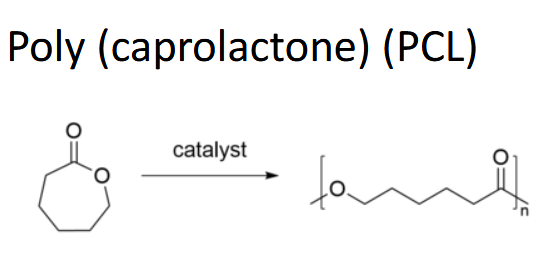
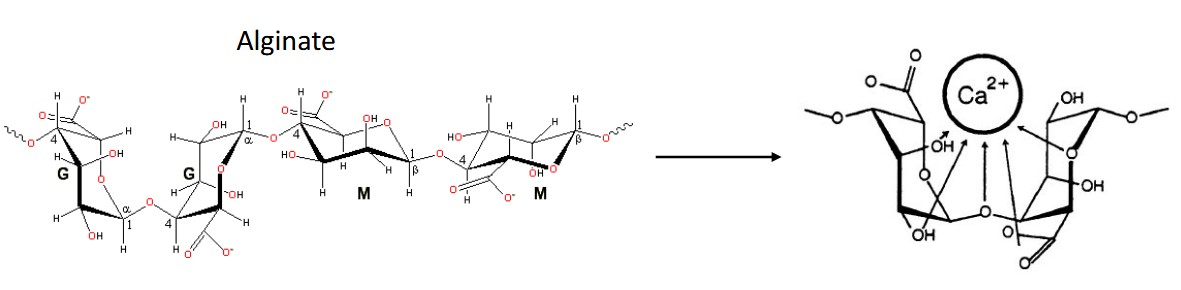
Everything
Lesson 1: Drug Delivery Methods + How to choose
Drug Delivery: administration of drugs through various routes to improve health
drug dispersion + solubility
drug formulation: what polymers
Conventional: controlled
oral = sustained
nasal = extended
rectal = site specific
injection = pulsatile
Oral (swallow): FPM
can’t give it to unconscious patients
low solubility/permeability
degraded by GI enzymes
irregular + food can mess w/ it
Buccal (dissolve in cheek)/Sublingual (dissolve under tongue): Bypass FPM
if you swallow you don’t get the effect
has to be small doses
fast absorption
Rectal: Bypass FPM
suppository (melty thing) or enema (liquid)
degrade by bacteria
Intravenous: Bypass FPM
100% efficiency + fast
Invasive + requires professional
Needs to be sterile + correct amount (avoid toxicity)
injection (bolus) or infusion (IV bag)
Subcutaneous (bolus under skin): Bypass FPM
can self administrate
slow + complete absorption
max dose of 2ml
Intramuscular (bolus into muscle): Bypass FPM
can self administrate
larger volume than subcutaneous (more than 2 ml, faster too)
Inhaler: Bypass FPM
gases are rapidly absorbed
have to be below 0.5 um
Transdermal (absorb through skin): Bypass FPM
local effect + easy
have to be below 10 mg/ml or Mw < 1000 Da
Low absorption sometimes, but it’s easy since it’s a patch
How should you choose which method for drug delivery?
what physicochemical properties the drug has
Drug molecular size (mw)
Dose size
Half life
Chemical stability
Loss of biological activity in aqueous solution (does it dissolve in water)
what effect you want the drug to have
Local: surface (topical/vaginal)
Systemic: everything that doesn’t diffuse from the skin
Immediate response (everything fast)
injection, inhalation, intramuscular, subcutaneous,
What affects the efficacy of the drug?
Dosage
How much was absorbed
Distribution to site
Rate/extent of elimination
Fastest to slowest:
injection > inhaler > intramuscular > subcutaneous > buccal/sublingual > transdermal > oral
Pathway of drug trying to get into your blood stream and do its thing
Drug delivery method
Absorption
Distribution to site
Elimination
First Pass Metabolism (FPM): drug concentration significantly reduced before systemic circulation bc of GI and liver
if you bypass = efficient
Lesson 2: Drug Solubility and Formulation
how much it dissolves and should we make the drug
Since we’re made of like 73% water and our blood is 92% water
hydrophilicity/hydrophobicity is important!
we have a lot of water and our proteins are carbon so balance
Strongest dipole dipole bond is hydrogen bonding
London dispersion (van der waals)
unequal distribution of electron = temporary induced dipole
weak
Dipole moment is how uneven distribution it is == how polar it is
if BIG it’s polar it’s soluble in water = better diffusion
if SMALL it’s non polar it’s soluble in lipids
Proteins have both hydrophilic and hydrophobic parts
AA build up proteins
20 Amino Acids
9 hydrophobic
5 charged (3 positive, 2 negative)
6 polar but not charged
Hydrophobic: nonpolar into core = most important protein stability
H bonds (wanna form water): form water + helix + sheets
Dipole-dipole (polar interaction): polar side chains
Coulombic (ionic): salt bridges but pH/ionic strength breaks it
Van der Waals (small temporary dipole): important for hydrophobic core
Chemical, covalent (disulfide bond): not in everything
Thermodynamics:
Enthalpy: heat
H = E + PV
Positive = endothermic, negative exo
heat of reaction = heat of
Entropy: disorder, if spontaneous +entropy
positive = more disorder, negative = less disorder
Gibbs Free Energy: energy that can be used to do work
What causes a protein to form?
nonpolar parts really do not like the water so they fold into a protein w/ nonpolar parts facing inwards to reduce free energy
it’s favorable (spontaneous) for proteins to fold
protein formation is exothermic and increases entropy
ice melting is spontaneous and endothermic
free energy o transfer from nonpolar solvent to water is proportional to hydrophobic surface area
Why does protein folding create MORE entropy?
surrounding water molecules are NOT bonding to the nonpolar parts (icy)
Dissolution: How to dissolve something
Separate solute particles from intermolecular forces
Separate solvent particles + have void spaces big enough for solute molecule
water has a high dielectric constant, so water is main solvent
Solute is put inside void space
If spontaneous, work is negative
How to overcome IMFS (break up the solute-solute bonds): Thermal Energy
if strength of interaction between solute-solute is bigger than kbT, it won’t dissolve
Boiling point of liquid is proportional to temperature and pressure
Every way to Increase Solubility:
Polarity: if same polarity, inc solubility
More branch chains = inc solubility, straight = dec solubility
Decrease molecular weight = inc solubility
If structurally similar = inc solubility
Crystal Structure: if more regular + uniformly packed = dec solubility
Ionizable (gain/lose proton to become base/acid)
Apparent Solubility = unionized + ionized form
If ionized = inc solubility
Cosolvent: added to solvent to inc solubility bc it is nonpolar so it lowers the polarity making the drug more easily dissolved
if not ionizable = hydrophobic = don’t react to pH
don’t mess with the drug, mess with the solvent (add co)
Surfactant: molecules that make up micelle like structures
hydrophilic head, hydrophobic tail (dec surface tension)
drug can be carried inside
Nanosizing: Make smaller = better dissolved
prevent sticking together by adding polymers to maintain SA to volume ratio
dissolution rate proportional to surface area in contact w/ solvent
Salt formation: change pH = better dissolved
Cocrystals: add w/ the drug (NOT changing the drug itself) by precipitation/grinding
Polymorph: Different structure of drug (salt, cocrystal,cosolvent) to make it more optimal for drug delivery
Lesson 3-4: Drug Release
What are the types of drug release?
Immediate
Delayed (tablets)
Sustained (over EXTENDED time)
Controlled (rate + location)
Stimulus release (trigger)
Targeted release (specific site)
How to control drug release:
Mechanism | What drives release | What limits rate | Common system type |
|---|---|---|---|
Dissolution control | Dissolving of drug or matrix | Solubility or dissolution rate or permeability of membrane | Membrane or matrix |
Diffusion control | Drug molecules diffusing through polymer | Diffusion rate through polymer | Membrane or matrix |
Osmotic control | Water entering by osmosis, pushing drug out | Membrane permeability & osmotic gradient | Osmotic pump |
Ion-exchange control | Ion swapping between drug–resin complex and body fluids | Ion concentration and resin properties | Ion-exchange resin beads or complexes |
How does reservoir work?
Water gets in, dissolves drug, drug gets out
Rate can be controlled by: dissolution profile or permeability of membrane
How does a diffusion drug release matrix work?
release drug at certain rate
Dependent on : porosity, swellability, dissolution rate
How does osmotic pressure work?
Drug in reservoir and coated w/ semipermeable membrane
Water diffuses into it and pushes drug through hole (oral)
Type | Polymer Nature | Key Mechanism | Water Interaction | Typical Control |
|---|---|---|---|---|
Homogeneous | Uniform solid | Diffusion through polymer | Minimal | Diffusion rate |
Porous | Solid with channels | Diffusion through pores | Moderate | Pore size/structure |
Hydrophilic | Swelling polymer | Diffusion + erosion | High (swelling & gel) | Polymer hydration/erosion |
Types of Solute Transport:
passive = no energy
diffusion: gradient, inc temp = move faster
convection: depends on flow of carrier fluid
What dictates diffusion?
intermolecular interactions between solute and solvent
Concentration gradient
Diffusional barrier
What is a diffusion coefficient: how fast it spreads out
affected by differences in solute size, noncovalent interactions, temp, viscosity
drag force from energy transferred by random collisions
Stokes Einstein Equation ****really important
if assume solute is really small, assume it’s a sphere —> f = 6pi (n) (R)
really important bc you can also find the radius (how big your particle is)
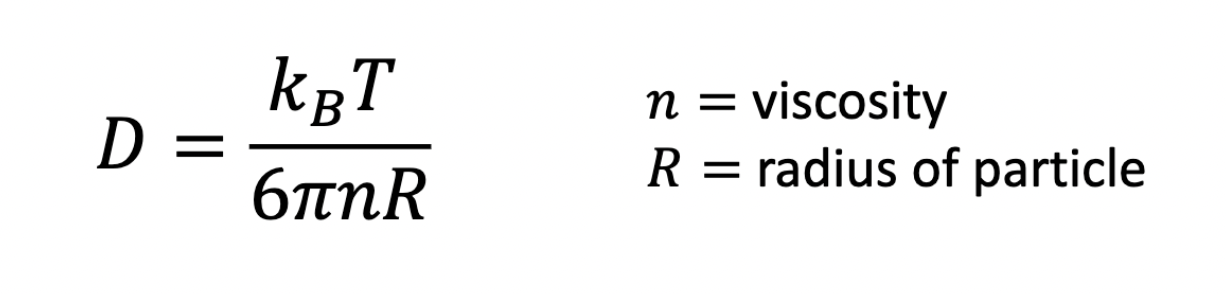
Fick’s 1st and 2nd Law

Steady state: dC/dt = 0
change in concentration over change in time
Nonsteady state (not at equilibrium): dC/dt = changing
means solute flux changes over time as drug reservoir is depleting
Permeability:
partition constant dependent on ionicity or hydrophilicity
Φ < 1: membrane surface concentration is less than solution
prefers to be in SOLUTION rather than in material
less solubility of solute in membrane
Φ > 1: membrane surface concentration greater than solution
prefers to be in MEMBRANE rather than in material
Φ =1: same surface concentration
Lesson 5: Modeling Drug Release
What are the different drug release mechanisms:
Dissolution, diffusion, osmosis, partitioning, swelling, erosion
How do we determine which drug release mechanism occurred?
given graph of time vs. cumulative release%
look at r² and if it’s similar to the model
Look at y = Ax + B —> K = 10^B —> n = A
n is what drug release mechanism
Fitting only valid up to 60% cumulative drug release
Types of Models:
Zero order
First Order
Hixson Crowell
Higuchi *
Korsmeyer Peppas **
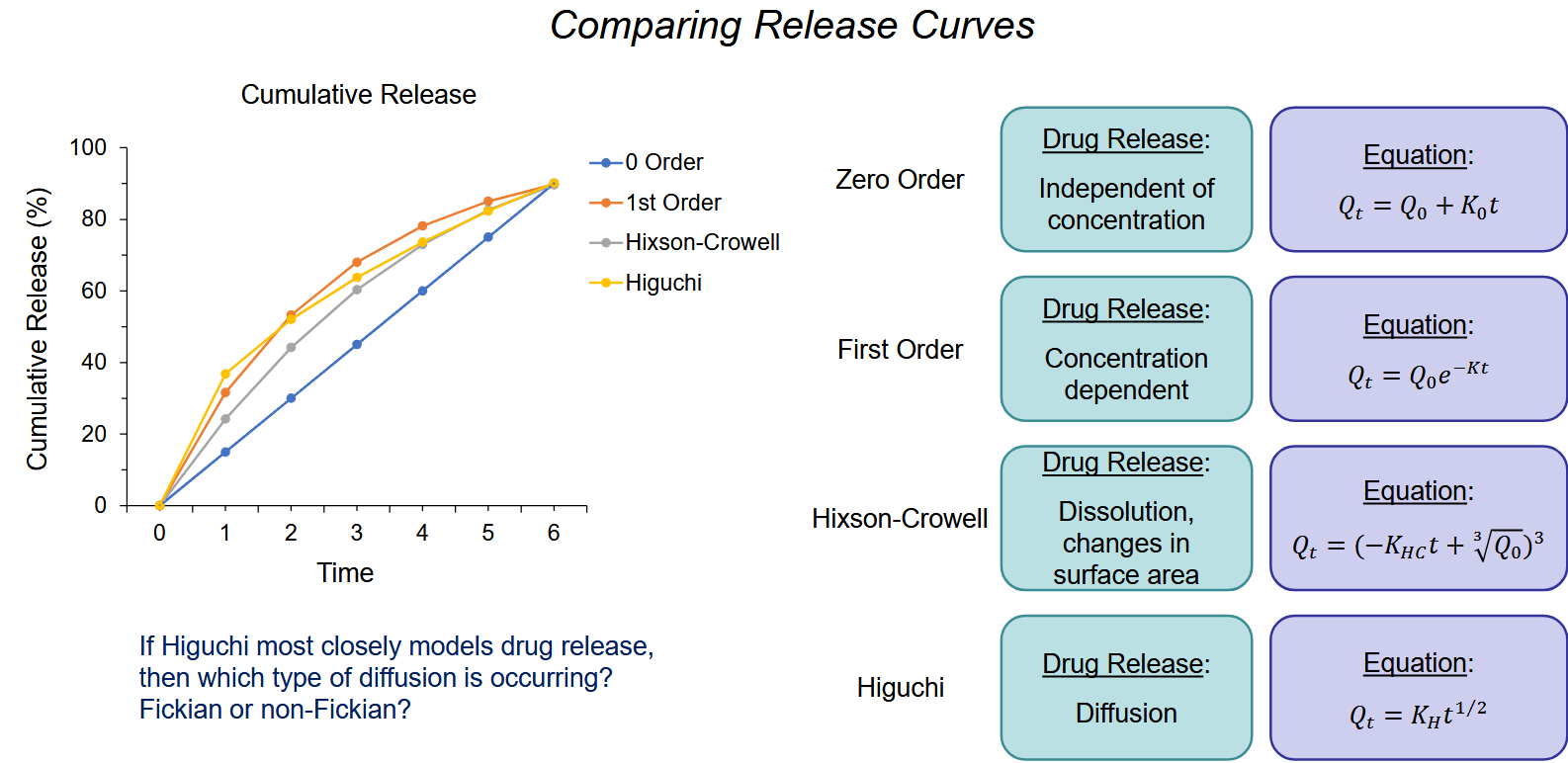
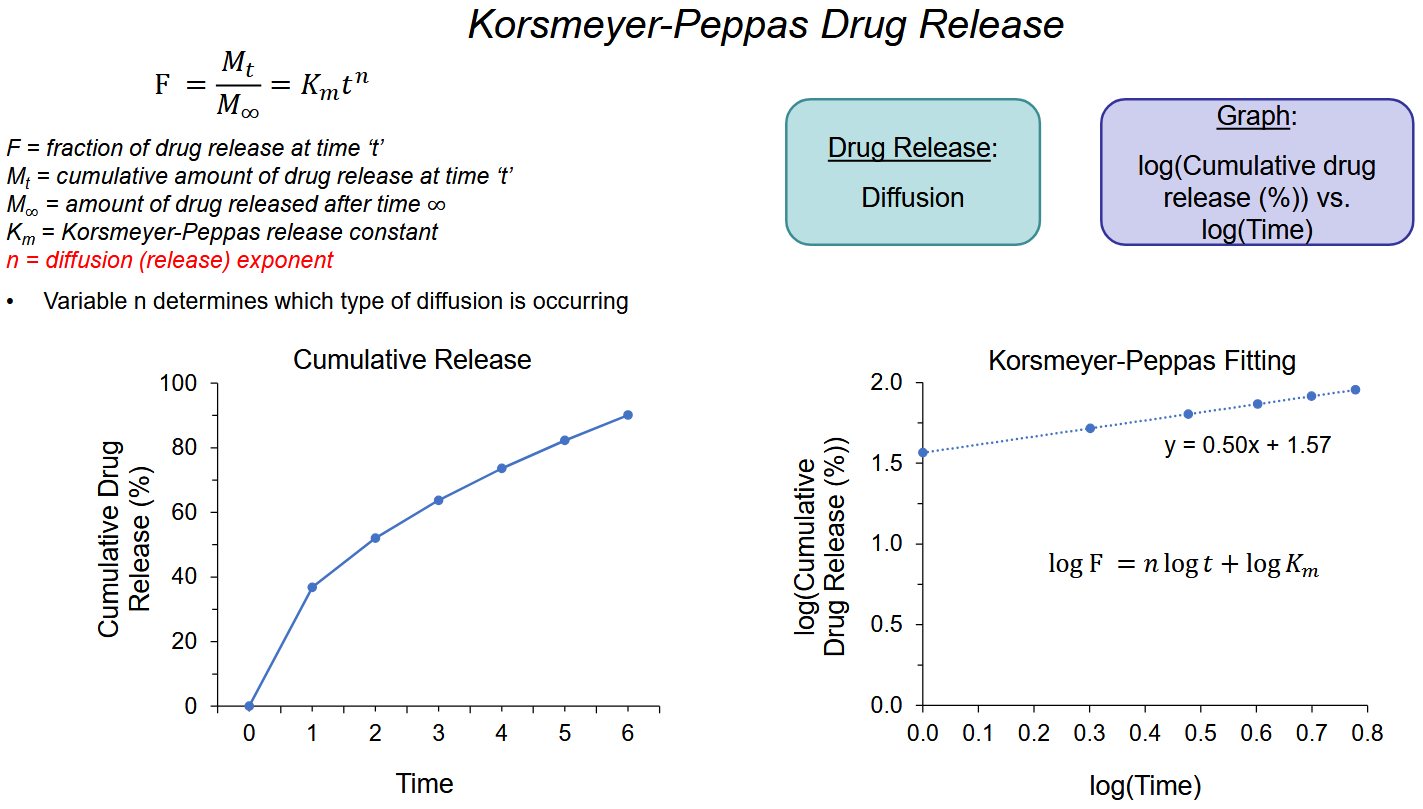
If it looks like higuchi —> korsmeyer peppas —> determine release mechanism

Fick’s Laws: How fast something dissolves
First law: how fast diffuses across barrior: flux (flow of solute) is proportional to concentration gradient
Second Law: concentration changes over time bc of diffusion
change in concentration with time in that region is proportional to change in concentration gradient at point in system
basically saying it goes from high to low until it becomes even concentration gradient then it slows down
Fickian: no deformation/stress to matrix; nonfickian: structual changes
Nonfickian: polymer properties make it release (erosion) NOT mass diffusion/concentration gradient
Miscibility: does it completely dissolve into a homogeneous mixture
important so drug releases uniformly + faster
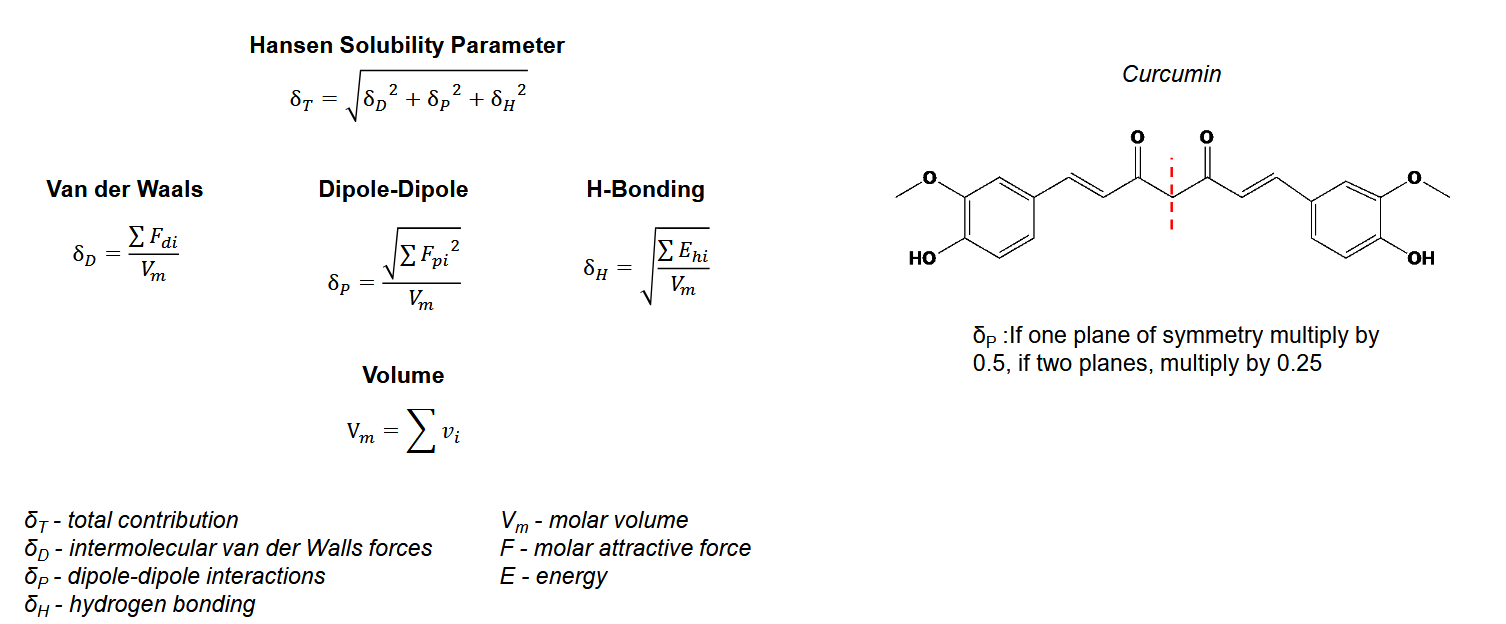
Flory HUggins: Is it miscible: x closer to 0 = more miscible the pair
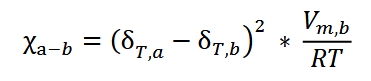
Lesson 6: Drug Delivery from Polymers
Porous Media:
can be regular array of lyndrical pores, foam, granular, fiber matrix
specific surface and porosity (0.9 in interstitial, 0.06 brain 0.3 skin, 0.6 tumor)
Solute transport behavior based on porosity (how much is pores) and pore connectivity
Nonpassing pore: one opening
Passing pore: two opening goes through medium
Isolated pore: just floating inside medium
Not all pores are accesible by solute (can be too small, connection is too small)
Size Exclusion Chromatograph: separates macromolecule by size through porous medium
Macromolecules can be flexible and can go through as a random coil
What influences pore permeability:
Size
Shape
Flexibility
Charge, charge-charge interactions in matrix
pH
Hydrophobicity/hydrophilicity
Ionic Strength
Dielectric Constant of Medium
Darcy’s Law: flow rate is directly proportional to the pressure gradient of medium and inversely proportional to the medium's resistance to flow
v = -K∇p
K is hydraulic conductivity constant, resistance to flow
inversely proportional to viscosity
p is pressure
Drug Eluting Polymers: norplant release hormone at constant rate (contraceptive)
Other release mechanisms: Triggered drug release, Hydrolysis, Polymer-drug interactions, Drug-drug interactions, Polymer relaxation, Pore closure, Heterogeneous degradation, Formation of cracks or deformation, Collapse of the polymer structure
Drug Release Profile:
zero order release profile: straight line
burst profile
moment it gets wet, drug comes out really quickly
NOT what you want unless you want fast acting
can put you over therapeutic window really quickly (bad), want sustained release so you can control it better
if you want to release burst release, don’t get it wet make it more hydrophobic
triphasic release:
initial first release, zero order, plateau (over)
phase 1: first release (looks like higuchi)
phase 2: steady state diffusion (constant zero order)
phase 3: polymer erosion (under)
hydrolyzes + pH change ==> degradation
bulk erosion: whole thing just bursts
surface erosion: outside dissolves, then only inside
biphasic release
inital first release, zero order (no plateau)
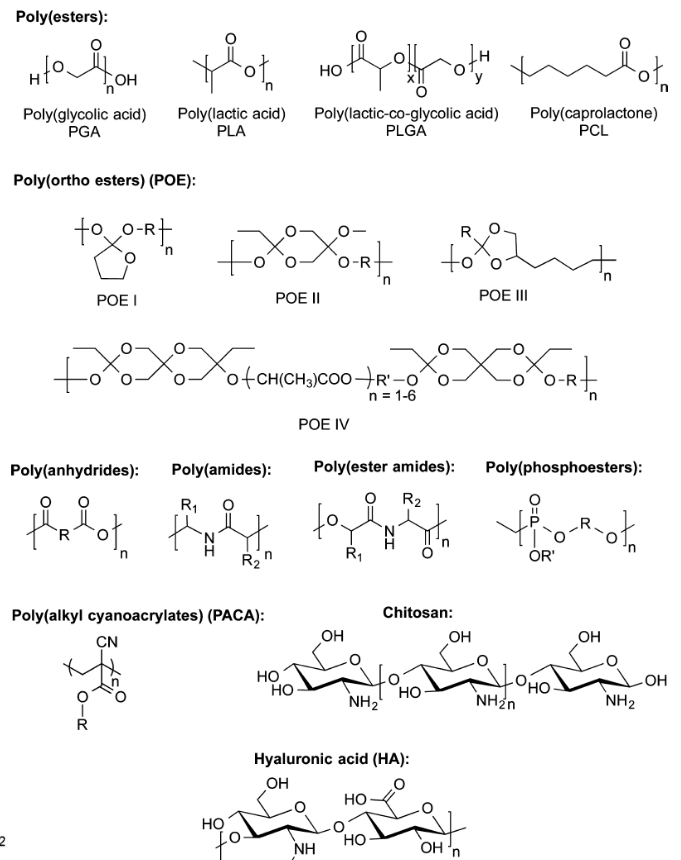
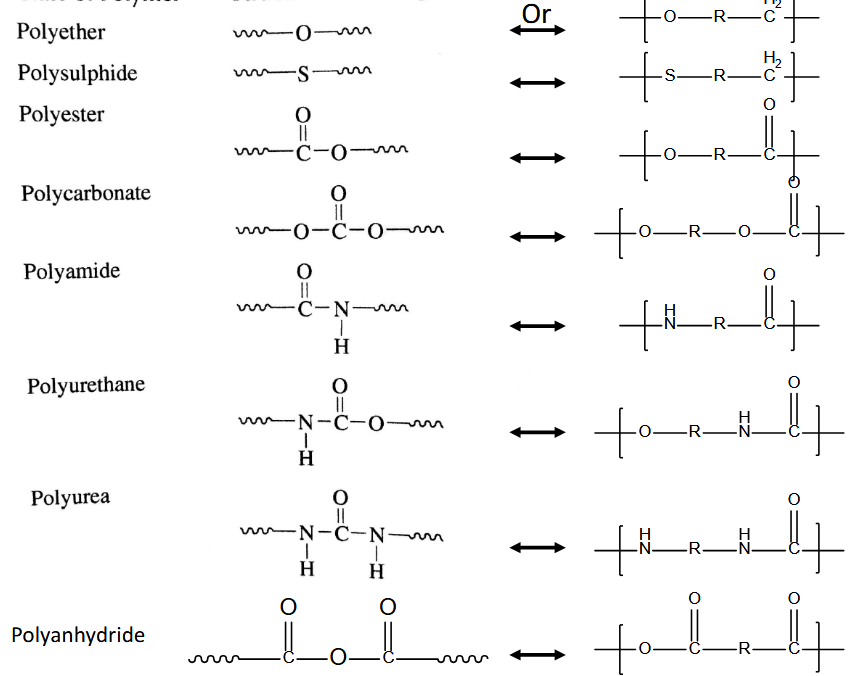
Lesson 7: Water Soluble and Degradable Polymers
Structures of Polymers:
Homopolymer: all monomer A
Copolymer: made of two monomers A and B
Polymer Blend: strands of only monomer A and strands only monomer B
Structure of Polymers:
Size and Structure
Polydispersity (nonuniformity)
Solubility
larger = less soluble
branched = more soluble
Stability (ph, temp, degrading)
Density
Crystallinity: strong IMF = high glass transition temp
reversible transition in amorphous materials from hard + glassy to viscous rubbery meaning it gets wiggly + elastic
Strength
Biodegradable Polymers:
Synthetic vs. natural
degradation through ester hydrolysis (more hydrophilic = faster)
crystallinity and water content
ester links (C=O-O)
What causes dehydration:
polymer hydration, crystalline, bond lability (how easy broken)
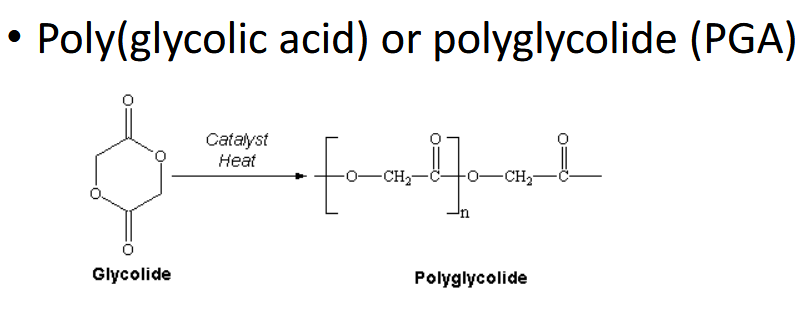
PGA - semicrystalline + more water soluble/degradable
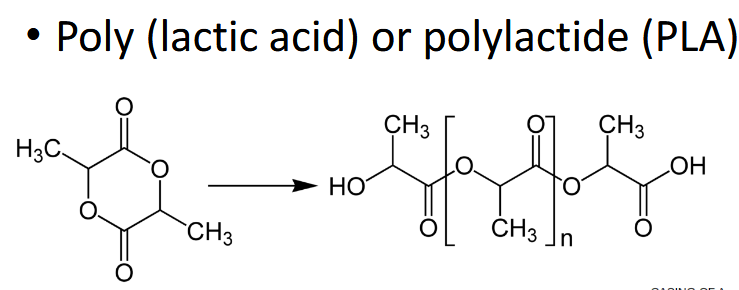
PLA is semicrystalline + more hydrophobic / stable
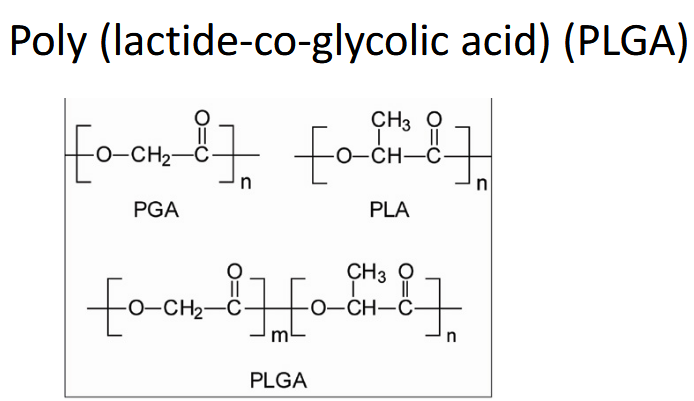
Copolymerization (combine) amorphous, inc in lactide/glycolide = inc crystallinity = slow drug release
If you change ratio, inc PLA degrades slower
If you add PEG blocks inc hydrophilicity of PLGA
Silicone (polysiloxanes)
easily cross linked at room temp, very flexible + elastic
Good as adhesive
If it looks wavy it’s a naturally derived water soluble polymer (ester)
Protein Polymers:
Collagen (gelatin)
Fibrin, elastin
Proteoglycan (filler)
Albumin (stability)
Decellularized ECM
Lesson 8 Polymer Synthesis and Characterization
Process for making polymer: Select monomer + distill (remove oligomers), polymerization, purification (manipulate solubility to leave behind), blending, casting, post processing
Polymerization:
step growth: add monomer or dimer or whatever fits (erosion profile), condensation (ester bond) and addition polymerization
chain growth: adding monomer to one end (burst)
1. Initiation, 2. propagation, 3. transfer, 4. termination
FRP (unpaired electrons) not very even, reversible deactivation has a step to slow it down to make it low polydispersity (RAFT)
living chain growth: zero order
Polycondensation: molecule is eliminated, not necessarily water (polyester, polyamide)
Nylon: alcohalide bonds w/ hydrophilic, monomer A + B not miscible, film and step growth at surface
Polyaddition: no elimination, glue/epoxy (polyurethane)
Ionic Polymerization: cationic/anionic electrophiles start and only uses vinyl monomers
Ring opening polymerization: polyether, polyester, polyamide
Mixing monomers:
block copolymer: AABB
link end groups, polymerize different monomer, end of first is macroinitiation for other reaction, end of first initiate next polymer chain
random copolymer: ABAAAB
alternate copolymer: ABAB
Polymer Characterization Methods
Size and Mw: Size Chromatography, Light scattering, mass spec
Chemistry: Nuclear magnetic spectroscopy, absorption spec (IR Raman)
Physical Properties: Mechanical (stress/strain), diff scanning calorimetry (glass temp), thermogravimetric analysis (mass over time as T inc for stability)
Lesson 10: Self Assembly
Surface free energy controls what happens at the biointerface
NEED positive energy to create unit area of surface
Equilibrium by minimizing surface energy —> curve
polar outside, nonpolar inside
Maximize entropy
Condensation: if greater than opp of dissolving occurs
u > 3/2 KT
Self energy (u): strength of interaction of molecule and neighbor
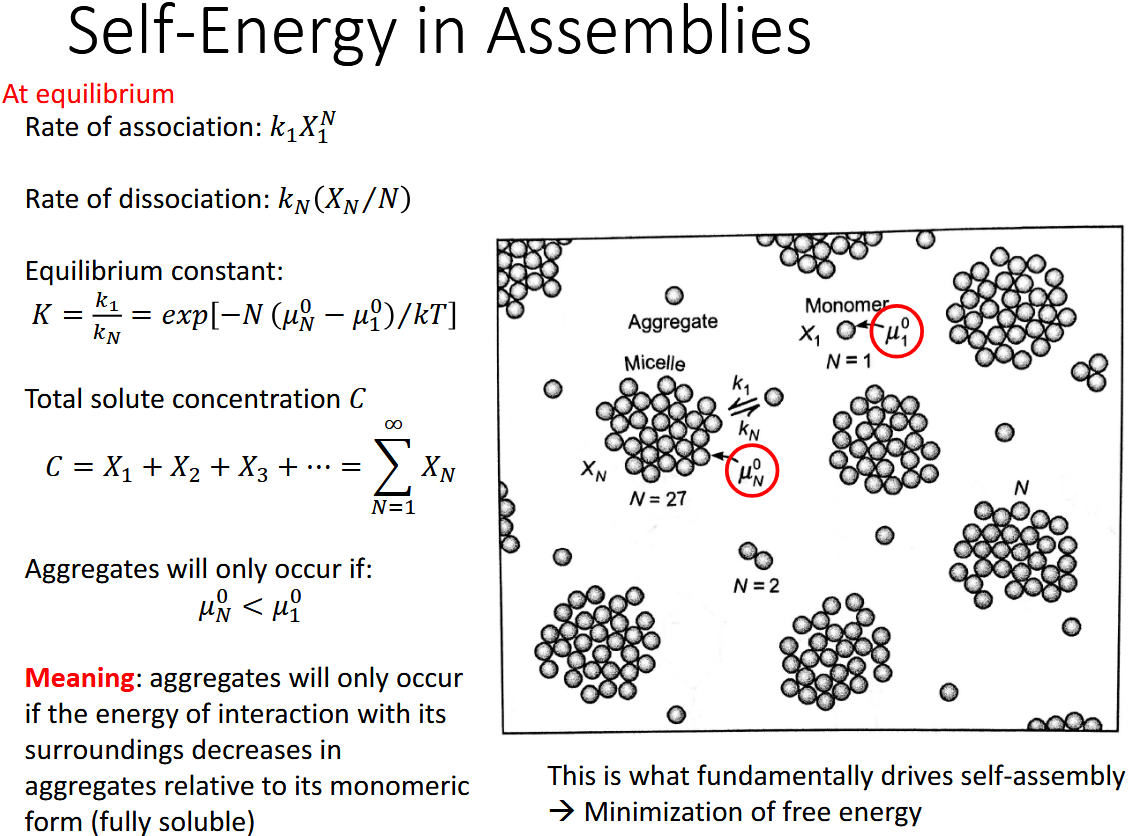
Aggregates form if u0n (interaction free energy per molecule) decreases with number of molecules
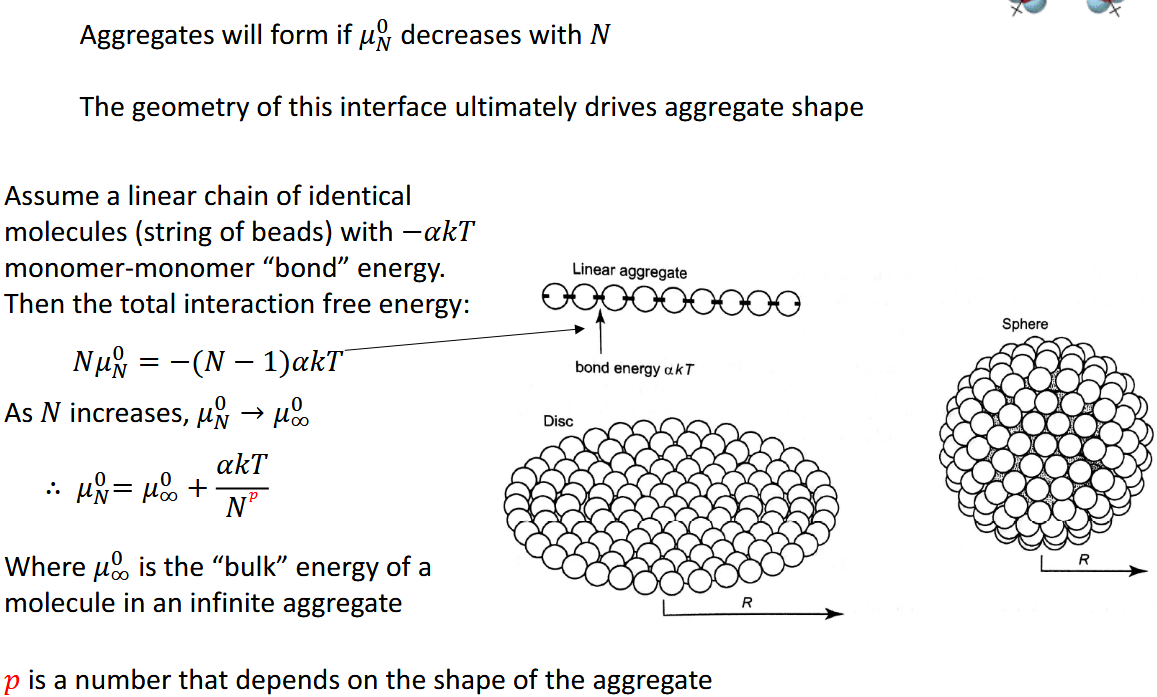
At low concentrations —> favorable to exist as monomers
Increase concentration until Critical Micelle Concentration (CMC) —> form aggregrate
Hydrophobic tails want to decrease a (interfacial area, a), reduce water exposure
Hydrophilic heads want to increase a (interfacial area), spread apart
C = v / (a * l)
C is critical packing parameter
v is volume of hydrocarbon chain
l is critical chain length
a is optimal area for headgroup
the bigger the C the more closely the heads wanna get together
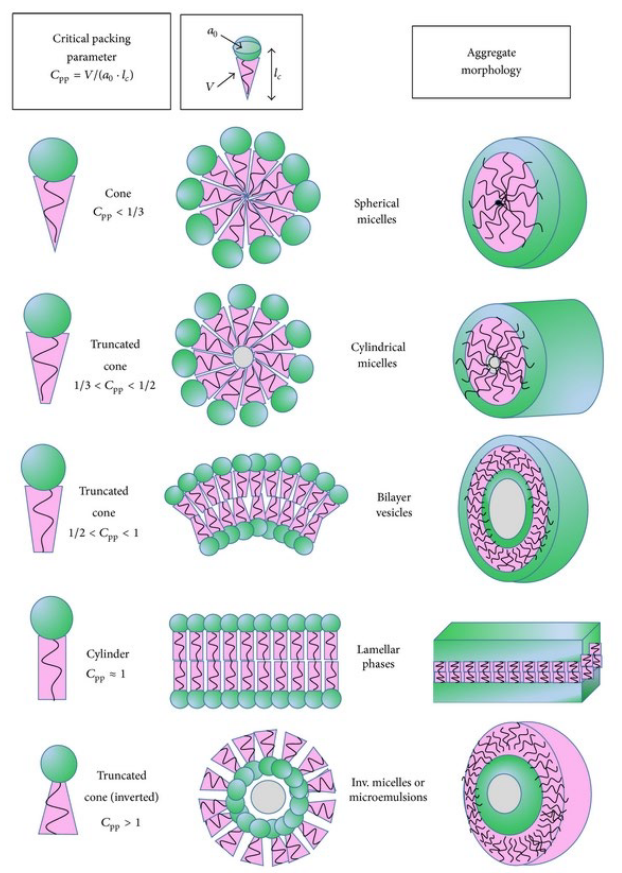
Also control self assembly:
Amphiphile concentration
solvent system
rate of addition
temperature
sonication
Higher Ordered Structures:
first create spherical micelle —> self assemble into rod —> stack on each other to make a group (silica condenses on outside) —> calcinate which removes micelles (solvent) —> fully silica (porous sand, you can put A LOT of drugs)
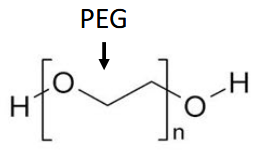
PEG (PEO) = Hydrophilic shield that gives stability, solubility, and stealth
PPO = Hydrophobic
The PEG PPO PEG sandwich is a triblock polymer
What are some ways that they can make specific interactions besides polarity
Host-Guest: specific size
host: cyclodextrine (ring of sugar) w/ hole in center
guest: hydrophobic (adamantane)
noncovalent bond so no catalyst required
How to get guest out: shear stresses
Organometallic:
Pi pi stacking: 2 aromatic groups noncovalent on top
Hydrogen bonding: DNA
Layer by Layer: coiled coils (alternating polymer peptide and silica templates can dissolve to get the capsules
Either lasagna or a ball
Lesson 11: Hydrogels
What is a hydrogel: network of cross linked polymer chains
spaces that can absorb/trap a lot of water
insoluble (swell and do not dissolve)
soft and flexible
Gelation: process where solution of polymers floating around become crosslinked and fills in a 3d space making a gel
Chemical Hydrogels
covalent bonds = generally permanent
Physical Hydrogels
noncovalent bonds (weaker) = reversible
How to make a hydrogel:
Inputs (solution phase)
Generation: put liquid into polymer matrix
Consumption: monomer consumed by substrate, agents consumed, chemical activation
Output: gel phase
How to measure hydrogel:
Chemical:
GPC/SEC
NMR
Plate Reader
Physical:
Mechanical Tester
Scale
Rheometer
Optical
Microscopy
Light Scattering
Increasing crosslinking = increase viscosity — stiffer = higher shear rate required for induced flow
More crosslinking = stiffer (high young’s modulus) = lower swelling
Fick’s Law: how fast drug can move THROUGH polymer network (hydrogel)
Dependent on pore structure, cross-linking, interaction w/ solutes
Hydrogels not just for storing water but can encapsulate (trap)
put drugs or actuators inside the pockets within a hydrogel
hydrogel protects it or controls release
Lesson 12: Drug Carriers
Liposomes: vesicle where aqueous volume on inside enclosed by phospholipid bilayer
from 20nm to several micrometers
can be multilamellar: concentric number of phospholipid bilayers each separated by an aqueous phase (way bigger)
Parts of a Liposome:
Coating
PEG (stealth cloak so it’s not destroyed immediately)
Targeting agent (bind to specific things so it releases at site)
Phospholipid
Cholesterol: stability
Payload
Hydrophobic drugs in the bilayer
Hydrophilic drugs in the core
Phosphatidylcholine (naturally in cell membrane) amphipathic
Hydrophilic heads
Phosphate group + choline group
Glycerol backbone
hydrophobic tail
2 fatty acid chains
kink (double bond) if unsaturated
10-24 carbons, 0-6 double bonds in chain
Drug Release Mechanisms: how does it get inside a cell
Endocytosis: carrier gets inside, whole thing eaten by endosome, lysosome degrade it and lets drug in **most common
Fusion: carrier fuses w/ membrane, letting drug in
Adorption: drug sticks to membrane and something else happens idk
Lipid exchange: drug slips between gap during exchange
Doxil: lipsome that carries chemotherapy drugs for cancer (hydrophilic)
when it dries it crystallizes as a rod
—
Lipid Nanoparticle (LNP)
Encapsulates DNA/RNA
It’s positively charged so the DNA stays on the inside
No bilayer, lot of lipids packed together (condensed) and DNA is at center
4 Lipid LNP System:
Helper Lipid: for bulk
Ionizable Lipid: makes it positive
Cholesterol: stability stiffen
PEG: makes sure it doesn’t get blown up and not stay floating around for too long
How lipid nanoparticles get inside and delivers mRNA:
put desired mRNA gene inside
Gets into cell membrane through endocytosis
Escapes endosome
mRNA can go downstream to create ribosome which expresses whatever tf
Micelles: kind of like a lipsome except it doesn’t fold on itself with head then tail inside, liposome has a bilayer with head, tail, head, core
How to get the drug inside a micelle:
chemical conjucation to polymer backbone
physical entrapment: dialysis or emulsification
How to release the drug from the micelle:
diffusion
dissociation
pH
temperature
cleavage biomolecules
unimer (strand) that cleaves (polymer drug bond) micelle
Gold nanoparticles:
really fucking small (wavelength of light)
inert and biocompatible
drug on the surface: covalently bonded
target specific if carrying ligands
Silica Nanoparticles:
either solid or porous
Drug or imaging agents inside pores
**can have gold outside silica inside
Quantum Dots:
made of cadmium (toxic)
extremely small, fluorescent (easily visible)
drugs, targeting ligands, polymers inside
Equations:
Thermodynamics:
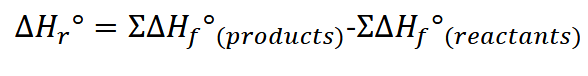
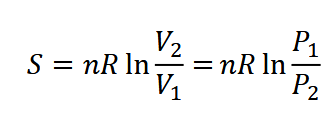
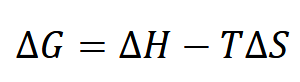
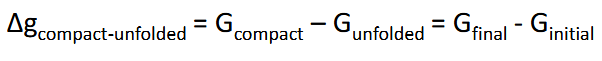

Dissolution: how fast it dissolves (assume stationary layer of thickness h)
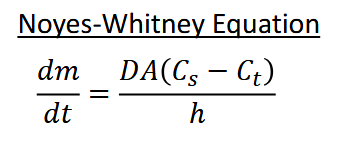

Will it dissolve:
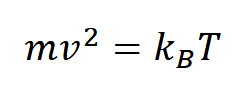
How does it spread out over time?
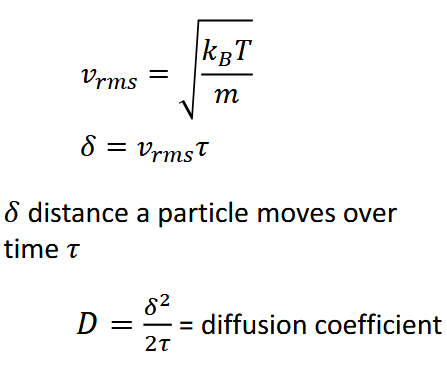
Will condensation occur? (opposite of dissolve)
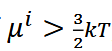
How to calculate Self Energy:
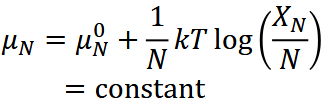
Permeability

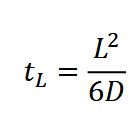

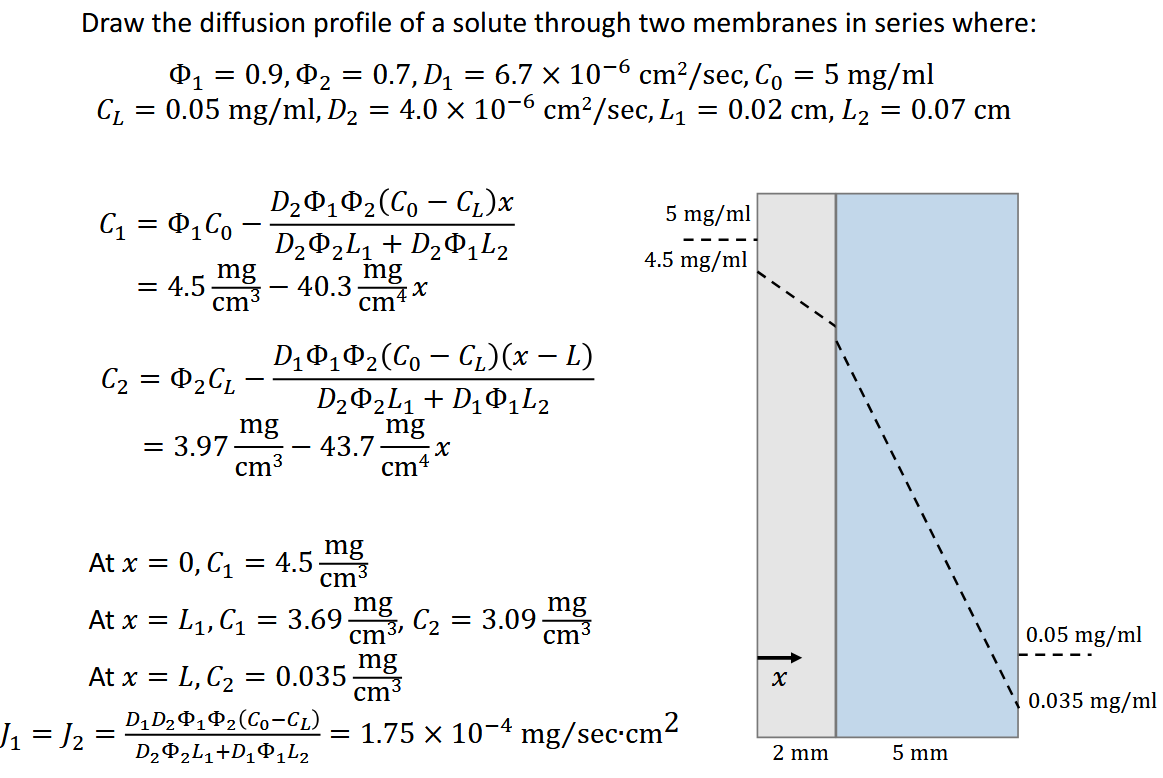


Porosity:

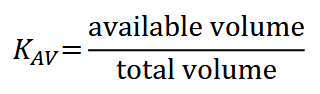

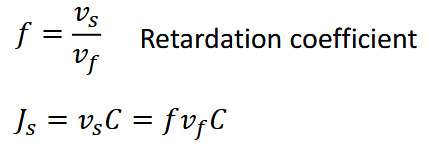

Structures: