M1L6 DNA replication defects
Enzymatic and non-enzymatic DNA-protein crosslinks (DPCs) are a poorly studied form of DNA damage
DPCs are a result of covalent attachment of proteins to DNA (high probability due to large number of proteins associated with DNA - histones, replicative enzymes, transcription factors, DDR proteins etc…)
Guanine and lysine are the strongest cross linkers
There are many lysine residues in histone tails, making them prone to attach to guanines in DNA
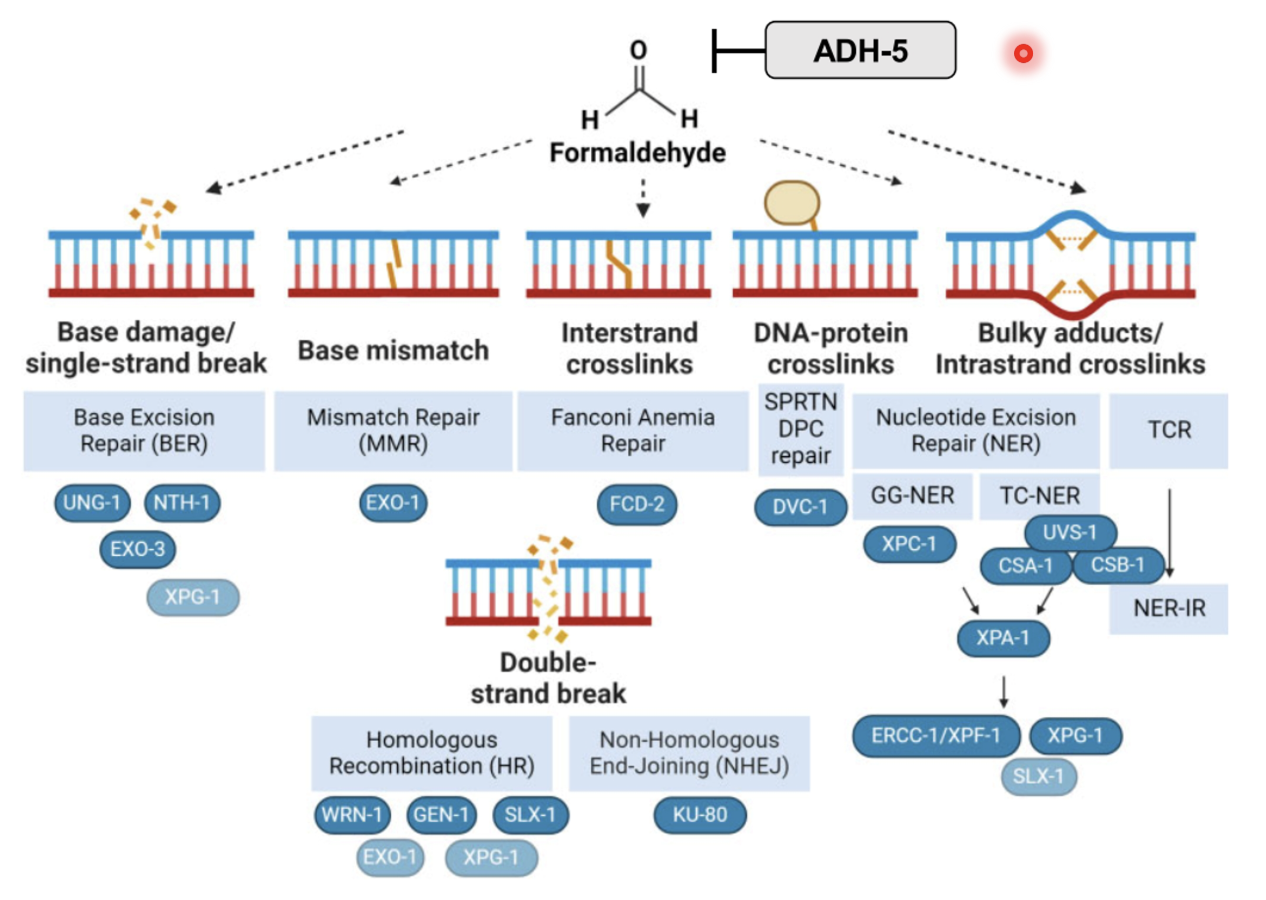
Formaldehyde is a major inducer of DPCs, as well as chemotherapy drugs (eg. topoisomerase inhibitors, cisplatin…)
Topoisomerase binds to DNA to make transient SSBs in DNA to relieve torsional strain, inhibitors stabilise this complex leading to propagation of DSBs
DPCs: non-enzymatic and enzymatic
Non-enzymatic: formaldehyde and methanol
Aldehydes (formaldehyde, acetaldehyde) can form covalent bonds between amino acid residues and DNA bases
UV or IR can also cross link nearby proteins to DNA
Endogenous metabolism can product reactive aldehydes and lipid peroxidation byproducts that contribute to this
Enzymatic: lesions caused by topoisomerase, DNA polymerase β etc enzymes
Stalls replication fork

Ubiquitination and SUMOylation mark DPCs for degradation
DPCs can be resolved through proteolysis or HR if fork collapse leads to DSBs or if there are DSBs resulting from topoisomerase activity
Proteolysis is carried out by the proteasome and SPRTN protease which degrade protein adducts
After partial proteolysis repair is completed by TLS polymerases which bypass the remaining peptide adducts and NER to remove residual protein fragments
Formaldehyde can cause many types of DNA lesions, mostly triggering covalent attachment of lysines of proteins to guanine in DNA (DPCs)
Ub and its chains coordinate spatial and temporal regulation of proteins at DNA damage sites
Eg. Ub at DNA DSB sites
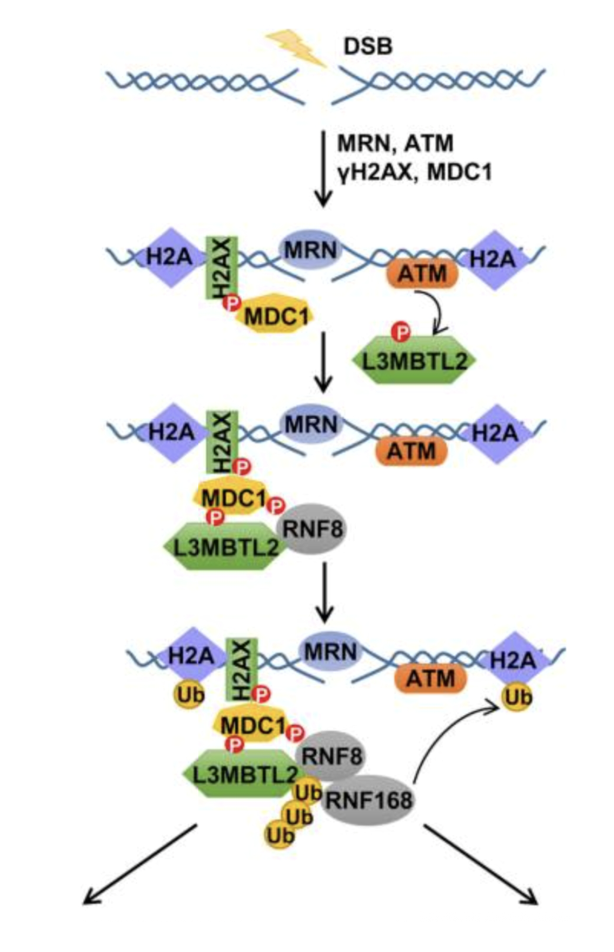
MRN detects the DSB and recruits/activates ATM kinase
ATM phosphorylates H2AX near the break
MDC1 binds to γH2AX, stabilising the repair focus
ATM phosphorylates L2MCTL2 which interacts with MDC1 and RNF8 (an E3 ubiquitin ligase)
RNF8 ubiquitinates histones and other local proteins
RNF8-generated ubiquitin chains recruits RNF168 which amplifies ubiquitin signalling my spreading ubiquitin marks on chromatin
Ubiquitin marks act as docking sites for repair proteins like 53BP1 (for NHEJ) or BRCA1 (for HR)
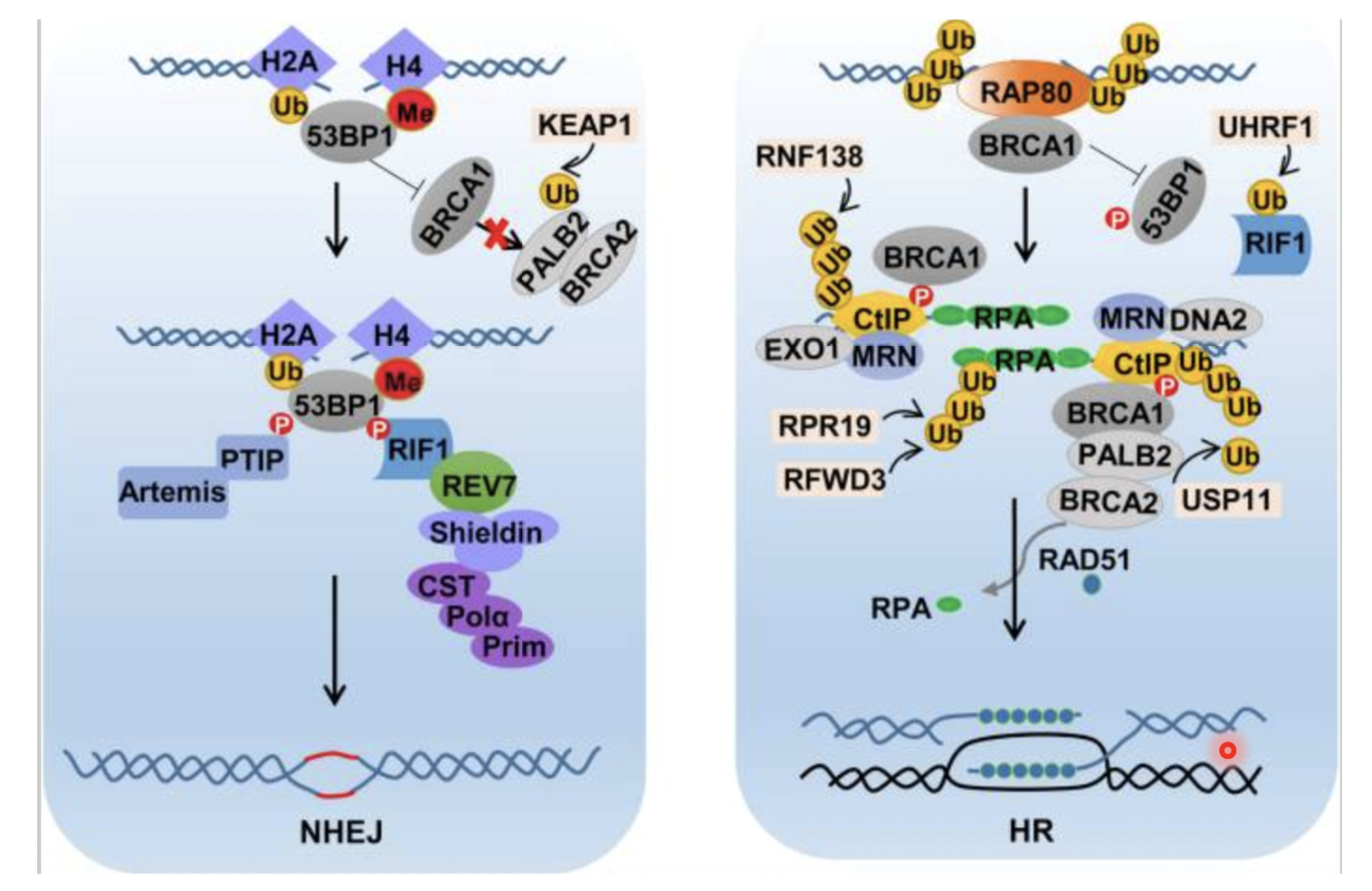
For NHEJ:
53BP1 binds to ubiquitylated H2A/H4
HR is blocked as 53BP1 and KEAP1 inhibits BRCA1 recruitment
PTIP FIF1, shieldin complex, CST–Polα–Primase, and Artemis assemble, processing and ligating DNA ends directly
For HR:
BRCA1 is recruited by RAP80, RNF8/RNF168
BRCA1 antagonises 53BP1 and promotes end resection for HR
MRN, CtIP, EXO1 and DNA2 create 3’ ssDNA overhangs
RPA coats ssDNA and BRCA1/PALB2/BRCA1 load RAD51. forming the nucleoprotein filament
RAD51 filament searches for homologous DNA template and promotes strand invasion and HR repair
Human genetic disorders associated with inherited mutations in various E3 ubiquitin ligases involved in DNA replication/repair - usually cancers, neurodegeneration, age-related pathologies but also others
Ubiquitination must also be removed for resolution of DSBs, eg. inhibition of p97 (which helps remove ubiquitination) does not allow for full resolution of DSBs
Chromatin associated degradation (CHROMAD)
p97 (aka VCP) is an ATPase in the ubiquitin system which recognises ubiquitinated substrates and uses cofactors with specificity to the substrate to bind and hydrolyse ATP and thereby extract substrates from chromatin
p97 is a homohexamer and physically extracts substrates through its central pore
Cofactors of p97 are implicated in human disease -
Ataxin-3 (mutated in ataxia/Machado-Joseph disease) which works with p97 to remove proteins by the proteasome, specifically RNF8
SPRTN (mutated in Ruijs-Aalfs Syndrome - premature ageing) - important for removing DPCs
TEX264 (colorectal cancer)
Formaldehyde (from the environment or due to our own metabolism) is a cause of ageing pathologies including neurodegeneration and cancer
Formaldehyde can be from smoke, industrialisation, environmental stressors but also a product of endogenous metabolism:
Synthesis of some amino acids or nucleic acids
Methyl groups removed by demethylases can produce formaldehyde in the presence of oxygen
Mitochondrial stress causes lipid peroxidation in its double lipid membrane, producing formaldehyde
Endogenous formaldehyde causes new inherited bone marrow failure syndrome (short stature, microencephaly, limb abnormalities)
Two-tiered protection from aldehyde (acetaldehyde and formaldehyde) toxicity
Detoxifying enzymes - aldehyde and alcohol dehydrogenases
DNA repair (DPCs)
Ethanol metabolism
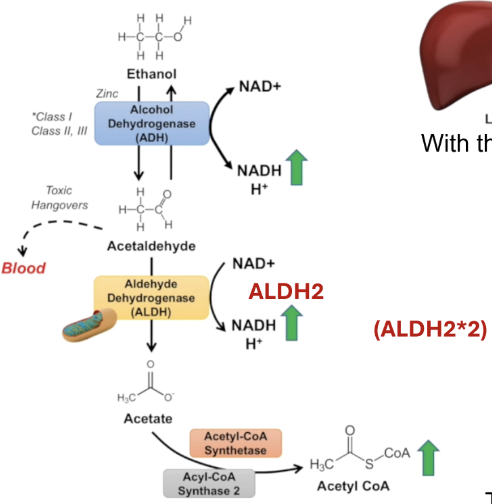
Alcohol dehydrogenase converts ethanol to acetaldehyde (toxic) by using NAD+ which gets converted to NADH + H+
Acetaldehyde is converted to acetate by aldehyde dehydrogenase using NAD+ which gets converted to NADH + H+
Acetate is converted to acetyl CoA by acetyl CoA synthetase 2
Methanol metabolism - methanol can come from consuming cheap alcohol (methanol mixed with ethanol) or even from your own microbiome via fermentation processes
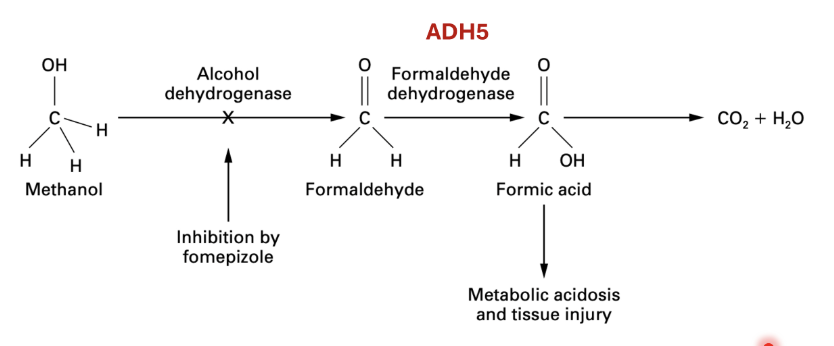
Methanol converted to formaldehyde by alcohol dehydrogenase (very toxic)
Formaldehyde converted to formic acid by formaldehyde dehydrogenase —> metabolic acidosis + tissue injury
Inhibiting ADH5 can lead to high levels of formaldehyde toxicity
Formic acid converted to CO2 + H2O
Formaldehyde induced DPCs
Tier 1 defence - ALDH2, ADH5
Tier 2 - DPC proteolysis repair, Ub system (SPRTN, p97, nucleophagy)
Human diseases caused by endogenous DPCs (likely due to formaldehyde toxicity)
AMeD syndrome (aplastic anaemia, intellectual disability, dwarfism) - digenic mutations in ALDH2 (SNP; on one allele) and ADH5 (biallelic mutations)
Ruijs-Aalfs syndrome - monogenic cause (biallelic mutations in SPRTN)
Cockayne syndrome - defect TC-NER, monogenic and biallelic mutations in CSA or CSB
SPRTN is key enzyme (metalloprotease) in DPC repair, needed for progression of replication fork
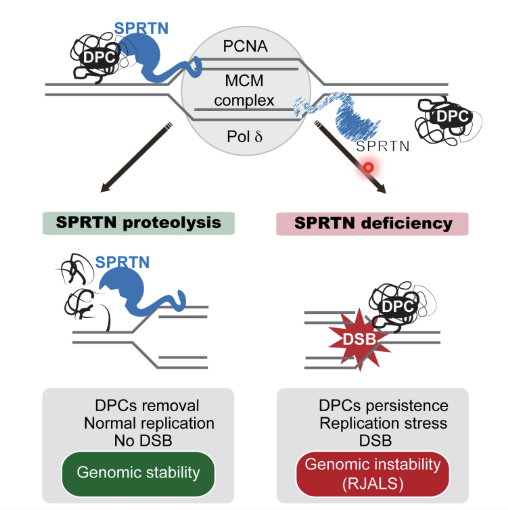
Cleaves a variety of DNA binding substrates (eg histones) and also itself
Cleaves free and covalently attached Top1 and Top2
Defects in SPRTN characterised by RJALS and liver cancer
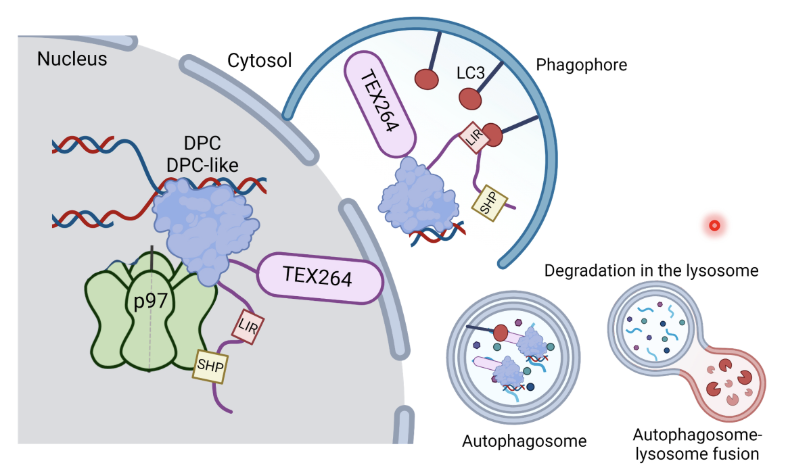
p97-SPRTN-TEX264 complex processes Top1-cleavage complexes (DPCs)
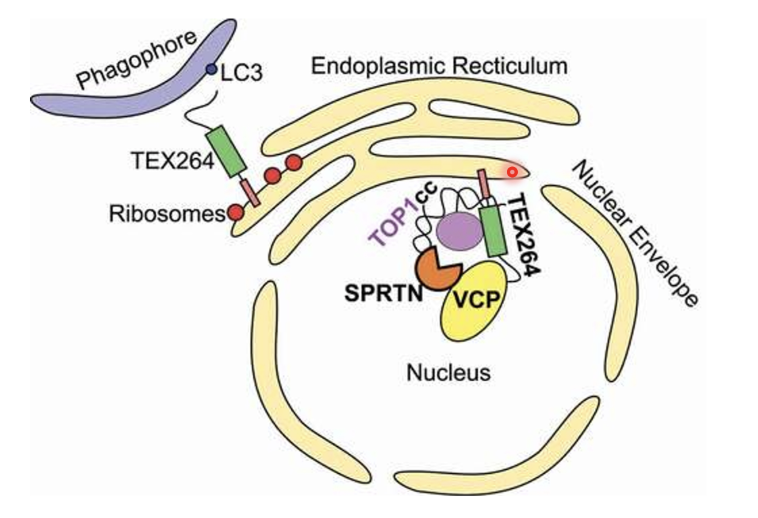
TEX264 is one of the main receptors for autophagy during starvation and it also plays a major role in DPC repair by acting as a p97 adaptor protein
p97-SPRTN-TEX264 can recognise DPC lesion (eg. Topo1cc or Topoisomerase 1-cleavage complexes which is an enzymatic DPC) on DNA
TEX264 recruits p97/SPRTN to the DPC and SPRTN proteolytically degrades the protein adduct while p97 proides unfolding and extraction energy to clear the protein remnants from chromatin
After DPC processing DNA can be repaired by DDR mechanisms
TEX264 also acts as an ER-resident autophagy receptor where it interacts with LC3 on phagophores for ER-phagy
Nucleotide excision repair
UV lesions, formaldehyde induced lesions…
Transcription-coupled NER:
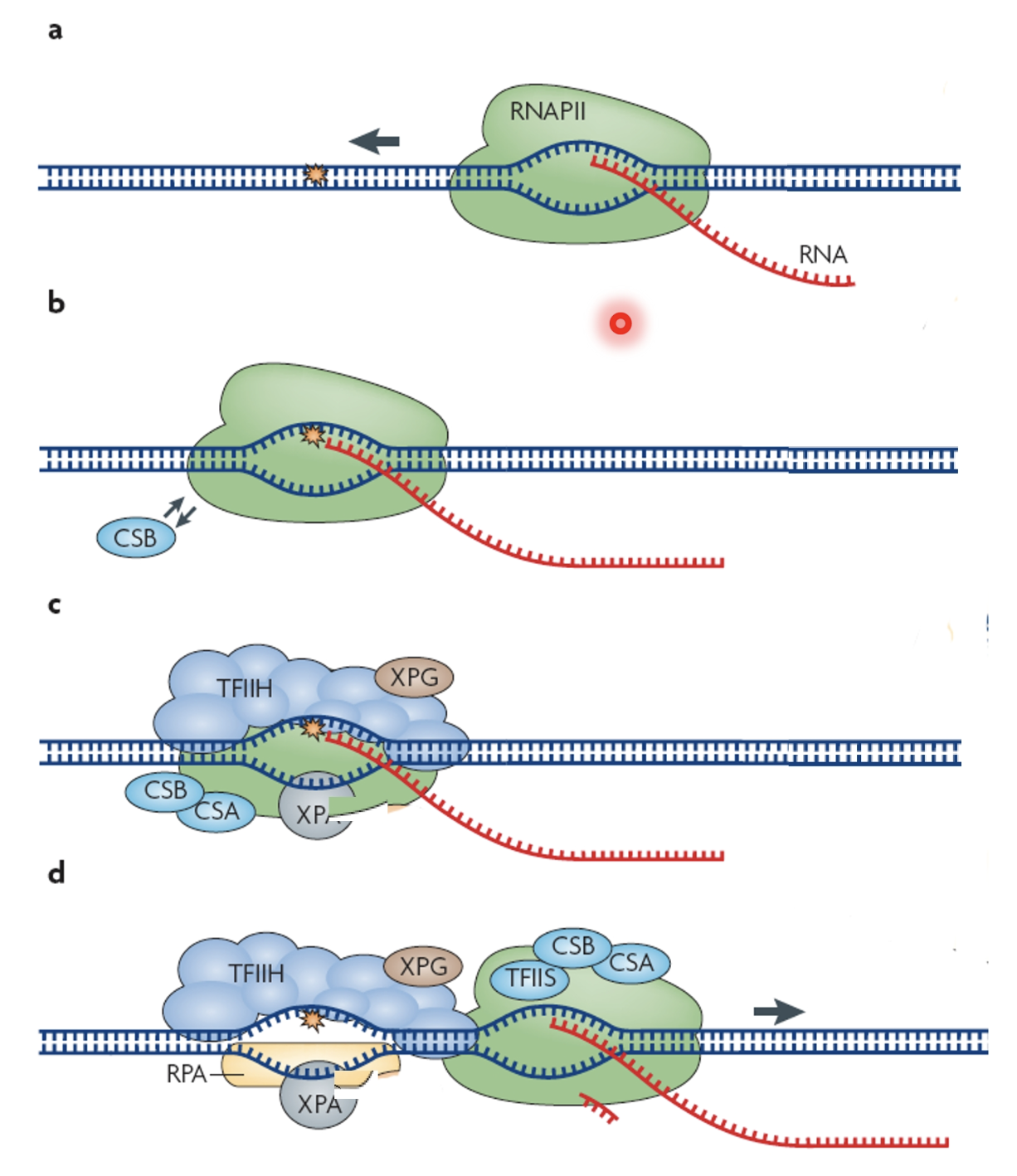
RNA pol II stalls at a lesion
CSB binds to stalled RNA pol II and recruits CSA (E3 ubiquitin ligase) and other repair factors, remodeling RNA pol II and forming the damage recognition complex that distinguishes transcription blocking lesions
TFIIH recruited to the site which containes the helicases XPB and XPD to unwind local DNA
XPG (endonuclease) and other XP proteins (XPA, XPB, DPD) join to form the NER core complex
SPG and XPF-ERCC1 make dual incisions flanking the lesion and the damaged oligonucleotide is excised
RPA and XPA stabilise the single stranded region\
XPA is also an E3 ubiquitin ligase which ubiquitinates TC-NER proteins like CSB and TFIIH to regulate their recruitment, retention or release at the lesion site
The gap is filled by DNA pol using the undamaged strand as a template and the nick is sealed by DNA ligase
After repair XPA and othe ligases promote proteasomal clearance of excess or damaged repair factors to prevent persistent stalling
TC-DPC repair begins as a TC-NER process
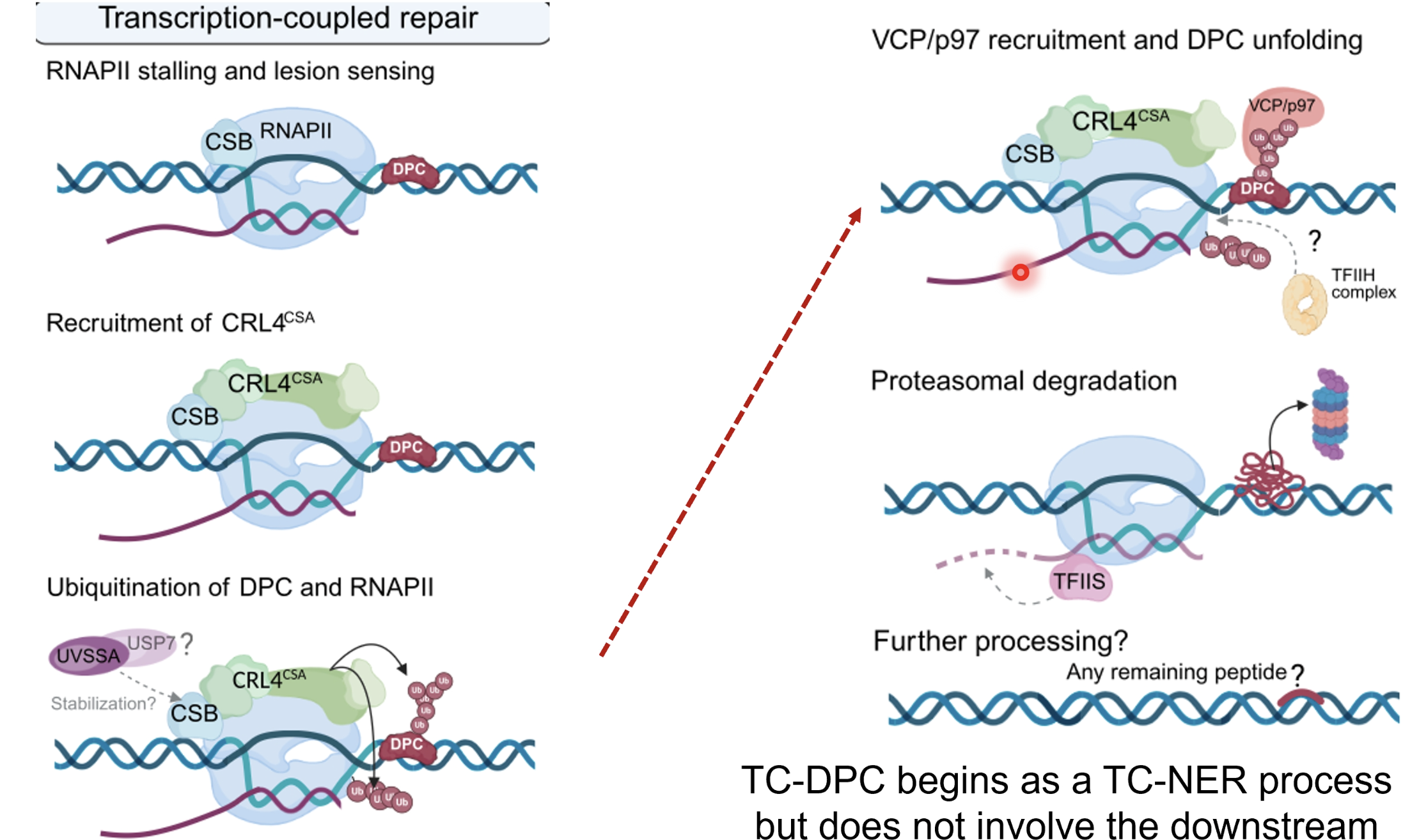
RNA pol II stalls at DPC and CSB binds and recruits CRLRCSA ubiquitin ligase complex which ubiquitinates the DPC to mark it for degradation and also RNA pol II to mark it to be removed or recycled after the lesion is repaired
p97 ATPase unfoldase is recruited to extract/unfold the ubiquitylated DPC protein from DNA
TFIIH likely facilitates or stabiilises p97 recruitment and activity but is not essential for unfolding itself, rather helps remodel RNAPII-DNA-DPC complex to improve access for p97
DPC protein is then degraded by the proteasome
TFIIS (transcription elongation factor) helps restart or rescue RNAPII
Remaining peptide adducts could be handled by SPRTN (like in RC-DPC repair)
Parallel DPC repair pathways
RC-DPC (SPRTN) -replication coupled
TC/DPC (CSA/CSB) - transcription coupled
Both converge on p97 (VPC)
Formaldehyde inhibits DNA replication
Top1 cleavage complexes induced by Top inhibitors and PARP1 complex induced by PARPi
Selective autophagy (nucleophagy) of Top1cc

Top1cc removal is strongly dependent on autophagy (DPC lesion-phagy)
May explain why many cancers upregulate autophagy
Part of the DPC lesion which can not be removed by SPRTN are becoming insoluble and forming aggregates
In this case, TEX264 removes the lesion and shuttles it to the lysosome for degradation
When there are DPCs autophagy proteins and lysosomes can be recruited to the replication fork even from outside the nucleus
There is interorganelle communication - replication fork recruits lysosomes and lysosomes can also recruit replication proteins
TEX264 expression can be an predictor of responsiveness to Topo inhibitors (higher expression = better responders)
Nucleophagy can also remove cytotoxic trapped PARP1
Inactivation of TEX264-selective autophagy resensitises BRCA-deficient cells that have acquired resistance to PARPi