lecture 5: structure and function of membrane proteins
introduction to membrane proteins
Membranes are highly selective permeability barriers that control information flow between cells. They can generate signals and harbour enzymes for energy conversion processes such as photosynthesis and oxidative phosphorylation. There are many jobs of membrane proteins and surround every organelle with differences between them. There are also differences between organisms, for example, gram negative bacteria have an inner and outer membrane which have different compositions such as the outer membrane having LPS and O polysaccharides. They can also have medical relevance such as antibiotic targets.
Membranes are made of different lipids. Phosphoglycerides have a phosphate group which can be joined to different head groups such as sereine to make different lipids. Glycolipids on the other hand have a glucose molecule that is attached to side chains of lipids to make different types - saturated and unsaturated polysaccharides. Thee can also be different lipid compositions in different organs and organisms. For example, the liver has only 22% composition of Phosphatidylethanolamine while the heart has 6.8% and E coli has 57.5%. In addition, eukaryotic cells also have cholesterol.
integral membrane proteins span through the lipid bilayer while peripheral proteins can be attached to the membrane or onto an integral protein. They can be involved in many processes including maintaining cell structure or for internal and external signalling. There are many different types of integral membrane proteins and they all have different jobs:
channels that can open and close to let molecules through. these are pores that run through the membrane
transporters that can change shape to switch the side of the membrane it is open to
receptors which bind onto molecules to have a downstream effect
membrane enzymes that convert substrate into product
structural proteins that maintain structure and make contact with both the extracellular matrix and cytoskeleton
studying membrane proteins can be very important as 30% of genes code for membrane proteins and 50% of drugs manufactured work on membrane proteins. structural biology of membrane proteins show that out of a total of 71,500 structures, only 3000 new ones are found in 2024. This statistic can change depending on the study but all show that there are fewer membrane protein structures discovered each year. This can be due to the difficulty of working with membrane proteins
structural studies of membrane proteins
step 1: obtaining from source
the first step in studying membrane proteins is obtaining it from its source and overexpressing the protein in a heterologous host. Some examples of proteins that have been purified from the source include:
photosynthetic reaction centre which was purified from purple bacteria
rhodopsin which was the first GPCR to be purified from bovine retina
Na+/K+ Atpase that was purified from pig liver
In general, membrane proteins are not abundant and they must be overexpressed
However, it is difficult to overexpress membrane proteins. This process can be done in bacteria however, this tends to only be useful for bacterial proteins and not eukaryotic proteins. They have different lipid composition in their membranes as well as no cholesterol or post translational processing.
Yeast or insect cells can also be used however, they are more difficult to work with than bacterial cells. However, thi is often preferred as there are some post translational modifications and it is easy to scale up
Mammalian cells are very often use as they have full post translational processing however, it can be difficult to scale this up but most times this is the only option. This is the most expensive and technically difficult workload
step 2: purification
The protein is expressed at this point and is sitting in the membrane and must be purified. The membrane protein has hydrophilic and hydrophobic regions and to get it out of the membrane, these interactions bust be kept in tact so that the protein does not aggregate. To get around this issue, the lipid id replaced with a detergent where it is then added to the membrane sample. The lipid is displaced and coats the protein in a micelle.
detergents
There are different groups of detergents and choosing a specific one is important. The larger the lipid chain of a detergents is, the better it is at retaining activity but it is less good at crystallisation. Shorter chains such as in octyl glucoside have better crystallization however sometimes denature the protein. Other detergents can also be ionic such as CHAPS which is zwitterionic
step 3: purification
Purifying the protein is the same for soluble domains but with detergent added. The protein may not be as stable as other proteins in the membrane sample.
membrane protein stability
the protein has to be stable enough to remain in solution during the purification process. You need high concentrations and quantities as well as high purity. Cryo-em has less stringent requirements than X ray crystallography but not by much as in Cryo-em you still need a high concentration (1-3 mg/ml) however, as there is generally less screening, you can get away with less pure and lower amounts
how to make sure the protein is stable in detergents suitable for structural studies.
Depending on the question being asked, a homologue of the protein may be more stable. This can often be other mammalian proteins but can also be other eukaryotic proteins or bacterial homologues. Depending on the question asked, using this could answer the desires question.
stabilizing the protein by mutating it has also been done before for GPCRs by testing the stability of every amino acid and combining the residues by making it more stable and to make a structure. This was the strategic use for Heptares and has been how many GPCR structures have been solved.
fusion proteins is more common now than using bacterial homologues but requires a lot of screening of them, binder and mutants.
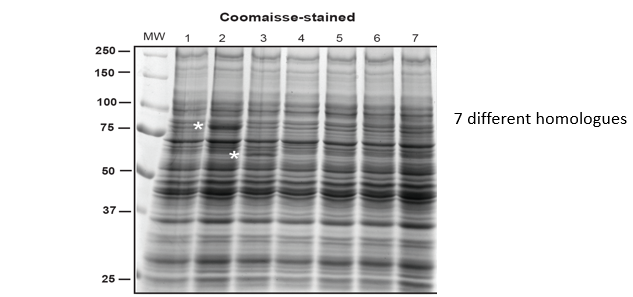
In the above example, there are 7 different homologues of the protein of interest. This is expressed and run in an SDS gel. As the protein is not expressing well, there are no distinct bands shown. To make it more clear, the protein can be probed with antibodies in a western blot or have green fluorescent protein fused to it. When the protein is fused with GFP, correct expression means that gfp also is expressed and so folds correctly and gives a signal. The gel is put under a fluorescence detector that shows any protein with the gfp added to it. This means is is easy to see where there is expression and if one is better expressed than another.
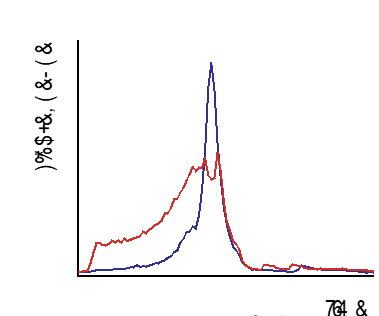
we can also screen for stability by size exclusion chromatography which separates the protein by size. The red line on the graph shows the distribution of the different sizes in the sample. The protein with gfp added is also showed by the blue line and a single peak indicates the protein has one size and is folded in a globular shape. Two or more peaks mean that it is big and probably aggregated. The presence of one line doesn’t mean that you WILL be able purify the protein but is likely
purifying the protein means that it can be used in kinetics or binding measurements as well as in structural biology. It can also be reconstituted into lipids
x-ray crystallography
The membrane protein in the membrane is sat in a protein detergent complex. A normal vapour diffusion method provides type II 3D crystals with detergent micelles. The detergent can also be replaced with lipids to reconstitute the protein into lipids. When the detergent is replaced with lipid cubs, you get lipids between the protein that make contacts with the hydrophilic groups. These are type I 3D crystals. 2D crystals can also be mad by electron microscopy however, this is not as common and is not used often
The point is to form crystals however, there can be problems with the size. If the detergent is too long, it will mask the crystal hydrophilic contacts and if it is too short, it will denature the protein. The minimum size that will keep the protein in solution is required.
crystallization 1: conventional vapour diffusion methods
conventional vapor diffusion methods can be used such as the 96 well plate method where there are 96 well plates with 80 uL precipitate in the well. As well as this, there is 100 nl of the protein and 100 nl of the solution and the equilibrium difference will form crystals. The chances for finding the right conditions for this to occur is minimal so 96 plates are used with 96 different conditions.
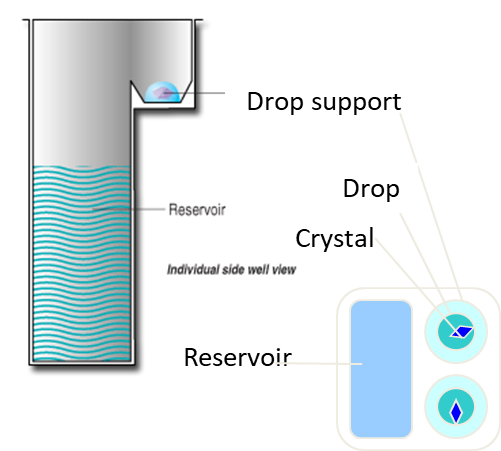
crystallization 2: cubic phase
The lipid detergent can be replaced with monoolein and this produces a cubic phase where the lipid is form large channels that are connected to where the protein sits in the bilayer. The protein can diffuse through the channels to form crystals. This is fiddly to set up but gives good results.
The cubic phase is formed by mixing lipids (monoolein) with water or an aqueous solution. This creates highly viscous, gel-like substance with a continuous 3D network of lipid bilayers. These resemble a natural membrane and so provide supportive environment for membrane proteins.
The membrane proteins are incorporated into the cubic phase and are often initially solubilised in detergent to keep them stable and then are mixed into the mesophase (the bilayer formed). This keeps the protein in their native state.
Once the membrane proteins are incorporated, conditions for crystallisation are optimised by adding precipitants, salts or other agents to promote the slow and controlled crystallisation of the protein. The cubic phase acts as a diffusion-controlled matrix, allowing gradual crystal formation. As the protein crystals form, they are usually small and embedded in the cubic phase. They can be analysed using x-ray diffraction analysis once they have been harvested.
crystallisation with fabs
To increase the hydrophilic surfaces of the protein to make crystal contacts, antibodies can be raised against a specific antigen in the hydrophilic region of the protein. the binders should bind tightly to fix it in a specific conformation and the best thing to use are antibodies such as FABs with the variable and constant light chain domains taken out or to just use the variable regions. Fvs can also be used as well as nanobodies from camels and sharks as they have specific IgG with just the heavy chain and a simpler variable region. As it is rigid and can bind to the protein, they are often used. Sybodies are synthetic domain antibodies and monobodies that are synthetic binding proteins can also be used and all of this is relevant for cryo-em. Fabs can also increase crystal contacts and mediates this. This can improve resolution from around 5 angstroms to 3.5. All crystal contacts are with the FAB and not with the transport itself and the addition if the FAB itself is what improves the resolution
Binders can also be used for GPCRs to replace the flexible loop which can be unstable.
data collection
often crystals are not well formed and diffract at low resolutions. there can be some very high resolution structures particularly in lipidic cubic phases. Data collection is helped by bright sources such as synchrontorons and free electron laser.
Cryo-em
electron microscopy
Electron microscopy itself can be used to give 2D or 3D structures. 2D crystals are rarely used nowadays and are planar crystals that can give information about structure. However, it is low res.
3D crystals on the other hand can still be used with cryo-em and achieves good data however is not mainstream. It is being pioneered by researchers and you still need to crystallise the protein first into small crystals
Single particle cryo-em came about the resolution revolution (see previous lectures)
Electron microscopy has similar problems to X-ray microscopy where a suitable detergent and buffer has to be found for the samples. This is less important if it masks the proteins but is more important that it doesn’t change the protein or reduce the contrast of the grids. Fabs can also be used as they can lock proteins in one particular state and as transporters can be floppy this is useful as it can lock them in a specific shape. They can also act as fiducial for assigning orientation and this increases the effective size of the protein
alternatives to detergents
alternatives to detergents can be used especially with cryo-em
nanodiscs
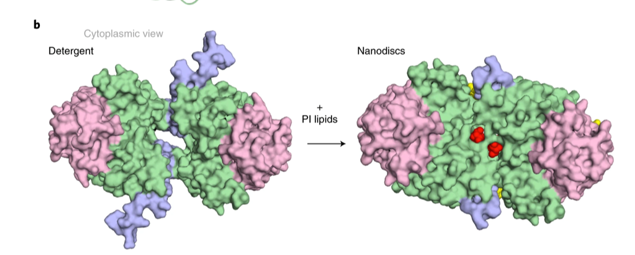
Nanodiscs are essentially a scaffolding protein which is a small protein that acts like a scaffold. It is mixed with lipids and forms a nanodisc or small discs that are full of lipid. The protein is added to this and the protein is therefore reconstituted into the lipid so is now back in the lipid bilayer as opposed to in the detergent.
Lipids can be important in terms of the structure and function of the protein. For example, the structure of Na+/H+ exchangers NHA2 was solved using cryo-em and nanodiscs. There is a subtle difference in the TM1 region when in a detergent compared when in the lipid bilayer of the nanodisc. This means that the presence of lipids can be important and change the structure and interaction of proteins
other examples of scaffold proteins are saposins and peptidiscs and SMALPs as well as amphipols which are similar to detergents in having both a polar and non polar group
NMR
structure determination by solution NMR is restirticed to small proteins less than 30 kDa as the protein tumbles too slowly in solution. Recent advances in technology however, have bought this up to 50 to 150 kDa. The bound detergent surrounding the protein effectively makes the particle even bigger but using NMR to solve structures is not possible for most membrane proteins with some exceptions such as solving bacterial transporter EmrE in lipid bilayers and the structure of human alpha 7 nicotinic acetylcholine receptor.