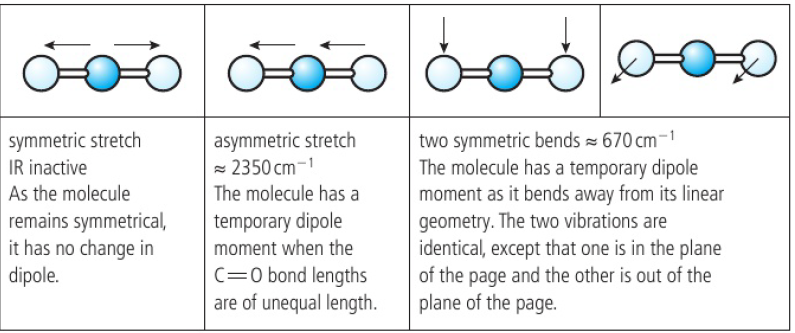
CO2 is a GHG as its molecules increase their vibrational energy by absorbing IR radiation
3 of the vibrational modes of CO2 are IR active → dipole changes as it vibrates
molecules then re-radiate the absorbed energy back to Earth’s surface → global warming
greenhouse effect increased as CO2 levels increased, causing (due to change in temperature):
changes in agriculture (crop yields)
changes in biodistribution due to desertification and loss of cold-water fish habitats
rising sea levels caused by thermal expansion and melting of polar ice caps/glaciers
Biofuels
photosynthesis converts light energy into chemical energy
chlorophyll (green pigment) absorbs solar energy which is used in this reaction:
6CO2(g) + 6H2O(l) → C6H12O6(s) + 6O2(g)
carbon dioxide + water → (using solar energy) glucose + oxygen
biofuels → produced from the biological fixation of carbon over a short period of time
ethanol is a liquid biofuel → used in internal combustion engines
made from biomass by fermenting plants high in starches and sugars
C6H12O6 → 2C2H3OH + 2CO2
process done at around 37C in absence of oxygen by yeast (provides enzyme)
advantages (when used in gasohol: 10% ethanol, 90% unleaded gasoline)
renewable, lower emissions of CO and nitrogen oxides, decreases dependance on oil)
disadvantages
ethanol absorbs water (it can form hydrogen bonds) so it seperates from the hydrocarbons
can cause corrosion
methane → made form bacterial breakdown of plant mateiral in absence of oxygen
C6H12O6 → 3CO2 + 3CH4
advantages of biofuels
cheap + readily available
renewable (if crops/trees are replanted)
less polluting than fossil fuels
disadvantages of biofuels
uses land → can be used for other purposes (ex: growing food)
high cost of harvesting and transportation
takes nutrients from soil / uses large amounts of fertilizers
lower specific energy than fossil fuels
Fuel cells
hydrogen fuel cell
H2(g) + ½ O2(g) → H2O(l)
this is a redox reaction (transfer of electrons from hydrogen to oxygen)
can produce an electric current if reactants are physically seperated
hydrogen fuel cell operates with either an acidic or alkaline electrolyte
in fuel cells, reactants are continuously supplied to different electrodes
hydrogen-oxygen fuel cell → alkaline electrolyte (most commonly used)
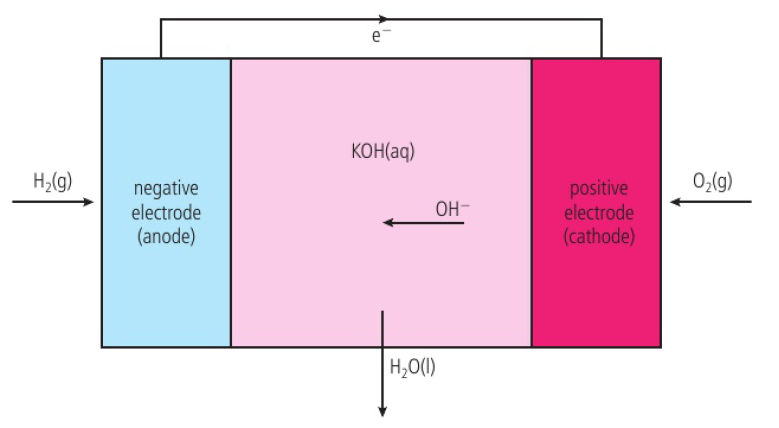
fuel cell will function as long as H2 and O2 are supplied
electrodes are often made of porous carbon with added transition metals (ex: nickel)
KOH (potassium hydroxide) provides the OH- ions that are transferred across the cell
problem: hydrogen gas must be extracted from other sources so might not be renewable
methanol fuel cell; DMFC → Direct Methanol Fuel Cell
methanol → stable liquid at normal environmental conditions, high energy density, easy to transport
DMFC → fuel is oxidized under acidic conditions on a catalytic surface to form CO2
H+ ions formed are transported across a proton exchange membrane from anode to cathode where they react with oxygen to form water
electrons are transported through an external circuit from anode to cathode
water is consumed at the anode and produced at the cathode
difference between fuel cells and primary voltaic cells:
fuel cells do not run out
fuel is supplied continuously to the cell as it is oxidized
1.4: Entropy and spontaneity
second law of thermodynamics: matter and energy tend to disperse and become more dispersed
entropy (S) → degree of dispersal of matter and energy of a system
spontaneous change → dispersion occurs naturally without work
Entropy
the natural tendency to change can be reversed if work is done
entropy → measure of dispersal/distribution of matter/energy in a system
ordered states with small energy distribution → low entropy
ex: gas particles concentrated in a small volume
disordered states with high energy distribution → high entropy
ex: gas particles dispersed throughout
as time moves forward, matter and energy become more dispersed → increases total entropy of universe
Predicting entropy changes
doubling number of particles also increases opportunity for matter/energy to be dispersed
doubling amount of a substance → entropy doubles
solid state → most ordered state with least dispersal → low entropy
increasing entropy: solid→liquid, solid→gas, liquid→gas
decreasing entropy: liquid→solid, gas→solid, gas→liquid
change due to number of particles (in gaseous state) is usually greater than any possible factor
Absolute entropy
entropy of a substance under standard conditions → section 13
all entropy values are positive
a perfectly ordered solid at absolute zero has an entropy of zero
Calculating entropy changes
calculated using differences between total entropy of the products and total entropy of the reactants
ΔS = ΔSproducts - ΔSreactants
calculations similar to enthalpy changes
entropy values are absolute values → always positive
Entropy changes of surroundings
to consider the total entropy change of a reaction, the entropy change in surroundings must also be considered
in an exothermic reaction, heat is transferred to the surroundings → general dispersal of energy
entropy of the surroundings increases as heat given out by reaction increases diispersal of surroundings
change in entropy of surroundings = enthalp ychange in the system x -absolute temperature
ΔSsurroundings = -ΔHsystem/T
exothermic reaction (-ΔH) increases entropy of surroundings
Calculating total entropy changes
second law of thermodynamics says that for a spontanous change:
ΔStotal = ΔSsystem + ΔSsurroundings > 0
ΔStotal = ΔSsystem - ΔHsystem/T > 0
endothermic reactions occur if change in entropy of system can compensate for negative entropy change of surroundings produced as the heat is transferred from surroundings to the system
strongly endothermic reactions are possible because there is a very large increase in dispersal of matter and entropy of the system
order may increase in local areas but only at the expense of greater disorder elsewhere
for chemical reactions, neither ΔHsystem or ΔSsystem can reliably be used to predict the feasability of a reaction
Gibbs energy
criterion or feasability of a reaction is given by:
ΔStotal = ΔSsystem - ΔHsystem/T > 0
ΔGsystem = ΔHsystem - TΔSsystem = -TΔStotal < 0
ΔGsystem → Gibbs energy
must be negative for a spontaenous process
for spontaneity, reaction must have ΔGsystem < 0
measure of quality of energy available
measure of energy free to do work rather than leave as heat
spontaneous reactions have negative Gibbs energy because they can do useful work
it is not essential for all heat to be transferred to surroundings to produce the necessary increase in the total entropy
enough energy must be transferred to surroundings to compensate for entropy decrease in the system, but the remaining energy is available to do work
this is the amount of energy that can be converted to electrical energy in a fuel cell
necessary energy transferred to surroundings = -TΔSsystem
energy available to do work = -ΔHsystem + TΔSsystem = -ΔGsystem
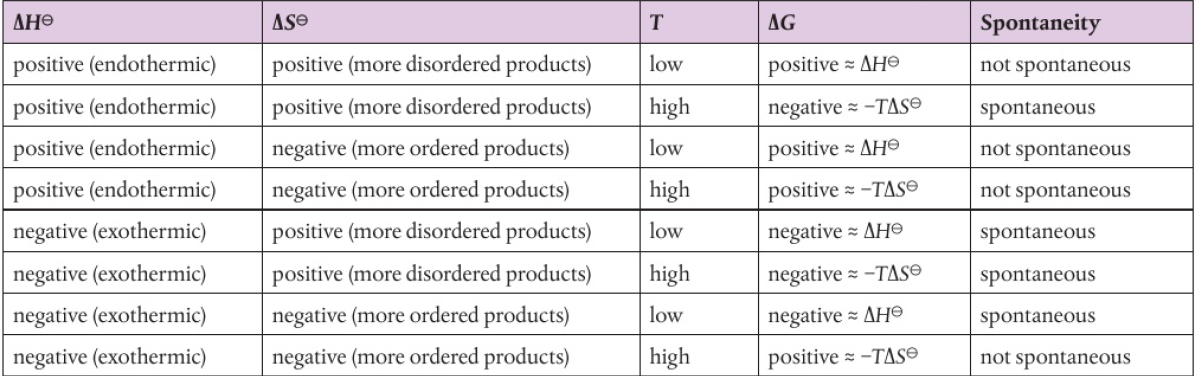
ΔG = ΔH - TΔS
ΔG is related to total energy change and this is just a reformulation of the 2nd law of thermodynamics
ΔG takes into account direct entropy change from transformation of chemicals in the system and indirect entropy change of surroundings resulting from the transfer of heat energy
ΔHsystem < TΔSsystem (T is always positive)
at low temperatures (TΔSsystem=0), this condition is met (exothermic) as ΔHsystem<0
endothermic reactions (positive ΔSsystem) can be spontaneous at higher temperatures
TΔSsystem > ΔHsystem
temperature Tspontaneous at which an endothermic reaction becomes spontaneous can be determined from:
Tspontaneous * ΔSsystem = ΔHsystem
Tspontaneous = ΔHsystem/ΔSsystem
The effect of ΔH, ΔS, and T on spontaneity of the reaction
ΔGsystem = ΔHsystem - TΔSsystem
if ΔG<0, reaction is spontaneous so:
if ΔHsystem > TΔSsystem, reaction is spontaneous
so if T is high, most likely not spontaneous makes ΔHsystem is high
GIbbs energy and equilibrium
only reactions where all reactants are formed into products have been considered
equilibrium mixture when ΔG=0
spontaneous reactions only occur when ΔG<0, so when ΔG=0
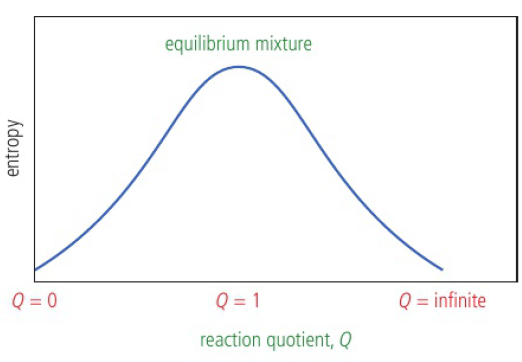
a mixture of reactant and product has higher entropy than pure samples
total entropy reaches a maximum when reactant = product
reaction quotient (Q) → ratio of products to reactants
ex: Q=[products]/[reactants] so at beginning, Q=0 and at the end, Q=infinity
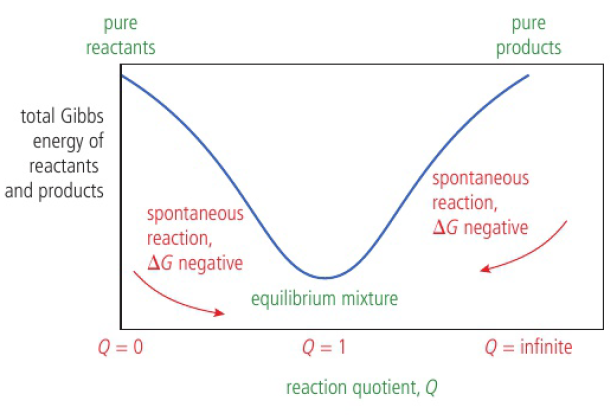
equilibrium mixture when ΔG<0 (negative)
at beginning of reaction, total Gibbs energy of reactants > products so reaction proceeds in forward direction and Q increases (products increase, reactants decrease)
as reaction proceeds, Gibbs energy (system) decreases until equilibrium is reached (Q=K)
once equilibrium is reached, all possible changes are not likely to happen (ΔG increases)
position of equilibrium corresponds to a mixture with more products than reactants
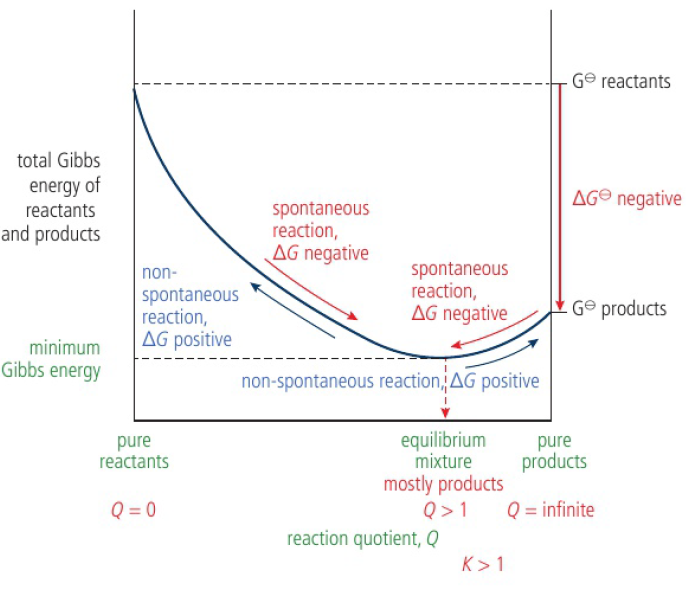
minimum Gibbs energy → equilibrium state, net reaction stops
relative amounts of reactants and products are at equilibrium
composition of equilibrium mixture is determined by the difference in Gibbs energy between reactants and products
K=[productseqm]/[reactantseqm] > 1 when ΔG<0
The equilibrium constant K
relationship between K (equilibrium constant) and ΔG (change in Gibbs energy)
so ΔG=-RT * lnK
useful when K is difficult to measure directly
ex: reaction is too slow to reach equilibrium/amounts of components are too small to measure
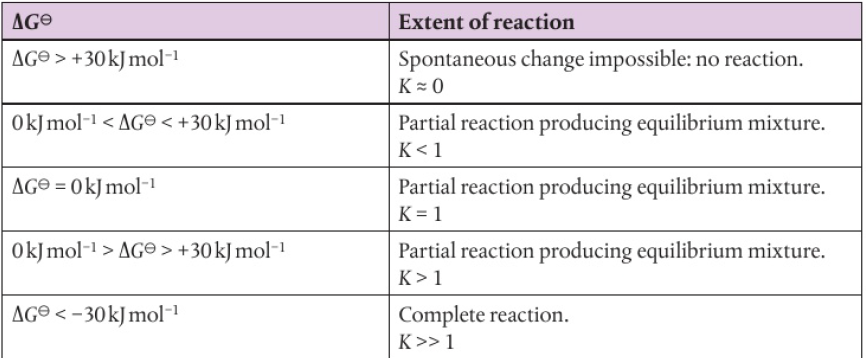
relationship between ΔG and extent of reaction:

2.1: How much? The amount of chemical change
Using chemical equations to find volumes of gaseous reactants and products
Avogadro’s Law → equal amounts of all gases measured under the same conditions of temperature and pressure contain equal numbers of molecules
equal number of particles of all gases occupy equal volumes
V has a direct relationship with n
volume occupied by one mole of any gas (molar volume, Vm) must be the same for all gases when measured under the same temperature and pressure
at STP, one mole of gas has a volume of 22.7dm³/mol
STP → OC (273K) and 100kPa
increase in temperature = increase in molar volume
increase in pressure = decrease in molar volume
number of moles of gas (n) = volume/molar volume
Titration
uses volumetric analysis to find unknown volumes or concentrations
pipette used to measure known volume into a conical flask
other solution put into a burette
point at which the two solutions have reacted completely → equivalence point
known when indicator changes color at the end point
titre → volume needed to reach equivalence point
Back titration
done in reverse by returning to the end point after it has passed
used when end point is hard to identify or when one of the reactants is impure
known excess of one reactant is added to reaction mixture, and unreacted excess is then determined by titration against a standard solution
reacting amount is determined by subtracting the amount of unreacted reactant from its original amount used
Limiting reactant and theoretical yield
limiting reactant → reactant that determines the quantity of product
always the one fully used up, other reactants are added in excess
theoretical yield → maximum amount of product obtainable (assuming 100% of limiting reactants is used)
usually expressed in grams or moles
Percentage yield
theoretical yield assumes that chemical reactions have no loss, waste, or impurities
experimental yield → actual yield with factors taken into account
factors that may cause experimental yield to be lower than the theoretical yield:
side reactions occuring
decomposition of reactants and/or products
loss of product during purification
reversible chemical reactions preventing process completion
factors that may cause experimental yield to be higher than the theoretical yield:
impurities in a product
when a product has not been fully dried
factors that impact experimental yield in both directions (depending on type of reaction):
an incomplete reaction
percentage yield = experimental yield/theoretical yield * 100
Atom economy
Green Chemistry → sustainable design of chemical products and chemical processes
aims to minimize use of chemical substances that are hazardous to human health / the environment
percentage yield does not give a quantity of waste produced
atom economy is maximized by turning as much reactant atoms into products
% atom economy = molar mass of desired product / molar mass of all reactants * 100
efficient processes have high atom economies → uses fewer resources and generates less waste
2.2: How fast? The rate of chemical change
Rate of reaction
rate of reaction → rate of change in concentration
as the reaction proceeds, reactants are converted into products
concentration of reactants decrease and concentration of products increase
rate of reaction (moldm³/s)= increase in product concentration / time taken = decrease in reactant concentration / time taken
if the line is a curve, use the gradient of the tangent
rate of reaction is not constant, but is greatest at the start and decreases over time
Measuring rate of reaction
change in volume of gas produced
used if one of the products is a gas
collecting the gas and measuring change in volume at regular time intervals
using a gas syringe or displacement of water in an inverted burette
displacement method can only be used if gas collected has low solubility in water
change in mass
if one of the products is a gas, this can be done by setting the reaction on a scale
does not work if the gas is hydrogen → too light
change in transmission of light: colorimetry/spectrophotometry
used if one of the reactants/products is colored (so gives characteristic absorption in the visible region)
sometimes indicator is added to make it a colored compound
colorimeter/spectrophotometer measures the intensity of light transmitted by reaction components
rate of product formation → change in absorbance
change in concentration → titration
quenching → a substance is introduced that effectively stops the reaction, obtaining a “freeze frame” shot
done to avoid chemically changing the reaction mixture
samples are taken from the reaction mixture at regular time intervals and analyzed by titration
titration takes time, during which the reaction would proceed → quench
change in concentration using conductivity
total electrical conductivity of a solution depends on the total concentration of its ions and charges
measured using a conductivity meter
non-continuous methods of detecting change during a reaction: ‘clock reactions’
measure time it takes for the reaction to reach a certain chose point
uses time as the dependent variable
limitation: only gives average rate of reaction
Collision theory
particles in a substance move randomly as a result of their kinetic energy
not all particles will have the same kinetic energy, but instead a range
therefore the measurement is an average
increasing temperature = increasing average kinetic energy of particles
kinetic theory of matter (S1.1)
Maxwell-Boltzman energy distribution curve
DIAGRAM HERE
the number of particles having a specific value of kinetic energy (or probability of that value occuring) against values of kinetic energy
area under the curve → total number of particles in sample
nature of collisions between particles
when reactants are placed together, their kinetic energy cause them to collide
energy from collisions may cause bonds to break and new bonds to form
as a result, products ‘form’ and the reaction stops
rate of reaction depends on the number of successful collisions which form products
successful collisions depend on:
energy of collision
geometry of collision
energy of a collision
particles must have the required activation energy (Ea) necessary for overcoming repulsion between molecules, and often breaking bonds in reactants
when Ea is supplied, reactants achieve the transition state from which products can form
activation energy is thus an energy barrier for the reaction → different for all reactions
Ea → threshold value
if you pass, you may react
DIAGRAM HERE → activation energy
particles with Ek>=Ea will collide successfully
particles with Ek<Ea may still collide, but unsuccessfully
therefore, rate of reaction depends on proportion of particles that has Ek>Ea
DIAGRAM HERE → Maxwell curve activation energy
generally, reactions with high activation energy will proceed more slowly as fewer particles will have the required energy for a successful collision
geometry of a collision
DIAGRAM HERE → different collisions
because collisions between particles are random, there are many likely orientations → only some are successful
therefore, rate of reaction is determined by:
values of kinetic energy greater than activation energy
appropriate collision geometry
Factors that influence the rate of reaction
temperature
increasing temperature increases average kinetic energy of particles
DIAGRAM HERE → Maxwell curve
area under both curves is the same → same number of particles
at higher temperature, more particles have higher kinetic energies so the peak of the curve shifts rightwards
as temperature increases, collision frequency increases due to higher kinetic energy → more collisions involving particles with necessary activation energy
therefore, more successful collisions (every +10K, reaction rate doubles)
concentration
increasing concentration increases frequency of collisions between reactants → more successful collisions
as reactants are used up, the concentration decreases and the rate of reaction decreases
pressure
increasing pressure “compresses” the gas, effectively increasing concentration
surface area
increasing surface area allows for more contact and a higher probability of collisions
instead of one big chunk, divide it into smaller sections to increase total surface area
stirring can increase total surface area by ensuring individual particles are spread
catalyst → a substance that increases rate of reaction without itself undergoing chemical change
most catalysts work by providing an alternative route for the reaction that has lower activation energy
DIAGRAM HERE → uncatalyzed reaction, catalyzed reaction
without increasing temperature, more particles will have Ek>Ea, so will be able to undergo successful collisions
catalysts equally reduce Ea for both forward and reverse reactions, so does not shift equilibrium or yield
DIAGRAM HERE → Maxwell curve
catalysts increase efficiency, and there are “best” catalysts for certain reactions → otherwise reactions move too slowly or are conducted at too high temperatures
Catalysts
every biological reaction is controlled by a catalyst → enzyme
there is a specific enzyme for every particular biochemical reaction
biotechnology → field that searches for possible applications of certain enzymes
catalysts can replace stoichometric reagants → greatly enhances selectivity of processes
therefore, important aspect of Green Chemistry
catalysts are effective in small quantities and can frequently be reused
therefore do not contribute to chemical waste → increases atom economy
Reaction mechanisms
most reactions that occur at a measurable rate occur as a series of simple steps, each involving a small number of particles
this sequence of steps is known as the reaction mechanism
the individual steps (elementary steps) usually cannot be observed directly
therefore this is only a theory → cannot be proved (but there are clues)
often the products of a single step in the mechanism are used in a subsequent step
exists only as reaction intermediates, not as final products
ex: NO2(g) + CO(g) → NO(g) + CO2(g)
mechanism follows these elementary steps:
NO2(g) + NO2(g) → NO(g) + NO3(g)
NO3(g) + CO(g) → NO2(g) + CO2(g)
overall reaction: NO2(g) + CO(g) → NO(g) + CO2(g)
reactants and products cancel out → reaction intermediates
NO2 in reactants in step 1 and products in step 2 cancel out
NO3 in products in step 1 and reactants in step 2 cancel out
molecularity → used in reference to an elementary step to indicate number of reactant species involved
unimolecular → elementary step that involves a single reactant particle
bimolecular → elementary step with two reactant particles
trimolecular reactions are rare → extremely low probability of >2 particles colliding at same time with sufficient energy and correct orientation
Rate-determining step
the rate-determining step is the slowest step in the reaction mechanism
products of the reaction can only appear as fast as the products of this slowest elementary step
rate-determining step therefore determines overall rate of reaction
DIAGRAM HERE → reaction coordinate, potential energy
two maxima represent the transition states
minimum represents the intermediate species
in this example, first maxima (first step) is higher, so more activation energy required → thus slowest step, so rate-determining
catalysts usually find an alternative for the slowest step to speed up the reaction (rate-determining step made faster or changes)
Rate equations
rate equations are determined experimentally and depend on the mechanism of a reaction
consider the reaction: C60O3 → C60O + O2
we can follow the reaction by recording the change in absorbance of light of a certain wavelength
absorbance is directly proportional to concentration of C60O3
rate of reaction is equal to the rate of change in concentration of C60O3
rate=- [C60O3]/t (negative because concentration is decreasing)
rate can be calculated by finding gradient of line’s tangent at a specific point
rate slows down as concentration of C60O3 decreases
similarities in concentration VS time and rate VS time graphs suggests that the rate must be related ot concentration at each time
straight-line graph between absorbance and rate confirms that the rate of reaction is directly proportional to concentration of C60O3
reaction rate is directly proportional, so reaction rate = k[C60O3]
k is the rate constant
this equation is a rate equation → first order rate equation because the concentration of the only reactant is raised to the first power
rate of all reactions can similarly be shown to depend on concentration of one or more of the reactants, and the particular relationship depends on the reaction
generally, rate is proportional to products of concentrations of reactants, each raised to a power
A+B → products so rate=k[A]m[B]n
m and n are known as the orders of the reaction with respect to A and B
overall reaction order is sum of individual orders (m+n)
orders can only be determined by experiment (empirically)
no connection between reaction equation (coefficients, moles) and rate equation
Rate equation and reaction mechanism
as the rate of reaction depends on the rate-determining step, the rate equation for the overall reaction must depend on the rate equation for the rate-determining step
because the rate-determining step is an elementary step, its rate equation comes directly from its molecularity:
A → products: unimolecular, so rate=k[A]
2A → products: bimolecular, so rate=k[A]²
A+B → products: bimolecular, so rate=k[A][B]
rate equation for rate-determining step, predictable from its reaction equation, leads to the rate equation for the overall reaction
when rate-determining step is not the first step, the intermediate cannot be used in the rate equation → instead, substitute
order of reaction with respect to each reactant is not linked to coefficients in overall equation, but is instead determined by their coefficients in the equation for the rate-determining step
Order of a reaction
reaction that is zero-order with respect to a particular reactant → the reactant is required for reaction but does not affect rate as it is not present in the rate-determining step
if a reactant is present in the rate-equation, it partakes in the rate-determining step
reaction order can be fractional or negative in more complex reactions
concentration-time graphs do not give a clear distinction between first and second order
rate-concentration graphs clearly reveal the difference
zero-order: rate=k[A]0=k
DIAGRAM HERE
concentration-time → straight line, constant rate
gradient of line = k
rate-concentration → horizontal line
first-order: rate=k[A]
DIAGRAM HERE
concentration-time → rate decreases with concentration
rate-concentration → straight line passing through origin with gradient k
second-order: rate=k[A]²
DIAGRAM HERE
concentration-time → curve, steeper at start than first-order graph but leveling off more quickly
rate-concentration → parabola (square function), gradient proportional to concentration and initially zero
order of reaction can only be determined experimentally, thus these graphs are required to distinguish them
Determination of the overall order of a reaction
methods for determining order of reaction depends on the reactants
two methods, but only initial rate method is covered
initial rates method
carrying out a number of separate experiments with different starting concentrations of reactant A, and measuring the initial rate of each reaction
concentration of other reactants are held constant to see effect of A on reaction rate
changing concentration of A but no effect on rate → zero order with respect to A
changing concentration of A produces directly proportional changes in rate of reaction → first order with respect to A (doubling concentration of A doubles reaction rate)
changing concentration of A leads to the square of that change in the rate → second order with respect to A (doubling concentration of A leads to a four-fold increase in reaction rate)
use of the integrated form of the rate equation
calculus is used to analyze the integral of rate equation
direct graphical analysis of functions of concentration against time
The rate constant, k
units of k vary with order of reaction
zero order: rate=k, k=moldm³/s
first order: rate=k[A], k=rate/concentration=s-1
second order: rate=k[A]², k=dm³/mols
third order: rate=k[A]³, k=dm6/mol²s
k is temperature dependent → general measure of rate of a reaction at a particular temperature
temperature dependence of k depends on value of activation energy
high Ea → temperature rise causes significant increase in particles that can react
low Ea → same temperature rise will have proportionally smaller effect on reaction rate
temperature dependence of k is expressed in the Arrhenius equation
The Arrhenius equation
Suante Arrhenius showed that the function of molecules with energy greater than the activation energy at temperature T is proportional to e-Ea/RT (R is gas constant)
reaction rate and therefore rate constant are also proportional to this value
k=Ae-Ea/RT
A → Arrhenius factor (frequency factor, pre-exponential factor)
A takes into account the frequency of successful collisions based on collision geometry
A is a constant for a reaction and has same units as k (so varies with order)
Arrhenius plot → lnk=-Ea/RT + lnA
rule of thumb → 10K increase doubles reaction rate
2.3: How far? The extent of chemical change
Dynamic equilibrium
reaction takes place at same rate as its reverse reaction, so no net change is observed
physical systems (ex: bromine in a sealed container at room temperature)
bromine is a volatile liquid (boiling point close to room temperature)
significant amount of Br2 molecules will have enough energy to leave the liquid state (evaporate)
container is sealed so bromine vapour cannot escape → concentration increases
some vapour molecules will collide with surface of liquid, lose energy, and become liquid
Br2(l) ⇌ Br2(g)
rate of condensation increases with concentration of vapour (more vapour particles)
eventually, rate of condensation will equal rate of evaporation
no net change → equilibrium (only occurs in a closed system)
DIAGRAM HERE → rate of condensation = rate of evaporation
chemical systems (ex: dissociation between hydrogen iodide (HI) and its elements (H2, I2)
2HI(g) ⇌ H2(g) + I2(g)
colourless gas ⇌ colourless gas + purple gas
there will be an increase in purple hue when the reaction starts (production of I2)
at some point, the increase in colour will stop
rate of dissociation of HI is fastest at the start as the concentration of HI is the greatest, then falls as the reaction proceeds
reverse reaction had initially zero rate (no H2 or I2 present) then starts slowly and increases in rate as concentrations of H2 and I2 increase
eventually, the rate of the forward and reverse reactions will equal, so concentrations remain constant
equilibrium → dynamic because both reactions are still occuring
if the contents of the flask were analyzed at this point, HI, I2, and H2 would all be present with constant concentrations → equilibrium mixture
DIAGRAM HERE → equilibrium
if the experiment were reversed (starts with H2 and I2), eventually an equilibrium mixture will again be reached
reactants ⇌ products
→ forward, ← backward
constant concentrations of products and reactants does not mean equal amounts
equilibrium position → proportion of reactant and product in equilibrium
predominantly products → lie to the right
predominantly reactants → lie to the left
Equilibrium Law
consider the reaction: H2(g) + I2(g) ⇌ 2HI(g)
if we were to carry out a series of experiments on this reaction with different starting concentrations of H2, I2, and HI, we could wait until each reaction reached equilibrium and then measure the composition of each equilibrium mixture
there is a predictable relationship among the different compositions of these equilibrium mixtures
[HI]²/[H2][I2] → concentration at equilibrium ([HI] is squared because that is its coefficient in the equation)
K → constant value, equilibrium constant (fixed value at specified temperature)
every reaction has its particular value of K
equilibrium constant expression for reaction: aA + bB ⇌ cC + dD
K = [C]eqmc[D]eqmd / [A]eqma[B]eqmb
[A] → concentration, a → coefficient in reaction equation, products → numerator, reactants → denominator
high value of K → at equilibrium, proportionally more products than reactants
lies to the right, reaction almost to completion
K values tells differing extents of reactions
higher value = reaction has taken place more fully
K » 1: reaction almost goes to completion (right)
K « 1: reaction hardly proceeds (left)
Le Chatelier’s principle
a system at equilibrium when subjected to a change will respond in such a way as to minimize the effect of the change
whatever done to a system at equilibrium, it will respond in the opposite way
after a while, a new equilibrium will be established with different composition
changes in concentration
equilibrium mixture disrupted by increase in concentration of a reactant:
rate of forward reaction increases: forward =/ backward anymore
equilibrium will have shifted in favour of products (rightward)
value of K remains unchanged
same happens with decrease in concentration of product
rate of backward reaction decreases → new equilibrium position will be achieved (rightward)
often in industrial processes, product will be removed as it forms
ensures equilibrium is continuously pulled rightward → increasing yield of product
changes in pressure
equilibria involving gases will be affected if there is a change in the number of molecules
there is a direct relationship between pressure exerted by gas and the number of gas particles
increase in pressure → system response: decrease pressure by favouring the side with less molecules
new equilibrium position, K remains unchanged (if temperature does not change)
ex: CO(g) + 2H2(g) ⇌ CH3OH(g)
increase in pressure shifts equilibrium rightward → in favour of smaller number of molecules
increase in pressure → increases yield of CH3OH
if number of molecules are the same for both sides, pressure will not change equilibrium
changes in temperature
K is temperature dependent → changing temperature affects K
ex: 2NO2(g) ⇌ N2O4(g) ΔH=-57kJ/mol (forward reaction exothermic)
decrease in temperature → system produces heat → favours forward exothermic reaction
new equilibrium mixture (rightward) → K increases (higher yield at lower temperature)
increasing yield takes too long→ decreasing temperature lowers rates of reactions
addition of a catalyst
catalyst speeds up rate of reaction by providing alternative reaction pathway with a transition state with a lower activation energy
increases number of particles that have sufficient energy to react (without increasing temperature)
because both forward and backward reactions pass through the same transition state, both rates will increase → no change in equilibrium position and K
will not increase equilibrium yield of a product
speeds up attainment of equilibrium state → products form more quickly
has no effect in equilibrium concentrations → not chemically changed
The reaction quotient, Q
K is calculated using concentrations at equilibrium
Q → calculated using concentrations when not at equilibrium
as time passes and reaction proceeds, concentrations will change and eventually reach equilibrium
Q changes in direction of K → used to predict direction of reaction
if Q=K, reaction at equilibrium, no net reaction occurs
if Q<K, reaction proceeds rightward in favour of products
if Q>K, reaction proceeds leftward in favour of reactants
Quantifying the composition at equilibrium
done by calculating equilibrium constant (K) or concentration of reactants/products
only homogeneous equilibria → all reactants/products in the same phase (gas or solution)
equilibrium law can be used to solve for K and initial/final concentrations
Measuring the position of equilibrium
Gibbs energy change can be used to measure the position of equilibrium
ΔG → measure of work available from a system calculated for a particular composition of reactants and products (ΔG=Gproducts-Greactants)
ΔG=negative → reaction proceeds in forward direction
ΔG=positive → reaction proceeds in backward direction
ΔG=0 → reaction is at equilibrium (Gproducts=Greactants)
at the start of a reaction, total Gibbs energy of reactants is greater than products (lot of work is available) → ΔG=negative, reaction proceeds in forward direction
as reaction proceeds, total GIbbs energy of reactants decreases but of products increase
ΔG less negative, less work is available
system reaches equilibrium when total Gibbs energy of reactants and products are equal
no work can be extracted from system (ex: dead battery)
total Gibbs energy decreases as reaction progresses as work is done by the system
occurs both when reaction starts with reactants and products
equilibrium state → net reaction stops → minimum value of Gibbs energy
DIAGRAM HERE → equilibrium
DIAGRAM HERE → equilibrium
decrease in total Gibbs energy appears as work done or increase in entropy
system has highest possible value of entropy when Gibbs energy at minimum (at equilibrium)
reaction with large and negative ΔG value → spontaneous, equilibrium with high products
reaction with large and positive ΔG value → non-spontaneous, predominantly reactants
ΔG=-RT*lnk
ΔG negative, lnK positive, K>1: equilibrium mainly products
ΔG positive, lnK negative, K<1: equilibrium mainly reactants
ΔG=0, lnK=0, K=1: appreciable amounts of both reactants and products
Rate of reaction and equilibrium
ex: reaction that occurs in a single step
A + B ⇌ C + D
rate of forward reaction: k[A][B]
rate of backward reaction: k’[C][D]
K=[C][D]/[A][B] (equilibrium constant)
at equilibrium, rate of forward reaction = rate of backward reaction
k[A][B]=k’[C][D]
rearranging gives: K=k/k’
if k»k’, K is large → reaction proceeds to completion
if k«k’. K is small → reaction barely takes place
increasing concentration of reactants speeds up forward reaction (vice versa)
shifts equilibrium rightward
equilibrium constant stays the same regardless
adding a catalyst increases k and k’ by same factor → K stays the same
k=Ae-Ea/RT → activation energies of forward and backward reactions are different
differently affected by temperature
for endothermic reactions (Ea(forward) > Ea(backward)), increasing temperature will have greater effect increasing k than k’, so K increases
3.1: Proton transfer reactions
H+ is equivalent to a proton
proton is transferred when:
reactant loses H+ so loses positive charge
product gains H+ so gains positive charge
this type of reaction can only occur between certain species: reactants that can release H+ and products with a lone pair that can accomodate an additional H+
Bronsted-Lowry acid-base behavior
Bronsted-Lowry acids and bases
acids donate H+, bases accept H+
Bronsted-Lowry acid is a proton (H+) donor
Bronsted-Lowry base is a proton (H+) acceptor
act of donating cannot act in isolation, must always have an acceptor
an acid can only be a proton donor if a base is present to accept it
ex: HA + B ⇌ A- + BH+
HA acts as an acid, donating a proton to B → B acts like a base, accepting it
in reverse reaction, BH+ is the acid while A- acts as a base
acid HA has reacted to form base A- and base B reacted to form acid BH+
HA and A-, B and BH+ → conjugate acid-base pairs
conjugate acid-base pairs differ by one proton
acid always has one more proton than its conjugate base (H+)
ex: H3O+ → H2O, NH3 → NH2-
most polyatomic ions can form Bronsted-Lowry acids by accepting H+ (ex: OH-, NO3-)
ammonium (NH4+) cannot accept H+ to form an acid, instead loses H+
acts as a Bronsted-Lowry acid and forms its conjugate base
Bronsted-Lowry bases are defined as any species which can accept a proton
a small group of Bronsted-Lowry bases (alkalis) are soluble bases which dissolve in water to release the hydroxide ion (OH-)
Amphiprotic species
some species can act as both Bronsted-Lowry acids and bases (ex: water)
these species are amphiprotic
depends on what the species is reacting with
amphiprotic substances must possess both a lone pair of electrons (to accept H+) and hydrogen that can be released as H+ (to dissociate)
moving left to right across a period: basic metal oxides through amphiprotic oxides to acidic oxides
ex: period 3
Na2O, MgO → basic
Al2O3 → amphoteric
SiO2, P4O10, SO2, Cl2O → acidic
The pH scale
measure of acid strength based off its H+ concentration
pH = log10[H+] or [H+] = 10-pH
ex: a solution with [H+] = 0.1mol/dm³ → 10-1mol/dm³ → -(-1)pH=1pH
most common acids and bases will have a positive pH in the range 0-14
pH has no units and is inversely related to [H+]
DIAGRAM HERE
for each increase of 10 times in [H+]. pH will decrease by 1 unit
pH is measured by estimating using universal indicator paper or solution
substance tested will give a characteristic colour → compared to indicator’s colour chart
pH is measured using a pH meter that directly reads the H+ concentration using a special electrode
must be calibrated before use (pH is temperature-dependent)
Ionization of water
majority of acid-base reactions involve ionization in aqueous solution
water does ionize, only very slightly at normal temperatures and pressures
H2O(l) ⇌ H+(aq) + OH-(aq)
K=[H+][OH-]/[H2O] → concentration of water is considered constant as so little of it ionizes
Kw=[H+][OH-] → ionic product constant of water
at 298K, Kw=1×10-14
H+ in aqueous solution always exists in H3O+(aq)
in pure water, [H+]=[OH-], so [H+]=√Kw
[H+]=1×10-7, pH=7 → neutral
[H+] x [OH-] is constant, so they have an inverse relationship
when one increases, the other decreases
acidic: [H+]>[OH-], pH<7
neutral: [H+]=[OH-], pH=7
alkaline: [H+]<[OH-], pH>7
Kw is an equilibrium constant → temperature dependent
increasing temperature shifts equilibrium rightward → increases Kw
[H+] and [OH-] increase → pH decreases
decreasing temperature shifts equilibrium leftward → decreases Kw
[H+] and [OH-] decrease → pH increases
pH of water is 7.00 only at 298K
still neutral regardless of temperature because [H+]=[OH-]
Strong and weak acids and bases
Bronsted-Lowry acids and bases dissociate in solution
acids produce H+ ions and bases produce OH- ions
aqueous solutions exist as equilibrium mixtures containing both undissociated form and its ions
ex: HA(aq) + H2O(l) ⇌ A-(aq) + H3O+(aq)
if this acid dissociates fully, its equilibrium is positioned at the right → exists virtually entirely of ions
this is said to be a strong acid
if the acid dissociates only partially, equilibrium lies to the left → undissociated form dominates
this is said to be a weak acid
ex: HCl(aq) + H2O(l) → H3O+(aq) + Cl-(aq)
strong acid + base → conjugate acid + conjugate base
strong acids are good proton donors
because their dissociation reaction goes almost to completion, their conjugate bases do not readily accept protons
in this example, the strong acid HCl reacts to form conjugate base Cl- which shows no basic properties
weak acids are poor proton donors
conjugate bases readily accept protons
acid dissociation reactions favour the production of the weaker conjugate
ex: NaOH(aq) → Na+(aq) + OH-(aq)
NaOH is a strong base because it ionizes fully
strong bases are good proton acceptors → react to form conjugates that show no acidic properties
base ionization reactions favour the production of the weaker conjugate
strength of an acid or base is a measure of how readily it dissociates in aqueous solution
TABLE HERE → common strong and weak acids and bases
since strength of an acid depends on the ease with which it dissociates to release H+ ions, it thus depends on the strength of the bond that has to be broken to release H+
longer bond = weaker bond = stronger acid
thus acid strength of hydrogen halides increase down the group (HF<HCl<HBr<HI)
distinguishing strong/weak acids/bases → strong acids/bases will contain a higher concentration of ions
comparisons only work when solutions of the same concenration are compared at the same temperature
electrical conductivity
depends on concentration of mobile ions (solution)
strong acids/bases will show higher conductivity
tested using a conductivity meter, probe, or pH meter with conductivity setting
rate of reaction
stronger acids would have an increased rate
pH
the stroner the acid, the lower the pH value
measured using universal indicator or pH meter
Neutralization reactions
reaction between an acid and base (H+ + OH- → H2O) is a neutralization reaction
during the reaction, an ionic compound (salt) forms → hydrogen in acid is replaced by a metal/positive ion
parent acid, parent base → relationship between an acid, a base, and their salt
when acids react with reactive metals, a salt is also formed (metal ion replaces hydrogen)
no H+ transfer, hydrogen reduced as gas (H2)
hydrogen ions are becoming electrically neutral by accepting electrons
metal is being ionized by electron loss
ex: Mg(s) + 2HCl(aq) → MgCl2(aq) + H2(g)
Mg(s) → Mg2+(aq) + 2e- (electron loss → oxidation)
2H+(aq) + 2e- → H2(g) (electron gain → reduction)
redox reactions, cannot be described by Bronsted-Lowry acid-base theory
acid + base → salt + water
metal oxides/hydroxides are bases which react with acids to produce a salt and water
ex: HCl(aq) + NaOH(aq) → NaCl(aq) + H2O(l)
ammonia solution is also a soluble base which reacts with acids to form a salt and water
ex: HNO3(aq) + NH4OH(aq) → NH4NO3(aq) + H2O(l)
when choosing a base in solution to make a specific salt, use solubility rules:
only soluble carbonates/hydrogencarbonates are: NH4CO3, Na2CO3, K2CO3, KHCO3, Ca(HCO3)2
only soluble hydroxides are: NH4OH, LiOH, NaOH, KOH
acid + carbonate → salt + water + carbon dioxide
metal carbonates/hydrogencarbonates react with acids to produce a salt, water, and carbon dioxide
ex: 2HCl(aq) + CaCO3(aq) → CaCl2(aq) + H2O(l) + CO2(g)
2H+(aq) + CO32-(aq) → H2O(l) + CO2(g)
H+(aq) + HCO3-(aq) → H2O(l) + CO2(g)
gas is given off, visibly produces bubbles → effervescence
neutralization reactions are exothermic
net reaction is formation of H2O from its ions (H+(aq) + OH-(aq) → H2O(l))
other ions (spectator ions) do not change during reaction → can be cancelled out
for reactions between strong acids/bases, enthalpy of neutralization → H=-57kJ/mol
expressed per mole of H2O formed → overall reaction is formation of water
pH curves
following examples all use (for better comparison):
0.10 mol/dm³ solutions for all acids and bases
initial volume of 50cm³ of acid in conical flask, base added with burette
monoprotic acids and bases → reacts in 1:1 ratio, equivalence is achieved at equal volumes for these equimolar solutions (i.e. when 50cm³ of base has been added to 50cm³ of acid)
acid-base titration → controlled volumes of one reactant are added gradually from a burette to a fixed volume of the other reactant that has been measured using a pipette and placed in a conical flask
reaction between acid and base takes place in flask until the equivalence/stoichometric point is reached
until they exactly neutralize each other
pH meter or indicator is used to detect the exact point equivalence is reached
when a base is added to an acid in a neutralization reaction, there is a change in pH
change is not linear (mostly due to logarithmic nature of pH) → shown on pH curve
in most titrations, a big jump in pH occurs at equivalence (point of inflection)
at equivalence, acid and base exactly neutralized each other → solution is only salt and water
ex: HCl(aq) + NaOH(aq) → NaCl(aq) + H2O(l) (strong acid + strong base)
full dissocation is assumed → both acid and base are strong
when base is added, volume also increases → changing concentration, changing pH
DIAGRAM HERE
initial pH=1 → pH of strong acid
pH changes only gradually until equivalence
very sharp jump in pH at equivalence (3→11)
after equivalence, flattens out at a high value → pH of strong base
pH at equivalence (pH=7)
measured by continuously using a pH probe
weak acid + strong base
DIAGRAM HERE
initial pH fairly high (pH of weak acid)
pH stays relatively constant until equivalence → buffer region
jump in pH at equivalence (not as much of a jump as strong acid)
after equivalence, curve flattens out at high pH (pH of strong base)
pH at equivalence > 7
anion hydrolysis releases OH-
point where 25/50cm³ of base is added → half-equivalence point (half of the acid has been neutralized)
mixture has equal quantities of a weak acid and its salt → buffer solution
buffer region → pH is relatively resistant to change
at this point, pH=pKa because [acid]eqm=[HA]initial so [salt]eqm=[A-]eqm
strong acid + weak base
DIAGRAM HERE
initial pH=1 → strong acid’s pH
pH remains relatively constant through buffer region until equivalence
jump in pH at equivalence (pH 3-7)
curve flattens out after equivalence at fairly low pH (pH of weak base)
pH at equivalence < 7
weak acid + weak base
pH at equivalence is difficult to define
DIAGRAM HERE
initial pH is fairly high (weak acid)
addition of base causes pH to rise steadily
change in pH at equivalence is much less sharp than other titrations
after equivalence, curve flattens out at fairly low pH (weak base)
there is no defined pH at equivalence because there are several equilibria involved
The pOH scale
pH scale simplifies [H+], pOH simplifies [OH-]
OH- ions are often found in low concentrations in solutions
pOH = -log10[OH-], pH = -log10[H+]
since [H+][OH-] = Kw = 1×10-14 at 298K:
pH + pOH = 14.00 at 298K
pKw = -log10(Kw)
Kw = 10-pKw
pH + pOH = pKw
Dissociation constants
weak acids and bases do not fully dissociate → concentrations of ions in solutions cannot be deduced from initial concentrations, depends on extent of dissociation
dissociation of weak acids and bases are represented as equilibrium expressions
ex: HA(aq) + H2O(l) ⇌ H3O+(aq) + A-(aq) (weak acid HA dissociating in water)
K=[H3O+][A-] / [HA][H2O] → concentration of water is constant
Ka=[H3O+][A-] / [HA] → acid dissociation constant
fixed value for a particular acid at a specified temperature
the higher the value of Ka at a particular temperature, the greater the dissocation, so the stronger the acid
because Ka is an equilibrium constant, it is not dependent on the acid’s concentration or other ions
ex: B(aq) + H2O(l) ⇌ BH+(aq) + OH-(aq) (weak base B dissociating in water)
Kb=[BH+][OH-] / [B] → base dissociation constant
the higher the value of Kb at a particular temperature, the greater the ionization, so the stronger the base
calculation rules involving Ka and Kb:
given concentration of an acid/base is its initial concentration → before dissociation
pH/pOH values refers to concentration of H+/OH- ions at equilibrium
concentration values for Ka/Kb must be the equilibrium values for reactants and products
when extent of dissociation / Ka and Kb value is very small, [acid]initial=[acid]equilibrium (same for base)
pKa and pKb
pKa = -log10Ka, pKb = -log10Kb → similar to pH/pOH scale
pKa and pKb numbers are usually positive and have no units
relationship between Ka/pKa and Kb/pKb is inverse
stronger acid → Ka increases, pKa decreases; stronger base → Kb increases, pKb decreases
pKa/pKb must be quoted at a specified temperature
Ka x Kb = Kw so pKa + pKb = 14.00 at 298K
the stronger the acid, the weaker its conjugate base will be
the higher the Ka, the lower the Kb for its conjugate
pH of salt solutions
salt formed in neutralization reaction is an ionic compound → cation is conjugate acid of parent base, anion is conjugate base from parent acid (MOH + HA → M+A- + H2O)
pH of a salt solution depends on whether/to what extent their ions react with water and hydrolyze it
strong acid + strong base
neither ion hydrolyses
neutral, pH=7
weak acid + strong base
anion hydrolyses
basic, pH>7
strong acid + weak base
cation hydrolyses
acidic, pH<7
weak acid + weak base
both ions hydrolyse
depends on relative strengths, pH cannot generalize
Acid-base indicators
indicators are weak acids/bases in which their undissociated and dissociated forms have different colours
only weak acids will be considered → HInd (generic acidic indicator)
HInd(aq) ⇌ H+(aq) + Ind-(aq) (different colours as HInd and Ind-)
increasing [H+] → equilibrium shifts leftward in favour of HInd
Ka=[H+][Ind-] / [HInd]
at the point where the equilibrium is balanced ([Ind-]=[HInd]), the indicator is exactly in th emiddle of its colour change: Ka=[H+][Ind-] / [HInd] = [H+] so pKa=pH
addition of a very small volume of acid/base will shift equilibrium → change colour
this point is the end point (pH=pKa(indicator))
different indicators have different pKa values so will have different end points
an indicator will be effective in signalling the equivalence point when its end point coincides with the pH at equivalence
different indicators must be used for different titrations, depending on the pH at equivalence
choosing an appropriate indicator:
determine what combination of strong/weak acids/bases are reacting together
deduce pH of salt solution at equivalence
consult data tables to choose an indicator with end point in range of equivalence point
Buffer solutions
buffer → something that acts to reduce the impact of one thing on another (shock absorber)
buffer acts to reduce the pH impact of added acid/base on a chemical system
buffer solution is resistant to changes in pH on the addition of small amounts of acid/alkali
acidic buffers (maintain pH<7)
made by mixing an aqueous solution of a weak acid with a solution of its salt of a strong alkali
ex: NaCH3COO(aq) → Na+(aq) + CH3COO-(aq) (soluble salt → fully dissociates)
mixture contains relatively high concentrations of CH3COOH and CH3COO- (acid and its conjugate base)
resovoirs, ready to react with added OH- and H+ in neutralization reactions
addition of H+ (acid): CH3COO-(aq) + H+(aq) ⇌ CH3COOH(aq)
addition of OH- (base): CH3COOH(aq) + OH-(aq) ⇌ CH3COO-(aq) + H2O(l)
basic buffers (maintain pH>7)
made by mixing an aqueous solution of a weak base with its salt of a strong base
ex: NH3(aq) and NH4Cl(aq) (weak base and salt of weak base with strong acid)
NH3(aq) + H2O(l) ⇌ NH4+(aq) + OH-(aq) (equilibrium lies to the left (weak base))
NH4Cl(aq) → NH4+(aq) + Cl-(aq) (soluble salt → fully dissociates)
resovoirs → NH4+ and NH3 to be used in neutralization reactions
addition of acid (H+): NH3(aq) + H+(aq) ⇌ NH4+(aq)
addition of base (OH-): NH4+(aq) + OH-(aq) ⇌ NH3(aq) + H2O(l)
buffer solutions are a mixture containing both an acid and base of a weak conjugate pair
buffer’s acid neutralizes added alkali, buffer’s base neutralizes added acid → pH change is resisted
has to be weak because strong acids/bases dissociate fully → do not exist in equilibrium, so cannot carry out reversible reactions to respond to both added H+ and OH- ions
Buffer composition and pH
pH of a buffer solution depends on the interactions among its components
ex: generic weak acid HA and its salt MA
HA(aq) ⇌ H+(aq) + A-(aq), MA(aq) → M+(aq) + A-(aq)
the dissociation of the weak acid is so small it is considered negligible
[HA]initial = [HA]equilibrium
salt is considered to be fully dissociated into its ions
[MA]initial = [A-]equilibrium
Ka=[H+][A-] / [HA] so [H+] = Ka[HA] / [A-]
[H+] = Ka[HA]initial / [MA]initial
[H+] = Ka[acid] / [salt]
pH = pKa + log10[salt]/[acid]
pOH = pKb + log10[salt]/[base]
pH of a buffer solution can be determined from:
pKa/pKb values of its component acid/base
ratio of initial concentrations of acid/base and salt used to prepare the buffer
when [acid] = [salt], pH=pKa / pOH=pKb
Factors that can influence buffers
dilution
dilution does not change Ka/Kb or ratio of acid/base to salt concentration → does not change pH
buffering capacity → amount of acid/base absorbed without significant changes in pH
decreases when concentrations are decreased with dilution
temperature
affects Ka and Kb so affects pH of the buffer
3.2: Electron transfer reactions
Redox reactions
oxidation is the loss of electrons
reduction is the gain of electrons
oxidation is hydrogen loss and reduction is hydrogen gain
oxidation is an increase in oxidation state and reduction is a decrease in oxidation state
oxidation state for molecules with more than one atom is an average value → can be a non-integer
oxidizing agent → reactant that accepts electrons (brings about the oxidation of the other reactant)
reducing agent → reactant that supplies electrons (brings about reduction and is oxidized)
Redox titrations
similar to acid-base titrations → redox reaction between two reactants to find equivalence point and thus titre
ex: Iodine-thiosulfate solution
several different reactions use an oxidizing agent to convert excess iodine ions into idoine:
2I-(aq) + oxidizing agent → I2(aq) + other products
liberated I2 then reacts with sodium thiosulfate (Na2S2O3)
oxidation: 2S2O32-(aq) → S4O62-(aq) + 2e-
reduction: I2(aq) + 2e- → 2I-(aq)
2S2O32-(aq) + I2(aq) → 2I-(aq) + S4O62-(aq)
starch can be used as an indicator → deep blue in presence of I2
Strength of oxidizing/reducing agents
strength depends on relative tendencies to lose/gain electrons
halogens react by gaining electrons and becoming negative ions → act as oxidizing agents
tendency to gain electrons decreases down the group (F2>I2 in reactivity)
the more reactive it is, the stronger it is as an oxidizing agent
ex: Cl2(aq) + 2KI(aq) → 2KCl(aq) + I2(aq)
K+ does not change, it is a spectator ion → can be disregarded
Cl is more reactive so “wins” over K because it is a stronger oxidizing agent (displaces K)
metals have a tendency to lose electrons and form positive ions → act as reducing agents
more reactive metals are stronger reducing agents
reactivity of group 1 metals increase down the group (K>Na as a reducing agent)
can be confirmed through displacement reactions
if the metal is displaced by another, it is a weaker reducing agent
Oxidation of metals by acids
acids react with metals to form salts and release hydrogen
ex: 2HCl(aq) + Zn(s) → ZnCl2(aq) + H2(g)
removing spectator ions: 2H+(aq) + Zn(s) → Zn2+(aq) + H2(g)
zinc is oxidized (0→+2) → acts as a reducing agent
zinc has the strength to “force” the hydrogen to accept the electrons as it is a strong reducing agent
no reaction occurs when copper is added to a dilute acid
copper lacks the strength to displace hydrogen
hydrogen is less reactive than zinc but more reactive than copper
this is why acids are so corrosive (except to low reactivity metals like Ag and Au)
Comparing voltaic and electrolytic cells
voltaic and electrolytic cells are known as electrochemical cells
voltaic cells convert chemical energy to electrical energy
a spontaneous chemical reaction drives electrons around a circuit
electrolytic cells convert electrical energy to chemical energy
an electric current reverses the normal directions of chemical change → non-spontaneous reactions occur
oxidation occurs at the anode and reduction at the cathode for both cells
anode is the negative terminal in a voltaic cell but the positive terminal is an electrolytic cell
Primary voltaic cells
if the half-reactions in a redox reaction occur at spatially seperated electrodes, the electrons must pass through an external circuit
electric current is produced → voltaic/galvanic cell
chemicals are not renewed during the process → primary cell
battery normally consists of several cells connected together
ex: Zn(s) → Zn2+(aq) + 2e-, Cu2+(aq) + 2e- → Cu(s)
half-cell made by putting a strip of metal into a solution of its ions
in the zinc half-cell, zinc atoms form ions by releasing electrons
makes surface of the metal negatively charged with respect to the solution
negative charge on electrode attracts zinc ions which can gain electrons in the reverse process (Zn(s) ← Zn2+(aq) + 2e-)
at a particular charge seperation, the rates of both processes will equal and equilibrium will be reached
Zn(s) ⇌ Zn2+(aq) + 2e-
position of equilibrium depends on reactivity of metal (the more reactive, the more formation of ions → greater negative charge, more electrons)
when the two half-cells are connected, electrons will have a tendency to flow spontaneously to the less negative half-cell
tendency of electrons to flow is quantified by measuring the potential difference between 2 electrodes
anode → where oxidation occurs (zinc), cathode → where reduction occurs (copper)
cell diagram convention
phase boundary → single vertical line between a solid electrode and an aqueous solution in a half-cell
salt bridge → double vertical line
aqueous solutions of each electrode are placed next to the salt bridge
anode on left, cathode on right (electrons flow from left to right)
Half-cells and voltage
any two half-cells can be connected to make a voltaic cell
direction of electron flow and voltage produced will be determined by the difference in reducing strength
most cases can be judged by the relative position of the metals in the activity series
larger voltage will be produced when there is a greater difference in electrode potentials
electrons flow from more to less reactive metal (ex: Zn→Cu, Zn→Ag)
anode (where electrons are produced) to cathode (where electrons are accepted)
Secondary (rechargeable) cells
involves reversible redox reactions using electrical energy
lithium-ion battery → benefits from lithium’s low density and high reactivity
can store a lot of electrical energy per unit mass
as lithium is reactive, steps must be taken to prevent it from forming an oxide layer (decreases contact with electrolyte)
lithium cathode placed in lattice of a metal oxide (MnO2), lithium anode mixed with graphite
non-aqueous polymer-based electrolyte is used
discharging:
lithium is oxidized at the negative electrode: Li(polymer) → Li+(polymer) + e-
Li+ ion and MnO2 are reduced at the positive electrode: Li+(polymer) + MnO2(s) + e- → LiMnO2(s)
the two half reactions are reversed when the battery is recharged
fuel cells → combustion reactions are redox reactions, can be used to produce an electric current if reactants are physically seperated
hydrogen fuel cell: H2(g) + ½O2(g) → H2O(l) ΔHf = -285.8kJ/mol
Gibbs energy gives maximum energy available for electrical output
H2(g) + ½O2(g) → H2O(l) ΔG = -228.6kJ/mol
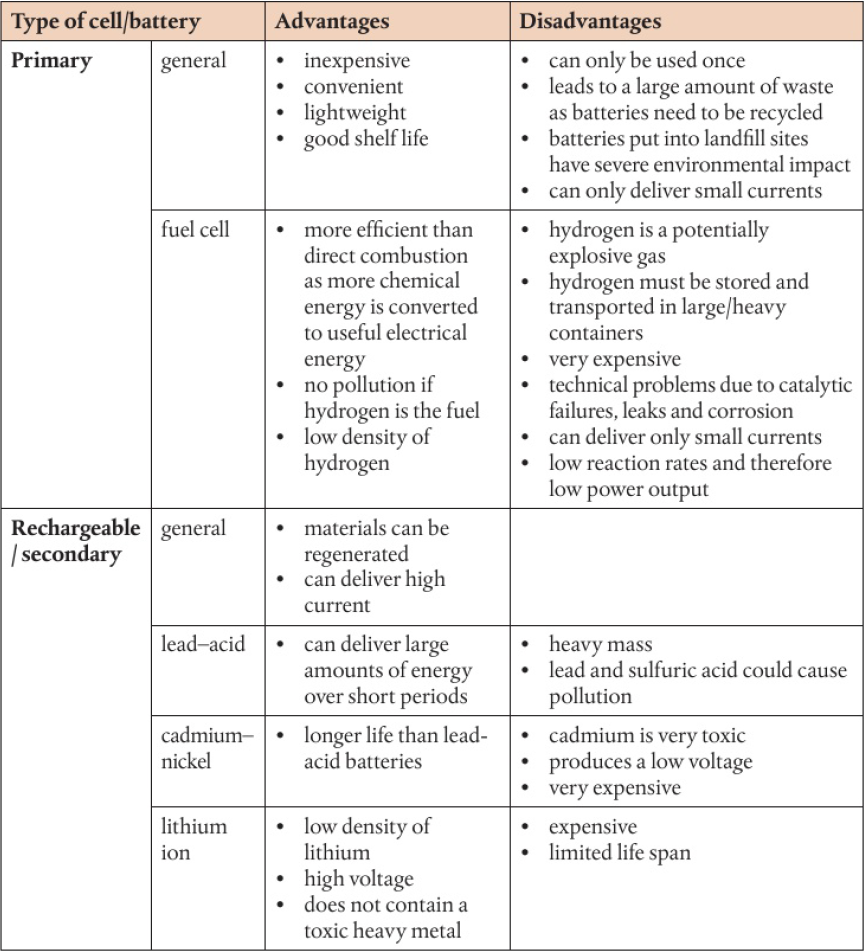
Advantages and disadvantages of fuel cells, primary cells, and secondary cells
Electrolytic cells
voltaic cell takes the energy of a spontaneous redox reaction and harnesses it to generate electrical energy
electrolytic cell reverses this process using an external source of electrical energy to bring about a non-spontaneous redox reaction that would otherwise not take place
electrolysis → process where electricity is used to bring about chemical reactions which break down substances
reactant must contain mobile ions which allow the currrent to pass through the electrodes
electrolyte → liquid, usually a molten ionic compound or an aqueous solution of an ionic compound
as an electric current passes through the electrolyte, redox reactions occur at the electrodes, removing the charges on the ions and forming products that are electrically neutral
ions are discharged during this process
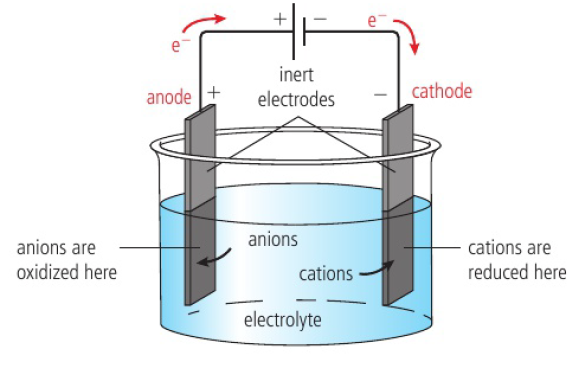
source of electric energy is a battery/DC power source
electrodes are placed in the electrolyte and connected to the power supply
electrodes are made from a conducting substance (metal/graphite)
electrodes are inert → do not take part in the redox reactions
electrodes must not touch each other → “shorts” the circuit, current would bypass the electrolytic cell
electric wires connect electrodes to power supply
ex: spontaneous reaction Na(s) + ½Cl2(g) → NaCl(s)
electrons are transferred from sodium to chlorine
process can be reversed if solid sodium chloride is first converted into the molten state: NaCl(s) → Na+(l) + Cl-(l)
ions are now mobile, ions can act as an electrolyte in an electrochemical cell
battery pushes electrons towards the negative electrode (cathode) → accepted by Na+ ions as they are reduced in the electrolyte
Na+(l) + e- → Na(l)
electrons are released at the positive terminal (anode) → chloride ions are oxidized
2Cl-(l) → Cl2(g) + 2e-
chemical reactions occuring at each electrode remove ions from electrolyte
compound has been split into its constituent ions → electrolysis has occured
Oxidation of functional groups in organic compounds
oxidation of alcohols
commonly the laboratory process uses acidified potassium dichromate (VI) → bright orange solution, reduced to green Cr3+(aq) as alcohol is oxidized on heating
when writing equations for these reactions, it is convenient to show the oxidizing agent simply as “+[O]”
primary alcohols → 2 H atoms attached to the carbon with the hydroxyl group, can be oxidized in a two-step reaction
removal of the first H atom leads to the formation of the aldehyde
the second H atom is removed under prolonged reaction, carboxylic acid is formed
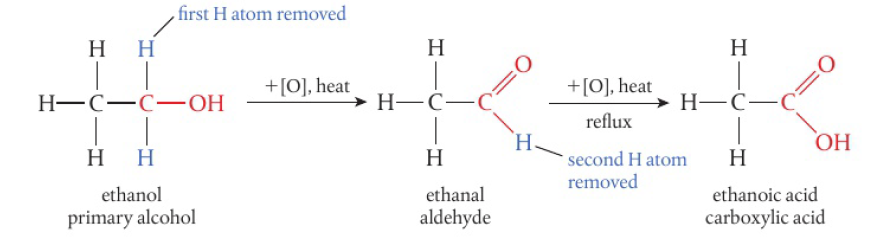
secondary alcohols → only 1 H attached to the carbon bonded to the hydroxyl group → one-step reaction
tertiary alcohols have no H atoms attached to the carbon bonded to the hydroxyl group → not readily oxidized without breaking the carbon chain
carbon skeleton of the molecule is broken in combustion
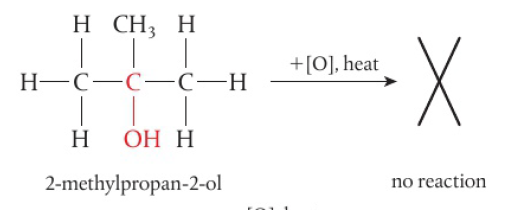
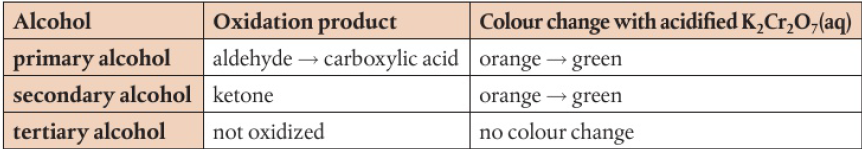
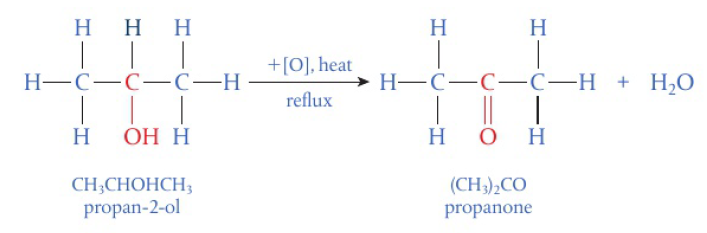
Reduction of functional groups in organic compounds
reduction of carbonyl compounds
redox reactions are more conveniently thought of as gain and loss of oxygen/hydrogen instead of electrons
oxidation of secondary alcohols to ketones involved the removal of 2 H atoms
this process can be reversed: ketones can be reduced to secondary alcohols by adding two H atoms
carbonyl group is polar with a partial positive charge on the carbon atom → can be attacked by species with a lone pair of electrons
hydride ion (H-) acts as a nucleophile on the electron-deficient carbonyl carbon
carboxylic acids can be reduced to aldehydes
reactions can be shown using [+H] like [+O] for oxidation

conditions: heat with LiAlH4 in dry ether, reaction cannotbe stopped at the aldehyde → reacts too readily with LiAlH4
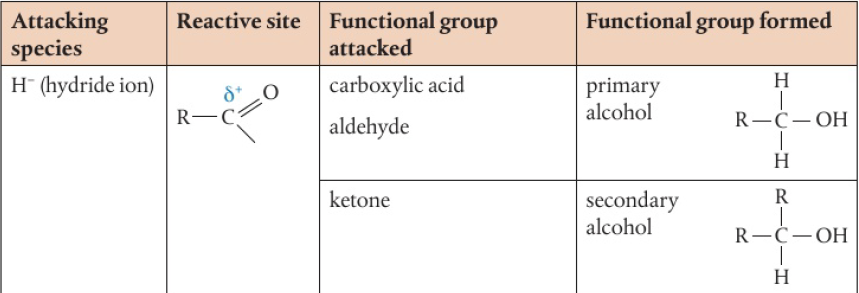
Reduction of unsaturated compounds
alkenes and alkynes are unsaturated hydrocarbons
they can undergo addition reactions so form a range of different saturated products
alkenes generally react by electrophilic addition
alkenes can be reduced to alkanes by reacting with hydrogen
CnH2n + H2 → CnH2n+2
alkynes are similarly reduces to alkenes and alkanes through addition of hydrogen
The standard hydrogen electrode
voltaic cell generates a potential difference → electromotive force (emf)
electrons tend to flow from more negative half-cell to more positive half-cell → potential generated is cell/electrode potential (E)
measured using a voltmeter: depends on difference in the tendencies of these two half-cells to undergo reduction
it is convenient to compare relative reducing power of different half-cells with the same fixed reference point
standard hydrogen electrode (SHE) → baseline for measuring and comparing electrode potentials of other half-cells
platinum is used as conducting metal → inert and will not ionize, catalyzes the reduction of H+(aq) ions
surface of metal is coated with very finely divided platinum (platinum black, “platinized platinum”) which increases the large surface area → helps in adsorption of hydrogen gas
increases rate of reaction of both forward and backward reactions
concentration of H+(aq) is 1mol/dm³ (pH=0), pressure of H2(g) is 100kPa at 298K
as the electrode is immersed in the acidic solution, an equilibrium is set up between the adsorbed layer of H2(g) and H+(aq) ions
2H+(aq) + 2e- ⇌ H2(g)
H+(aq) ions are reduced in the forward reactions, H2(g) molecules are oxidized in the backward reaction
position of equilibrium depends on electrode potential of the other half-cell to which the SHE is connected to in a circuit
hydrogen half-cell is assigned an electrode potential of zero volts (0V)
Measuring standard electrode potentials
standard conditions (standard half-cells):
all solutions must have a concentration of 1.0 mol/dm³
all gases must be at pressure 100kPa
all substances used must be pure
temperature is 298K
if the half-cell does not include a solid metal, platinum is used as the electrode
when the SHE is connected to another standard half-cell by an external circuit, the emf generated is the standard electrode potential (EꝊ)
ex: Cu2+(aq) / Cu(s) half-cell: +0.34V
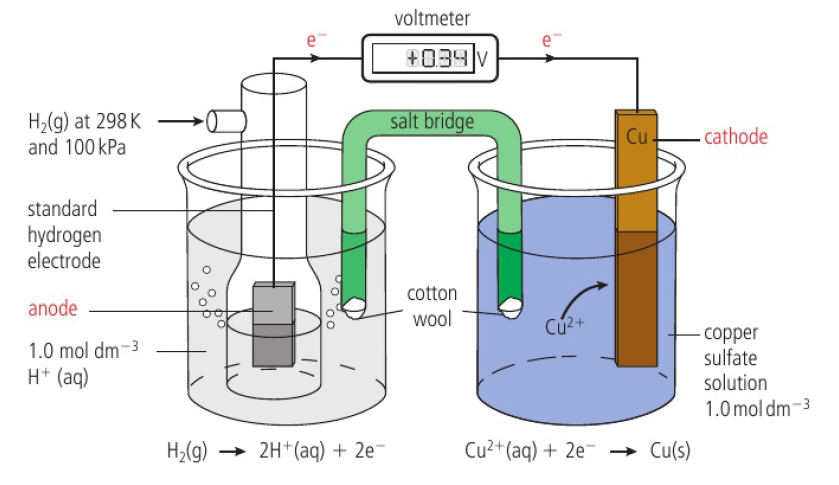
positive value for E shows that electrons tend to flow from hydrogen half-cell to copper half-cell → electrons are produced by oxidation of H2(g) and used to reduce Cu2+(aq) ions
hydrogen half-cell is the anode, copper half-cell is the cathode
positive cell potential → Cu2+ ions have a higher tendency to be reduced than H+ ions → low reactivity of copper
standard electron potential is always given for the reduction reaction
standard electrode potential values can be known as standard reduction potentials
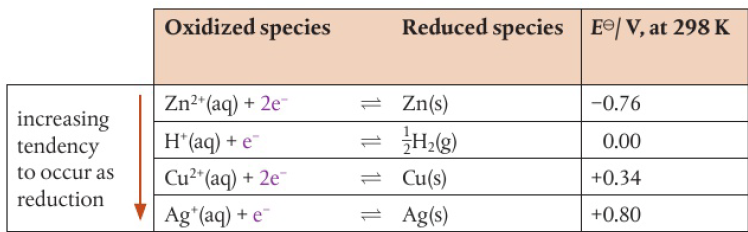
EꝊ values do not depend on total number of electrons (is not scaled up/down with stoichometry of equation for a reaction)
the more positive the EꝊ value for a half-cell, the more readily it is reduced
electrons always flow through the external circuit in a voltaic cell from the half-cell with the more negative standard electrode potential to the half-cell with the more positive standard electrode potential
EꝊcell = EꝊhalf-cell where reduction occurs - EꝊhalf-cell where oxidation occurs
EꝊ values must be the reduction potentials as supplied in data tables
the voltaic cell will always run in the direction that gives a positive value for the EꝊcell
if EꝊcell is positive, the reaction is spontaneous
if EꝊcell is negative, the reaction is non-spontaneous and reverse reaction is spontaneous
metals with the most negative EꝊ values are the strongest reducing agents
non-metals with the most positive EꝊ values are the strongest oxidizing agents
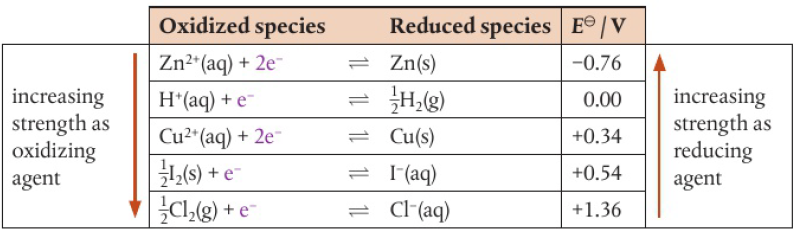
Electrode potentials and Gibbs energy changes
ΔGꝊ = -nFEꝊcell
n = number of moles of electrons transferred in the reaction
F = the charge carried by 1 mol of electrons (Faraday constant, 96500C/mol)
reaction is spontaneous when:
EꝊcell is positive
ΔGꝊ is negative
the more positive the value of EꝊcell, the more energetically favourable the reaction
voltmeter can be considered an indirect measure of Gibbs energy change and electrode potential
electrons always tend to flow towards the half-cell with the highest EꝊ value
Electrolysis of aqueous solutions
predicting the products at the electrodes in electrolysis of aqueous solutions is more difficult because the water present can also be oxidized/reduced
ex: electrolysis of sodium chloride
cathode: sodium cannot be produced as it would react immediately with water to produce hydrogen
Na+ + e- → Na
Na + H2O → Na+ + OH- + ½H2
water is preferentially reduced to hydrogen
anode: water can be oxidized to oxygen
2H2O(l) → 4H+(aq) + O2(g) + 4e-
when a solute M+A- is in an aqueous solution, there is more than one redox reaction possible at each electrode
anode: either A- or H2O can be oxidized
cathode: either M+ or H2O can be reduced
discharge of an ion at the electrode is known as selective discharge
in the example, water is reduced in preference to sodium because sodium is highly reactive → negative electrode potential
outcome can be determined generally:
relative EꝊ values of ions compared to water
relative concentrations of ions in the electrolyte
nature of the electrodes
identify all ions present in electrolyte, determine which will migrate to which electrode
write the half-equation for the possible reactions at each electrode
the half-reaction with the higher electrode potential is the oxidation reaction at the cathode (higher electrode potential = gains electrons most readily)
the half-reaction with the lower electrode potential is the reduction reaction at the anode (lower electrode potential = loses electrons most readily)
to deduce overall reaction, balance the electrons lost and gained at the anode and cathode then add the two half-equations
consider what changes would be observed in the cell as a result of the redox reactions (colour changes, precipitation of solid, gas discharge, pH changes)
Electrolysis examples
electrolysis of water
water is not a good conductor of electricity, so some sulfuric acid or sodium hydroxide is usually added
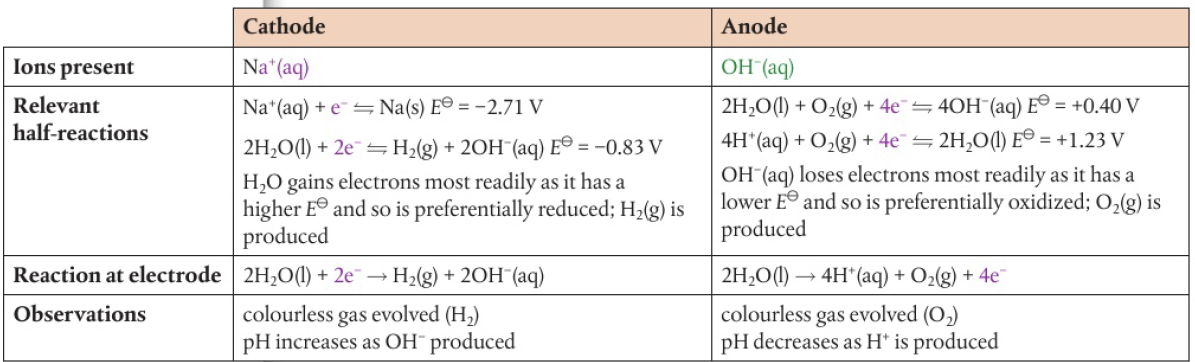
electrolysis of NaCl(aq) (brine)
electrolysis of CuSO4(aq) with carbon(graphite) or other ‘inert’ electrodes
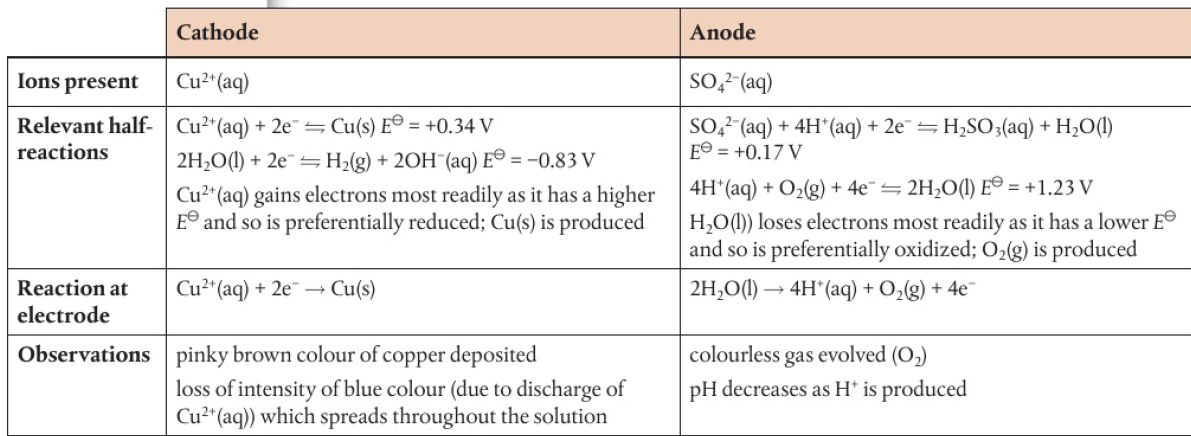
electrolysis of CuSO4(aq) with copper electrodes
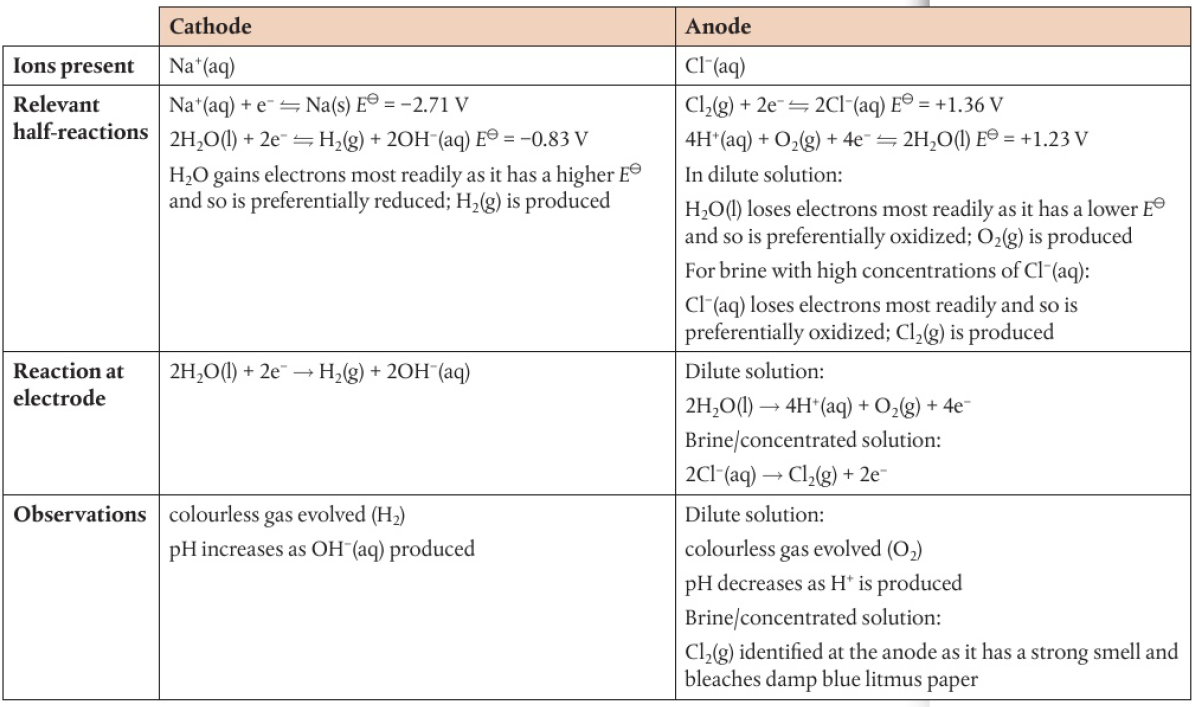
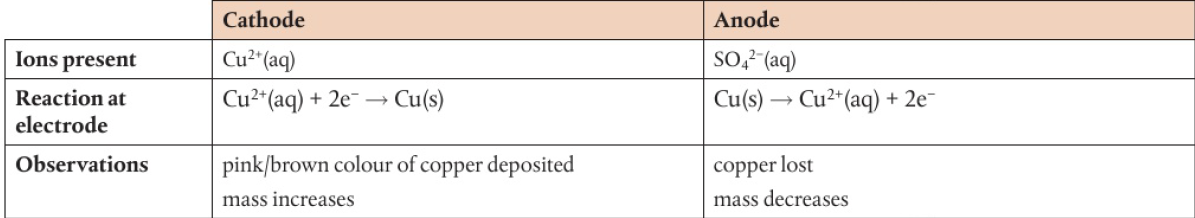
Electroplating
process of using electrolysis to deposit a layer of metal on top of another metal or other conductive object
electrolytic cell used for electroplating has following features:
object to be plated forms the cathode
electrolyte contains the metal ions which are to coat the object
anode is sometimes made of the same plating metal → as it is oxidized, it replenishes the ions in the electrolyte which are discharged at the cathode
reduction of metal ions at the cathode leads to deposition on its surface
process can be controlled by altering current and time accordingly to thickness of metal layer desired
purposes of electroplating:
decorative
corrosion control: metal covering the surface is oxidized preferentially to metal underneath
improvement of function
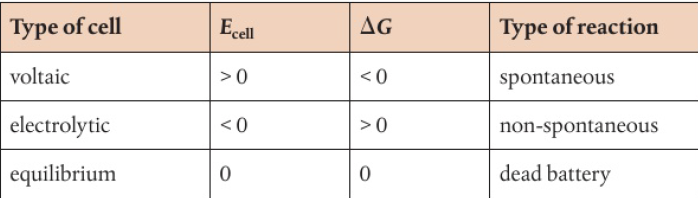
3.3: Electron sharing reactions
Radicals
radical → chemical species that contains an unpaired electron
presence of unpaired electron results in high enthalpy
radicals are very reactive → energetically favourable for them to take an electron from other species or combine with other radicals to form a covalent bond (both results in products with lower enthalpy)
radicals have short lifetimes (reactivity) and do not exist for very long
exception: radicals formed in the upper atmosphere, can persist for a significant length of time (low probability of collision)
radicals can be atoms, molecules, or ions
Homolytic fission
homolytic fission → covalent bond breaks to form two radicals
breaking bonds is endothermic → homolytic fission requires energy
amount of energy depends on strength of covalent bond
weaker bonds, sufficient energy by heating compound → thermolytic fission
stronger bonds, absorption of UV light → photolytic fission
termination reactions → two radicals combine, sharing unpaired electrons and forming a covalent bond (reverse process of homolytic fission)
chlorofluorocarbons (CFCs) → when in the stratosphere, the UV radiation breaks them down, releasing chlorine atoms (reactive radicals)
chlorine radicals released then catalyse the decomposition of ozone
Cl(g) + O3(g) → O2(g) + ClO(g)
ClO(g) + O(g) → O2(g) + Cl(g)
Radical substitution reactions of alkanes
alkanes are relatively unreactive compounds → strong C-C and C-H bonds, non-polar, high activation energies
at regular temperatures, reactant molecules lack sufficient KE to overcome activation energy, so alkanes are kinetically stable
alkane reactants have a higher enthalpy (exothermic) and are thermodynamically unstable compared to the products
alkanes are saturated, so undergo substitution reactions (another reactant takes the place of a hydrogen atom in the alkane)
products between alkane and halogen: halogenoalkane and hydrogen halide
once formed, radicals will start a chain reaction → mixture of products including halogenoalkane is formed
initiation
formation of radical species, which can then react with the alkane
propagation
radicals formed in initiation stage react with other species present to form new radicals
there are many possible propagation steps, all of which allow the reaction to continue (chain reaction)
termination
radicals are removed when they reach with each other and pair up electrons to form a covalent bond (many possible termination steps)
3.4: Electron-pair sharing reactions
Nucleophiles
nucleophile → reactant that forms a covalent bond to its reaction partner (the electrophile) by donating both bonding electrons
tend to be electron rich, having 1+ lone pairs of electrons and may also carry a negative charge
ex: H2O, NH3, alcohols, amines, OH-, F-, Cl-
forms a coordination bond
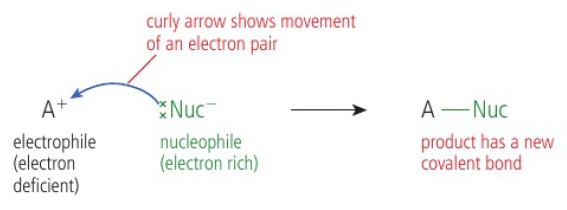
Nucleophilic substitution reactions
halogenoalkanes are polar compounds → halogen atom is more electronegative than carbon, exerts a stronger pull on shared electrons
halogen gains a partial negative charge and carbon gains a partial positive charge and is said to be electron deficient
nucleophiles are attracted to electron-deficient species
leads to reactions which substitution of the halogen occurs → nucleophilic substitution
CH3Cl + OH- → CH3OH + Cl-
overall reaction between a nucleophile and an electron-deficient substrate molecule (like a halogenoalkane):
for the nucleophile to donate an electron pair to the substrate molecule and form a new bond, another bond must break
leaving group → species that breaks away from the substrate molecule
halogens generally make good leaving groups as they form relatively weak bonds with carbon
halogens have a higher electronegativity meaning it draws the bonded electrons towards itself, making the carbon atom electron deficient → susceptible to nucleophilic attack
the bond that forms and the bond that breaks must both involve the carbon atom that is bonded to the leaving group
if nucleophile is a neutral species (ex: water), the initial product formed will be positively charged
subsequently deprotonates and loses an H+ to give a neutral product
if the leaving group was a halide ion, the H+ and halide ion combine to form a hydrogen halide
overall equation: H2O + RX → ROH + HX
Heterolytic fission
bond-breaking in nucleophilic substitution reactions involves heterolytic fission → a covalent bond breaks such that one of the products gains both of the shared electrons
formation of oppositely charged ions (the more electronegative atom will gain the negative charge)
ex: Cl-Cl → Cl+ + Cl-
ex: H-Cl → H+ + Cl-
bond breakage occurs through movement of an electron pair (use double-headed curly arrows instead)
opposite process: nucleophile donates a pair of electrons to an electrophile, forming a coordination bond
Electrophiles
electrophile → reactant that forms a covalent bond to its reaction partner (the nucleophile) by accepting both electrons
tend to be electron deficient or contain an electron-deficient region
often have a positive charge / partial positive charge
ex: hydrogen halides, halogens, halogenoalkanes, H+, NO2+, CH3+, carbocations
Electrophilic addition of alkenes
alkenes are unsaturated hydrocarbons
more reactive than alkanes: double bond has a high electron density which makes it attractive to electrophiles
reaction occurs at site of double bond (pi bond is selectively broken) which creates two new bonding positions on the carbon atoms
enables addition reactions with electrophiles (range of different saturated products)
addition of water (hydration)
converts alkene into an alcohol
water is a poor electrophile, so requires a strong acid catalyst (ex: concentrated sulfuric acid)
ex: CH2CH2 →(H2O, H2SO4) CH3CH2OH
addition of halogens
halogens react with alkenes to produce dihalogeno compounds
reactions happen quickly at room temperature, accompanied by loss of color of reacting halogen
addition of halogen halides
halogen halides (ex: HCl, HBr) react with alkenes to produce halogenoalkanes
reactions take place rapidly in solution at room temperature
reactivity is in order of the decreasing strength of the hydrogen halide bond down group 17
HI (weakest bond) reacts most readily
Lewis acids and Lewis bases
a Lewis acid is a lone pair acceptor
all Bronsted-Lowry acids are Lewis acids, but Lewis acids also include any species capable of accepting a lone pair of electrons (molecules with an incomplete valence shell)
a Lewis base is a lone pair donor
Lewis bases and Bronsted-Lowry bases are the same group of compounds: must have a lone pair of electrons
Lewis acid-base reactions result in the formation of a covalent bond (always coordination bond, both electrons come from the base)
once a coordination bond is formed, it is no different from a regular covalent bond
same properties (bond length, bond strength, chemical reactivity)
Lewis acids are electrophiles, Lewis bases are nucleophiles
nucleophile (‘likes nucleus’) is an electron-rich species that donates a lone pair to another reactant to form a new covalent bond
an electrophile (‘likes electrons’) is an electron-deficient species that accepts a lone pair from another reactant to form a new covalent bond
Complex ions
transition metals can act as Lewis acids and accept lone pairs of electrons when they bond with ligands to form complex ions
ligand donates both electrons, coordination bond is formed
ligands are therefore Lewis bases
ligands can be neutral molecules/anions but must always contain at least one lone pair of electrons
ex: H2O, NH3, CO, I-, Br-, F-, OH-
transition metals typically bond to more than one ligand when they form complex ions
number of coordination bonds between ligands and central ion in the complex ion → coordination number
charge on complex ion is dependent on:
charge on central transition metal ion
charge on ligands
coordination number
Nucleophilic substitution mechanisms
shorthand notation: SN (substitution nucleophilic) is used to describe this type of reaction
polar carbon-halogen bond in halogenoalkanes means the carbon atom is electron deficient, thus attacked by nucleophiles (like OH-)
during these reactions, the carbon-halogen bond breaks and the halogen atom is released as a negative ion (halide)
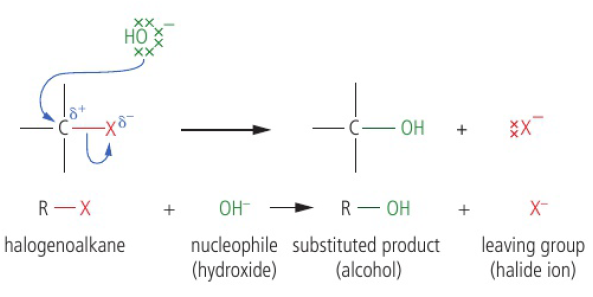
exact mechanism of reaction depends on whether the halogenoalkane is primary, secondary, or tertiary
primary halogenoalkanes react via SN2
primary halogenoalkanes have at least 2 hydrogen atoms attached to the carbon of the carbon-halogen bond
ex: chloroethane (C2H5Cl)
overall reaction with NaOH:
C2H5Cl + OH- → C2H5OH + Cl-
as hydrogen atoms are small, the carbon atom is relatively open to attack by nucleophile
unstable transition state is formed in which carbon is weakly bonded to both halogen and nucleophile
carbon-halogen bond then breaks heterolytically (electrons go to the same species)
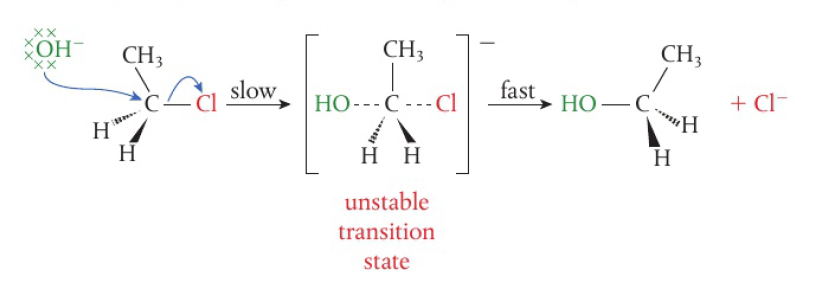
effectively a one-step concerted reaction with an unstable transition state
mechanism is dependent on concentration of both the halogenoalkane and the hydroxide ion, so it is a bimolecular reaction (second order)
therefore the mechanism is fully described by SN2: substitution nucleophilic 2nd order
nucleophile attacks the electrophilic carbon on the opposite side from the leaving group, causing an inversion of the arrangement of atoms around the carbon atom
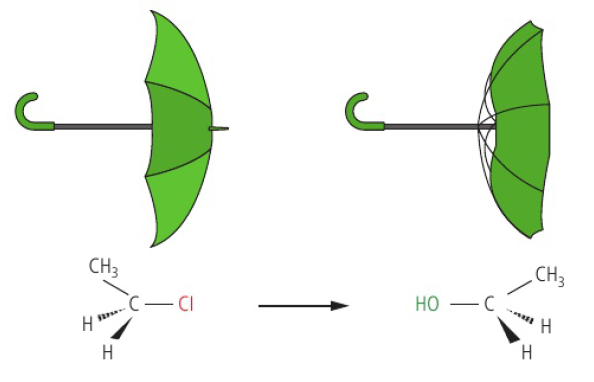
configurations are a result of different spatial arrangements around tetrahedrally bonded carbon atoms
SN2 mechanism → stereospecific, three-dimensional arrangement of reactants determines three-dimensional configuration of products
bond formation comes before bond cleavage (breaking) in transition state, so stereochemistry of the carbon attacked is not lost
optically active reactants will react via SN2 to give optically active products (stereospecific)
tertiary halogenoalkanes react with SN1
tertiary halogenoalkanes have 3 alkyl groups attached to the carbon of the carbon-halogen bond
ex: 2-chloro-2-methylpropane
overall reaction: (CH3)3CCl + OH- → (CH3)3COH + Cl-
presence of 3 alkyl groups causes steric hindrance → bulky groups make it difficult for an incoming group to attack the carbon atom
SN2: nucleophile attacks electron-deficient carbon, SN1: heterolytic fission of the carbon-halogen bond and halide ion leaving
creates a carbocation intermediate → positive charge centered on carbon
intermediate is then attacked by the nucleophile in the second step of the reaction, leading to a bond being formed
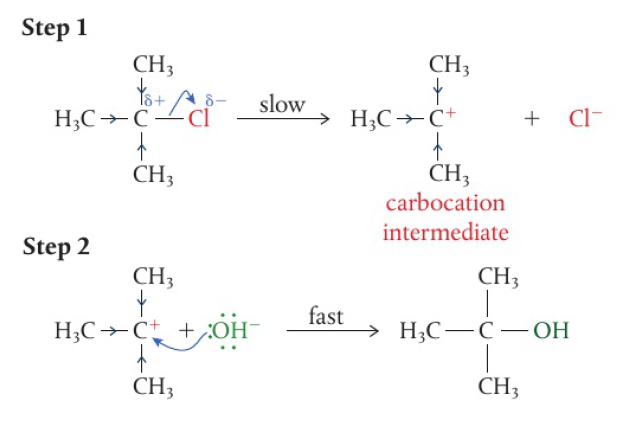
slow (rate-determining) step is determined by concentration of only the halogenoalkane → unimolecular reaction, first order
SN1: substitution nucleophilic 1st order
carbocation intermediate has a planar shape → nucleophile can attack from any position
SN1 is not stereospecific, optically active reactants react via SN1 to form racemic mixtures (50:50 mixture of 2 enantiomers)
mechanism is favoured by tertiary halogenoalkanes because carbocation is stabilized by presence of 3 alkyl groups → each alkyl group has an electron-donating/positive inductive effect
stabilizing effect helps carbocation to persist long enough for second step to occur
secondary halogenoalkanes
it is not possible to be precise about the mechanism of nucleophilic substitution in secondary halogenoalkanes
usually undergo a mixture of both SN1 and SN2 mechanisms
influence of leaving group (halogen)
rate of nucleophilic reactions for halogenoalkanes (with same carbon chain length and branching) is: iodo>bromo>chloro>fluoro
the weaker the bond, the easier it is to break and the better the halide is as a leaving group
SN1 and SN2 mechanisms have different rate expressions + reaction profiles
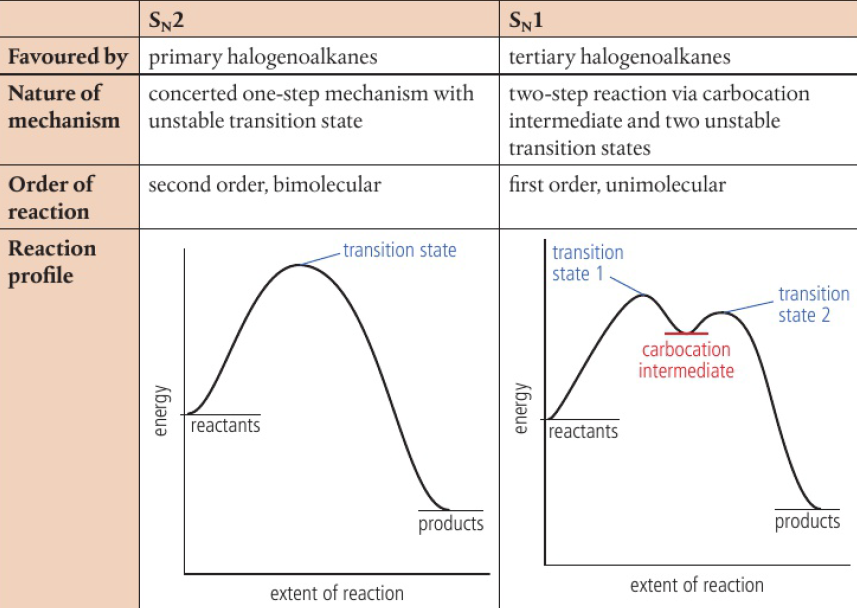
Electrophilic addition reaction mechanism
electrophilic addition reactions of alkenes require the breaking of a pi bond
carbon atoms of carbon-carbon double bond are sp2 hybridized, forming a planar triangular shape with bond angle 120
fairly open structure, relatively easy for incoming groups to attack
pi bond represents an area of electron density above and below the plane of bond axis
pi bond is attractive to electrophiles (area of electron density)
electrons in pi bond are less closely associated with the nuclei so it is a weaker bond than a sigma bond, so breaks more easily
when pi bond breaks, reactants attach at each carbon atom through formation of 2 sigma bonds
reactions between reagants and alkenes are therefore known as electrophilic addition reactions
addition reactions of alkenes involve a two-step mechanism
ex: ethene + bromine → 1,2-dibromoethane
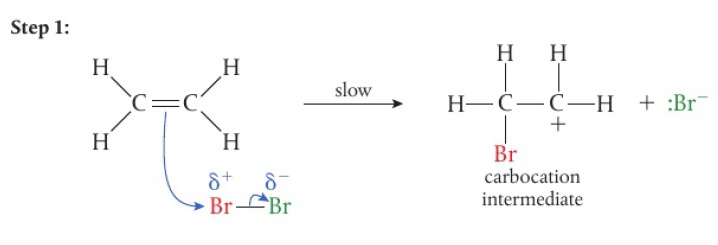
bromine is neutral, but becomes polarized due to electron repulsion as it approaches the electron-rich region of the alkene and acts as an electrophile with an electron-deficient bromine atom
electrons in pi bond of alkene are attracted to electrophilic bromine so causes Br-Br bond to break heterolytically
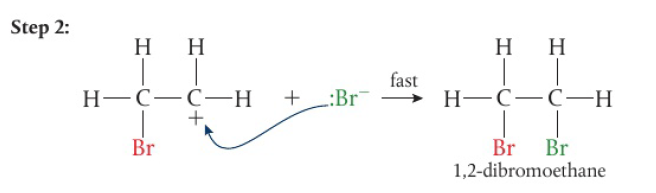
this step is slow, resulting in an unstable carbocation intermediate
unstable carbocation reacts rapidly with negative bromine ion (Br-), acting as a nucleophile and attacking the positively charged carbon in the carbocation, forming a covalent bond
ex: water
water is a weak electrophile so does not directly undergo addition reactions with alkenes, but with strong acids (catalysts), H3O+ can act as the electrophile
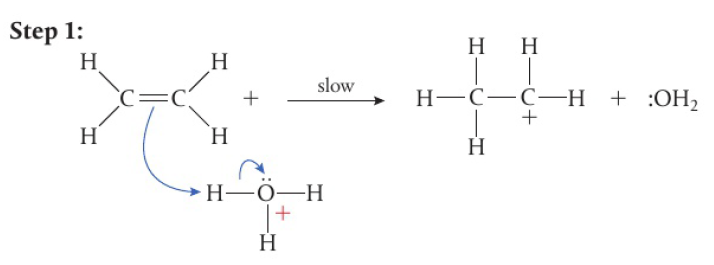
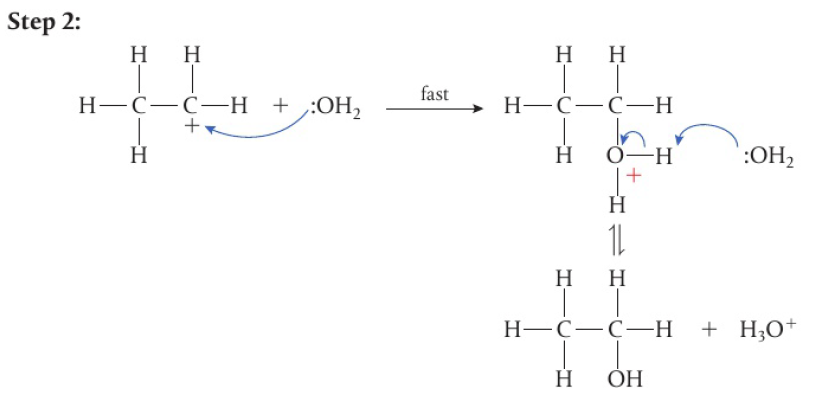
Addition of hydrogen halides to unsymmetrical alkenes
addition reaction of an unsymmetrical alkene has two potential products but only one is formed exclusively
ex: propene + hydrogen bromide
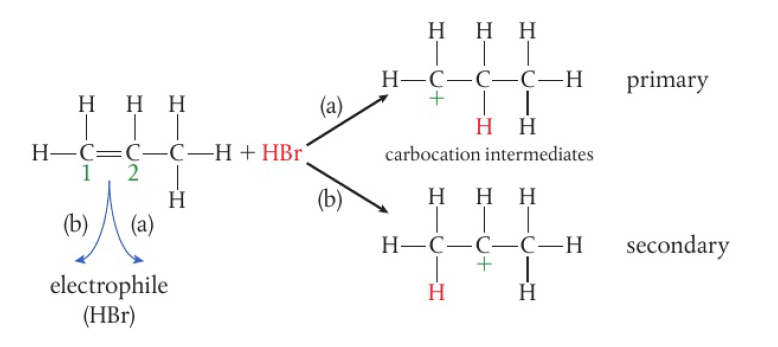
two different carbocation intermediates, one primary (a) and the other secondary (b)
alkyl groups around a carbocation stabilize it somewhat due to their positive inductive effects (push electron density away from themselves so lessen the density of the positive charge)
(a) only stabilized by one, whereas (b) is stabilized by two, so (b) has greater stabilization
more stable carbocation will be more likely to persist, so 2-bromopropane is the main product of the reaction
Markovnikov’s rule: the hydrogen atom will attach to the carbon that is already bonded to the greater number of hydrogens
the more electropositive part of the reacting species bonds to the least highly substituted carbon atom in the alkene (one with smaller number of carbons attached)
Electrophilic substitution of benzene
benzene molecules contain 3 pi bonds and pi electrons are delocalized due to resonance → minimizes repulsion between pi electrons so benzene molecule is stabilized compared to regular alkenes
despite high unsaturation, benzene does not behave like alkenes in its characteristic reactions
unusual and highly stable aromatic ring determines that substitution, not addition, is its favoured reaction
benzene is attractive to electrophiles because its ring is a region of high electron density
delocalized cloud of pi electrons attracts electron-deficient species and can form a new bond as a hydrogen atom is lost → electrophilic substitution
reaction has high activation energy, proceeds rather slowly (first step)
electron pair from benzene is attracted to electrophile leading to a distruption of the delocalized pi system
unstable carbocation intermediate that forms has both entering atom/group and leaving hydrogen temporarily bonded to the ring

incomplete circle inside the ring → delocalized pi system disrupted (positive charge distributed over carbon atoms in the ring)
heterolytic breaking of C-H bond results in the two electrons moving to regenerate the aromatic ring and thus formation of a neutral substitution product
product is more stable
nitration of benzene
substitution of -H by -NO2 to form nitrobenzene
electrophile: NO2+ (nitronium ion), generated using a nitrating mixture (mixture of concentrated nitric acid and concentrated sulfuric acid at 50C)
sulfuric acid (stronger acid) protonates nitric acid (nitric acid acts as a Bronsted-Lowry base) and forms (with loss of water) the electrophile
NO2+ is a strong electrophile → attracts pi electrons of benzene ring to form carbocation intermediate
loss of a proton from intermediate leads to reformation of arene ring in the product nitrobenzene