CHEM 153L Midterm+Final Learning Objectives
Discriminate between biofuels and fossil fuels.
- Fossil fuels- a fuel such as coal, oil, or natural gas formed in the earth from plant or animal remains; a finite resource. Huge contributor to carbon emissions
- Biofuels- generated by treating fresh biomass. The energy is stored as chemical energy in the main storage forms starch or sucrose previously converted by light energy through photosynthesis; from these the microbes/plants/animals can convert this stored chemical energy into biofuels via transesterfication or fermentation. They are easy to regenerate.
Understand the strategies on how biofuels can be produced. How do the generations of biofuels differ from each other?
- First generation uses food crops for feedstock. This uses agricultural resources and creates competition within the food industry and will cause food prices to increase.
- Second generation uses waste plant material as feedstock -> no competition with food production.
- Third generation uses algal cells grown in bioreactors. This does not compete with the agricultural resources and utilizes less land. Algae doesn’t need a feedstock, just sunlight and CO2 because they can perform photosynthesis (autotroph)
- Oils can undergo transesterificationby reacting with alcohol to form fatty acids
- Carbohydrates have to go through glycolysis and then we can utilize the carbon skeletons to make biofuels
What are typical carbohydrate storage forms of energy?
- Sucrose- composed of glucose and fructose
- Starch- made up of glucose; 1,4 glycosidic bonds also 1,6 glycosidic bonds create branching (amylopectin)
- Glycogen- glucose storage w/ lots of 1,6 glycosidic bonds
What kind of molecule is isobutanol? Draw an isobutanol molecule
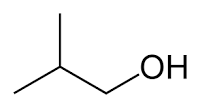 Isobutanol is an alcohol
Isobutanol is an alcohol
What is the advantage of isobutanol as a biofuel molecule over ethanol?
- Isobutanol has a higher energy density and is less miscible with water which makes it easier for transport as addition to fuel. Ethanol, on the other hand, is miscible with water, so if you transport gasoline with ethanol and water, some of that ethanol dissociates and converts to water. Thus you lose some fuel which is a loss of money
How can microorganisms be utilized to generate ethanol?
- Alcoholic fermentation leads to ethanol production. This is an anaerobic process, but bacteria can be genetically engineered to perform fermentation in aerobic conditions. Fermentation not only produces ethanol but it also regenerates NAD+ for glycolysis (usually done by the TCC)
Recapitulate the isobutanol production pathway used in this course?
- The overexpression of isobutanol production pathway is done by hitchhiking on the valine biosynthesis route
- From glycolysis, pyruvate is produced. Two pyruvate molecules are fused together (while CO2 leaves). We utilize acetolactate synthase and this forms 2-acetolactate.
- SLIDE 21 OF LEC 1
Relate between the transmittance and absorbance of a sample
- The higher the concentration of the sample, the higher its absorbance
- Transmittance decrease exponentially when the sample concentration increases
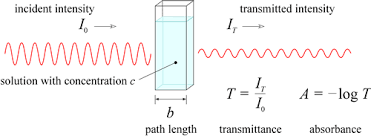
Recall the Beer-Lambert law

Predict the change in absorbance, when the parameters are changed
- Absorbance correlates linearly with change in concentration
- Transmittance and concentration have an decreasing exponential relationship while absorbance and concentration have an increasing linear relationship
Explain why pathlength of samples may vary in our lab
- When using a cuvette spectrophotometer, the light is directed horizontally so the pathlength will be the same for all samples so long as the same cuvette is used
- Using a microplate reader, the light is directed through the well top to bottom so it is dependent on the volume of the sample loaded into the well.
Discriminate between reduced and oxidized forms of nicotinamide adenine dinucleotide and their phosphorylated counterparts
- NAD+ does not absorb light at 340 nm while NADH does. They both absorb at 260 nm
- NADP+ is NAD+ with a third phosphate group
- NAD(P)+ to NAD(P)H is reduction
Discriminate between Accuracy and Precision:
Accuracy: how close the measurement is to the reference data. Calculated using percent error
Precision: repeating a measurement several times and all the data is very close together. Calculated using standard deviation
Write down the reaction of alcohol dehydrogenase
Ethanol + NAD+ -> acetaldehyde + NADH
Describe the hydride transfer that happens during the alcohol dehydrogenase reaction
Zinc in the active site of the alcohol and the NAD+ abstracts a hydride ion from the ethanol
Describe the Michaelis-Menten equation
- One assumption is that [ES] stays constant since the rate of formation is the same as the rate of consumption of [ES]
- When [substrate] is much below Km, rate will be low and therefore more dependent on the [substrate]
- When [substrate] is at Km, then the reaction rate is at Vmax
- When [substrate] is much above Km, the rate will be close to Vmax
Michaelis-Menten Graph
- At lower substrate concentrations (initial rate of reaction), the reaction is first order meaning that the rate of reaction depends more heavily on the concentration of the substrate
- At higher substrate concentrations, the reaction is zero order meaning that the rate of reaction is approaching Vmax and the reaction rate is not dependent on the concentration of substrate, but rather on the enzyme concentration
- Is a plot of substrate concentrations vs reaction velocities

Define the Michaelis-Menten constant Km
- Km is the substrate concentration at which half of active sites have substrate bound and catalyze the reaction; it is a measurement of an enzyme’s affinity for its substrate
- The lower the Km, the higher the affinity of the enzyme for its substrate
How will we analyze/estimate/calculate Vmax and Km from the lab?
- We will use the Michaelis-Menten equation, Lineweaver-Burk graph, and analysis of values with non-linear regression analysis (least squares analysis)
- We must estimate Vmax and Km from the Michaelis-Menten graph
- Lineweaver Burk is double reciprocally analyzed data the x-intercept negative reciprocal represents Km and the y-intercept reciprocal represents Vmax
- The third approach is using raw data and non-linear least squares analysis to get an exact estimate of Vmax and Km
- You first must estimate Km and Vmax from the dataset by looking at the michaelis-menten graph
- Then using the michaelis-menten equation, we can calculate the expected rate at each substrate concentration
- Now take the least square difference
Specific Enzyme Activity
- Measures how many µmol of product per minute that 1 mg of enzyme produce under optimal conditions
Turnover Number Kcat
- Measures how many µmol of product per minute that 1 µmol of enzyme can produce under optimal conditions
- Can be calculated using Vmax/total enzyme concentration
Why are bacterial cells beneficial for protein overexpression?
- Bacterial cells are cheap to cultivate in large numbers and small space
Which growth phase will bacterial cells be most efficient in expressing foreign proteins?
- Growth conditions are optimal in the log (exponential) phase with the most ribosomes per cell
- Best metabolic state to mass produce protein of pinterest
What factors affect induction efficiency?
- [inducer], temperature, duration of IPTG treatment
What is bacterial transformation?
- The uptake of foreign DNA by bacteria
What are the essential components of a plasmid for protein expression?
- Needs amp resistance gene which serves as the selectable marker (we use B-lactamase) which allows us to select for only those bacteria to grow that have the plasmid; all bacteria without the plasmid will die
- Needs origin of replication which is “F1 ori” (rich in AT bonds) to start the process; without, the bacterium cannot replicate/copy the plasmid
- LacI gene encodes for the lac repressor which interacts with the lac operator and keeps the expression of the his6-yqhd off
- T7 Promoter which is recognized by the T7 RNA polymerase
- Multiple cloning site which allows for the insertion of foreign DNA without disrupting the rest of the plasmid
- lacO operator which is where the lac repressor binds and prohibits the RNA polymerase from binding to the promoter and initiating transcription
- Transcription termination sequence for RNA polymerase to stop transcribing
- Ribosome binding sequence
- Histidine6 tag (later purified by affinity chromatography)
What is the natural function of the lac-operon?
- Digestion of lactose through the production of lactase
What is the importance of normalization for cell volume using OD600?
- It ensures that each lane has the same number of cells so that the only thing that influences the protein movement is the amount of protein
What is an SDS page used for?
- It is used to separate proteins based on their molecular weight
- Sodium dodecyl-sulfate polyacrylamide gel electrophoresis
- Used an electric field
What is the role of using the detergent SDS with simultaneous boiling of the sample?
- The addition of SDS with simultaneous boiling of the sample will disrupt non-covalent bonds that stabilize secondary, tertiary, and quaternary structures (BUT DO NOT DISRUPT PRIMARY PROTEIN STRUCTURE WHICH IS AA SEQUENCE)
- It will also cover the proteins with a negative charge, about 1 negative charge per 2 aa
- Disulfide bridges cannot be disrupted by SDS treatment (we must use reducing agents)
What is the role of adding B-mercaptoethanol or dithiothreitol?
- These are reducing agents that can reduce disulfide bonds
- We usually add reducing agents to protein sample buffer (loading buffer)
What is the role of adding bromophenol blue to the mix?
- We add bromophenol blue as tracking dye to our sample/loading buffer to essentially see when to turn off the run; it represents the migration of the smallest proteins
What are the materials in the gel itself and how does this relate to pore size?
- Acrylamide alone will form linear polymers called polyacrylamide which has low stability
- We add bisacrylamide to the gel mix for stability because it can be radicalized at both ends and is therefore used to connect two linear polymers with one another
- Increasing the concentration of acrylamide leads to larger pores while increasing the bisacrylamide will lead to smaller pores
What is needed to initiate the polymerization reaction of acrylamide/bisacrylamide monomers?
- Radical initiation is needed to start polymerization
- Acrylamide and BIS will only polymerize if BOTH ammonium persulfate and TEMED are added
Bromophenol Blue vs Coomassie Blue Purpose
- Coomassie is used for protein staining after SDS/PAGE running and bromophenol blue is used as a migration front indicator (and pH indicator too) during running
Why is it important to add a pH buffer in the gel? What pH buffer is used?
- TRIS buffer is used (hydroxymethyl aminomethane)
- The use of a buffer is important because it ensures glycine is in the correct protonation state for the respective part of the gel
What is the stacking effect and what would happen in a gel run without it?
- The purpose of stacking gel is to line up all the protein samples loaded on the gel, so that they can enter the separating gel at the same time
- Without the stacking gel, you will not obtain a sharp band for a single protein
- The glycine zwitterions further back in the stacking gel have a greater resistance (greater voltage) which means that these proteins will move faster than the proteins that are closer to the leading ion fronts (Cl-) which have less resistance (lower voltage)
- Under this strategy, the proteins will be compressed to a single line before entering the separating gel
What are the pHs for the stacking and separating gels?
- There is a pH gradient between the stacking and separating gels
- The stacking gel has a pH of 6.8 where glycine will exist as a zwitterion after being protonated
- The separating gel has a pH of 8.8 where glycine will
Explain the effect of zwitterions in the stacking gel.
- Glycine zwitterions will not move in an electric field which leads to an increase in resistance which will lead to an increase in local voltage in this part of the gel
What is the structure of the SDS-page gel?
- The negative electrode buffer (pH 8.3-8.5) is on top and the buffer consists of TRIS (keeps the glycine negatively charged) and glycine
- Next is the stacking gel which consists of TRIS-Cl with pH 6.8
- Next is the separating gel which consists of TRIS-Cl with pH 8.8
- Finally we have the positive electrode buffer (pH 8.3-8.5)
When would happen if the pH of the running buffer was not correct? What if it were instead set to a pH of 6?
- If the running buffer had a pH of 6, the glycine ions would exist as zwitterions and these would not travel in the electric field
- We need the glycine to travel from the negative electrode into the stacking gel
- If glycine doesn’t enter the stacking gel, the proteins will run into the gel but not in focused bands since there was insufficient stacking
What is the purpose of the protein standard ladder?
- It is used to estimate the molecular weight of proteins of interest
What are the two components of the interaction between an antibody and antigen?
- Affinity can be measured by the free energy
- Specificity can be measured by looking at the dissociation constants across different ligands
Define the terms antigen and epitope
- While the antigen evokes the antibody response in the host, the antibody doesn't bind to the entire protein, but only to that segment called the epitope
- The epitope is the part that is complementary to the antibody
Describe what type of molecule an IgG antibody is and how large it is.
- It has 2 heavy chains and 2 light chains
- It is a protein that is about 150,000 Da and contains 2 specific antigen-binding sites
- The antigen-binding sites are variables regions while the heavy chain is a constant region
Why do we need to transfer proteins from gel to membrane before detaching the protein of interest with a specific antibody?
- The membrane is sturdier than the gel
- The proteins are contained within the gel and the antibodies might have a hard time reaching the proteins with the material of the gel (acrylamide and bisacrylamide)
Why does nitrocellulose membrane have to bind all proteins equally well?
Why is the blocking step necessary?
- It prevents antibodies from sticking to the free membrane non-specifically
What is the purpose of secondary antibody incubation?
- After rinsing the membrane to remove unbound primary antibody, the membrane is exposed to a specific enzyme-conjugated secondary antibody. The chosen secondary antibody will bind to the primary antibody, which has already reacted with the target protein
Why is it important to wash unbound protein before adding the next antibody or before detection?
- To remove any loosely bound antibodies that are not bound to protein so that the secondary antibodies bind to primary antibodies bound to protein and not loosely bound primary antibodies that might be washed away in the second wash.
- 1 dalton is 1 g/mol
- The alkaline phosphatase takes off a phosphate from the X-P so that it can participate in a redox reaction and this creates the color change we are wanting to see
- If we have the molarity of the enzyme, we can determine its turnover rate
- If we have the concentration of the enzyme, we can determine its specific enzyme activity
Dr. Hong Study Guide
2. Describe the difference between first, second and third generation biofuels.
First generation: use food crops for feedstock. This competes with the food industry for resources. This causes food prices to skyrocket
Second generation: utilization of waste plant material to make biofuels. This won’t be eaten so it won’t compete with the food industry. Waste plant material has a ton of carbon to be converted to biofuels.
third generation: utilization of algal cells grown in bioreactors on land that isn’t used for agriculture. It’s a 1 stop shop bc it can do fermentation and transesterification and we can collect the products and use them for biofuels.
3. Describe two advantages of biofuels as compared to fossil fuels.
they are renewable, while fossil fuels are finite, and the require less land and resources than fossil fuels (you’re digging for fossil fuels). They also don’t pollute the air as much as fossil fuels when burned
4. How can modern biotechnology be applied to produce biofuel molecules?
we can grow algae in tanks or bioreactors by feeding them the respective feedstock. They convert this food into fuel molecules which can be harvested. We can also engineer these bacteria to overexpress a specific protein or we can engineer a direct pathway to produce a desired lipid.
5. While we discussed the advantage of using longer chain alcohol molecules for biofuels, what could be one reason for industry to still produce large amounts of ethanol as biofuel.
it’s easy and cheap to produce!
6. What experimental steps did you do in the laboratory to produce biofuels?
we are over expressing a specific gene that codes for an Alcohol Dehydrogenase in E. coli. The last step of our pathway to produce isobutanol is utilizing an alcohol dehydrogenase to convert isobutyaldehyde to isobutanol. We are using a plasmid so that our bacteria can overexpress this enzyme so that we can produce the max amount of isobutanol.
Study conditions
Find the optimal conditions for the protein of interest (temperature/pH), Km could allow us to figure out which enzyme is the most efficient (the less substrate used, the better), find which induction time that would maximize our yield. We will also use SDS PAGE to separate proteins by molecular weight. We can see that with the thickness of the bands. We used Comassie blueto make sure that we actually loaded the correct amount of cells per well (this is the top of the separating gel). Comassie blue binds to our proteins so we could see them moving. We then cut the gel above 50 kDa, and we are able to see how much of our protein of interest we have at 42 kDa. We use immunoblotting to isolate our protein of interest via detecting the His tagged proteins. We are also seeing the effect of signal amplification with two different kinds of antibodies. AB includes a primary and secondary antibody, while C is a hybrid, where it is one antibody with one enzyme attached. We could also compare the band thickness by comparing the induction time.
We use affinity chromatography to purify our protein of interest
Bromophenol blue doesn’t bind to the proteins, it’s just so that we can see how much we add to each.
Blue bands = thickness means more proteins
You want the proteins of the same molecular weight to end up
Why do we normalize w cell density?
- Loading more cells per volume. If we ignored it, we will be adding more cells to each well. This doesn’t mean anything about the induction. If we load the same amount of cells per mL, then we can say that the induction time and treatment with IPTG affected the amount of protein that was produced.
- The bands should all have the same band intensity in the stacking gel, so this is how we can tell that we added the same amount of cells per well. This is when, on the separating gel we can compare the amount of protein of interest that our cells produced.
7. Write down all experimental steps (like your flowchart) and understand the purpose of each step. Which result for this experiment would you expect, if one of the steps was omitted (one by one)?
8. Remember the purpose of using relevant chemicals for this method (those that we covered in lecture). Which results would you expect if the incorrect molecule was used instead.
9. Why is protein overexpression useful?
10. What is a trial overexpression? Why is a trial overexpression advised? Which parameters do you adjust to find optimal overexpression conditions.
11. Which components do you need on the plasmid for protein expression to work? List all components with their respective function.
12. What is the function of the T7 promoter on the plasmid DNA?
13. What is the function of the operator sequence on the plasmid DNA?
14. What is cell density and how do you measure it?
15. What does inducing protein expression really mean? And how does inducer contribute to the control of the expression of the gene of interest?
16. What happens if you induce a culture in the stationary phase?
17. What happens if you induce a culture in the lag phase?
18. Why do you need to monitor the cell density of various samples?
19. How is a mutation in the Lon protease (one particular protease) helpful for protein overexpression?
20. What are the different phases to a microbial growth curve, and what would it look like as a curve? SDS PAGE/Immunoblotting
21. Write down all experimental steps (like your flowchart) and understand the purpose of each step. Which result for this experiment would you expect, if one of the steps was omitted (one by one)?
22. Remember the purpose of using relevant chemicals for this method (those that we covered in lecture). Which results would you expect if the incorrect molecule was used instead.
23. What is the purpose of running SDS-PAGE?
24. What is the purpose of immunoblotting?
25. What molecules are involved in gel formation? What are their respective roles?
26. How do you treat the proteins before loading them onto the polyacrylamide gel? Why? (Think about what would happen, if you missed any of the chemicals or steps necessary.)
27. Why is it advisable to use a stacking gel in SDS PAGE? What would we see if a stacking gel was not used? Would proteins migrate?
28. Explain what is different in the separating gel (compared to the stacking gel) that allows proteins of different sizes to migrate at different speeds.
29. How do you adjust the pore size in an acrylamide gel?
30. What are the options of visualizing proteins following separation by SDS-PAGE?
31. How do you stain all proteins in the gel?
33. What are the two types of ladders you used for immunoblot and SDS-PAGE? Why did we use one over another for each respective technique?
34. Why do all proteins migrate in the same direction in SDS PAGE, irrespective of their intrinsic charge?
35. Why can we ignore the various protein conformations when comparing their migration in SDS PAGE?
36. Why does the molecular weight of proteins not affect their speed of migration in the stacking gel but in the resolving gel?
37. What would happen if the pores in the stacking gel were as small as those in the separating gel? What would the gel look like?
38. Explain why we need a stacking gel and how the stacking effect works.
39. Is it possible to use SDS PAGE to determine if a protein is found as a monomer, dimer, trimer, etc in the cell? Explain your answer.
40. Describe the rationale of how immunoblotting works.
41. Remember why we needed to transfer proteins onto the membrane. How do you get proteins to move from gel to membrane without losing their position relative to each other?
42. Why do you need a loading control?
43. What are loading controls?
44. Draw an IgG antibody molecule.
45. What is the difference between primary and secondary antibody molecules?
46. Can a primary and its secondary be from the same species?
47. What are the options of visualizing your protein of interest on the membrane?
48. Which detection method did you use in the course?
49. Recognize the structures of all molecules necessary here: acrylamide, bisacrylamide, TEMED, ammonium persulfate, sodium dodecyl sulfate, ß-mercaptoethanol,
TRIS, bromophenol blue, Coomassie brilliant blue,..... Literature-based questions
1. SDS-PAGE: While we use antibodies in the lab to label our protein of interest on the nitrocellulose membrane, scientists separated antibody molecules themselves via SDS-PAGE here. Remember what you learned about SDS PAGE and antibody molecules in lecture and follow the questions below. Guiding questions for your analysis: a. Why is Coommassie staining used here to visualize the proteins after SDS-PAGE rather than immunoblotting? b. The same antibody was loaded in lanes 2 and 3, but the samples were not treated the same prior loading. Which component of the protein sample buffer, when added or left out, could affect the band pattern of antibody molecules in this way? Explain and specify the different treatment of samples in lanes 2 and 3. c. Remembering the structure of an IgG antibody, label the three bands you see in lanes 2 and 3. Which band(s) contain(s) the variable region of the antibody that binds to the epitope of the antigen molecule?
50. Write down all experimental steps (like your flowchart) and understand the purpose of each step. Which result for this experiment would you expect, if one of the steps was omitted (one by one)?
51. Remember the purpose of using relevant chemicals for this method (those that we covered in lecture). Which results would you expect if the incorrect molecule was used instead.
52. Write down substrates and products of an alcohol dehydrogenase reaction.
53. When you set up this reaction in the lab, what else (not listed in 53) do you need to add? Why?
54. Recognize the structure of these molecules (including cofactors).
55. Indicate how you can measure the enzyme reaction (i.e. which molecule are you actually measuring in the spectrophotometer). Keep in mind, if the absorbance is expected to rise or fall in the reaction you wrote above.
56. Differentiate what we mean by 0th order and 1st order reaction conditions? What would you do in the lab to set up one or the other?
57. Which graph would you pick to illustrate what 1 st order or 0 th order reaction means? Label axes.
58. Draw a graph to illustrate what “initial rate” means. Label the axes. What is the issue with assays that are not in the initial rate?
59. What do the Km and the Vmax indicate about an enzyme reaction?
60. You are working in a biochemistry lab and are analyzing an enzyme. If you know the molarity of the enzyme, would you be able to determine its specific enzyme activity or its turnover number? Describe the difference between the two.
61. Specific enzyme activity. You are measuring the maximum velocity of a dehydrogenase enzyme by looking at the absorbance of 340 nm. During the initial rate phase, you measure an absorbance change of negative 0.5/min. The molar absorptivity coefficient is 6.22 mM-1 cm-1 and the cuvette length is 1 cm. The kcat of this enzyme is 1300 min -1 . The original undiluted enzyme stock concentration is unknown. You dilute the enzyme by the dilution factor of 15 before adding it to the assay. You then used 18 µL of this diluted enzyme solution for the enzyme reaction in a total volume of 1.3 mL. What is the original molarity of the enzyme?
62. Could you answer the above question, if you were given the specific enzyme activity instead of the kcat (and no other change or additional info)?
63. What could be the issue if we used a first order enzyme assay for measuring enzyme concentration?
64. When you plot the Michaelis-Menten graph, are all data points initial rates? Explain your answer.
65. How do you transfer the experimental data of product vs time to the Michaelis-Menten graph?
66. Why is it essential that nitrocellulose binds equally well to all proteins?