Genomics and Bioinformatics Comprehensive Bioinformatics Guide
Learning Objectives for Genomics and Bioinformatics
Definition Proficiency: Recall and provide precise definitions for genomics and bioinformatics.
Technological Context: Explain how the exponential development in various technologies enabled the field of genomics.
Interdisciplinary Integration: Illustrate how genomics specifically benefits from the integration and use of bioinformatics.
Key Inquiry: List the fundamental questions that can be answered through genomic and bioinformatic research.
Biological Understanding: Explain why genomic information is a critical key to understanding life itself.
Collaborative Dynamics: Recognize the essential role that international collaboration plays in modern genomics.
Definitions and Core Concepts of Genomics and Bioinformatics
Genomics: The study of genomes—defined as the entirety of an organism's genetic material—and how that information translates into biological function.
Methods have been developed to accumulate and understand genomic information.
Genomics involves processing, storing, and sharing large amounts of high-complexity data.
Bioinformatics: The application of mathematics, statistics, and computer science to better understand the genome. It serves as the analytical engine of genomics.
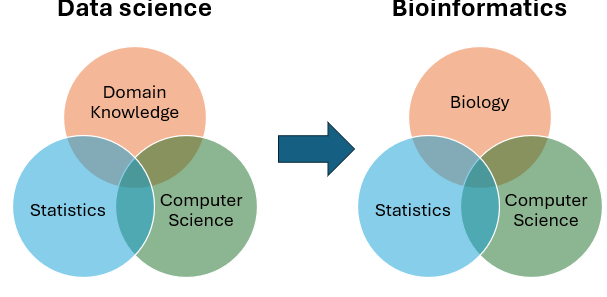
Bioinformatics as Data Science: It is specifically the application of data science to biology. It exists at the intersection of three pillars:
Domain Knowledge: High-level understanding of Biology.
Computer Science: For algorithmic development and processing power.
Statistics: To ensure data validity and extract patterns.
The Co-Evolution of Genomics and Computing
Scaling Together: Genomics and computer science scaled in tandem. As the number of available sequences increased, the field required:
Better algorithms to process genetic strings.
Increased computing power for complex calculations.
Expanded storage space for massive data repositories.
Biotechnological advances were inversely made possible by better computing power.
Role of the Internet: The Internet evolved alongside computation and genomic technology.
Development timeline: From ARPANET to the modern multi-billion user Internet.
Enabled the dissemination of genomic information.
Facilitated bioinformatic development through the sharing of software.
Supported large-scale international collaboration (e.g., the Human Genome Project).
Comprehensive Timeline of Genomic Discovery (1869–2024)
1869: DNA (originally called 'nuclein') was isolated by Friedrich Miescher.
1944: DNA was identified as the hereditary material by Avery, MacLeod, and McCarty.
1953: The DNA double-helix structure was solved by Watson, Crick, Franklin, and Wilkins.
1965: The first nucleic acid sequence was completed (Yeast tRNA).
1977: Sanger Sequencing was developed, and the first DNA genome () was sequenced.
1983: PCR (Polymerase Chain Reaction) was invented.
1984: First ancient DNA (aDNA) was sequenced from the extinct Quagga.
1990: The Human Genome Project (HGP) was officially launched.
1995: Sequence of the first bacterial genome (H. influenzae).
1996: Sequence of the first eukaryote genome (Yeast).
1998: Sequence of the first animal genome (C. elegans).
2000: Blair/Clinton Joint Statement declaring human genome data free and public. First plant (Arabidopsis) and fruit fly genomes sequenced.
2001: Publication of the first draft of the Human Genome.
2002: Release of the Mouse Genome Draft.
2003: Human Genome declared 'Complete' () and ENCODE Phase 1 launched.
2007: Development of Next Generation Sequencing (NGS).
2008: Launch of the 1000 Genomes Project and the NIH Roadmap Epigenomics Project.
2009: Genome 10K (G10K) launched to sequence vertebrate diversity.
2010: Drafts of the Neanderthal and Denisovan genomes released.
2011: BLUEPRINT Epigenome (EU) and 10k Bird Genome (B10K).
2012: High-coverage Denisovan genome and ENCODE Phase 2 results.
2013: SCOTUS (Supreme Court of the US) bans patents on human genes.
2014: Long read sequencing technologies emerge.
2015: Final results of 1000 Genomes Project and NIH Roadmap Epigenomics.
2016: All of Us project launch.
2017: Vertebrate Genomes Project (VGP) launch and gnomAD release.
2018: Earth BioGenome Project, Darwin Tree of Life, and Genomics England 100k Genomes / UK Biobank Exome release.
2020: PCAWG (Pan-Cancer Analysis of Whole Genomes) results.
2021: African BioGenome Project launch and sequencing of million-year-old Mammoth DNA.
2022: T2T Human Genome (Telomere-to-Telomere, complete) and ENCODE Phase 4.
2023: Zoonomia Project ( mammals comparison) and Human Pangenome Reference Draft.
2024: UK Biobank 500k WGS (Whole Genome Sequencing) and T2T Bread Wheat genome assembly.
Reference Genomes of Key Model Organisms
Bacteriophage MS2: Virus (RNA); sequenced in 1976; size ; First RNA-based genome.
Bacteriophage : Virus (DNA); sequenced in 1977; size ; First DNA-based genome.
Haemophilus influenzae: Bacterium; sequenced in 1995; size ; First free-living organism sequenced.
Saccharomyces cerevisiae: Yeast; sequenced in 1996; size ; First eukaryotic genome.
Methanococcus jannaschii: Archaea; sequenced in 1996; size ; First Archaea genome.
Caenorhabditis elegans: Roundworm; sequenced in 1998; size ; First multicellular animal.
Arabidopsis thaliana: Plant; sequenced in 2000; size ; First plant genome.
Drosophila melanogaster: Fruit fly; sequenced in 2000; size ; First insect genome.
Homo sapiens: Human; sequenced in 2001 (Draft); size ; First mammal sequenced.
Mus musculus: Mouse; sequenced in 2002; size ; Primary mammalian model.
The Human Genome Project (HGP) and Assembly Challenges
Competition vs. Collaboration:
The International Human Genome Sequencing Consortium (launched 1990) represented a public effort.
Craig Venter, CEO of Celera (founded 1998), led a competing private effort.
Initial planning occurred at the Banbury meeting in 1989.
Genome Assembly Puzzle: Assembling a genome requires piecing together millions of short fragmented reads like a puzzle.
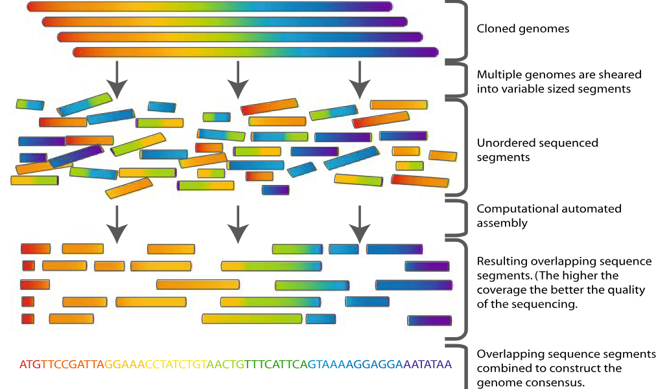
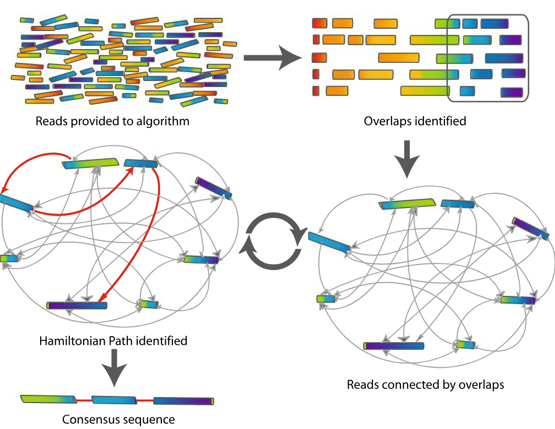
Evolution of the Human Genome Assembly:
The first release was in 2002, but it was not truly "complete" until 2022.
Frequent version updates require researchers to reprocess old data on new assemblies.
Difficult Regions: Centromeres, telomeres, and other highly repeated regions were the hardest and last to be sequenced.
Long-read technology: Technologies like Nanopore and PacBio were essential to fill the final gaps in the assembly.
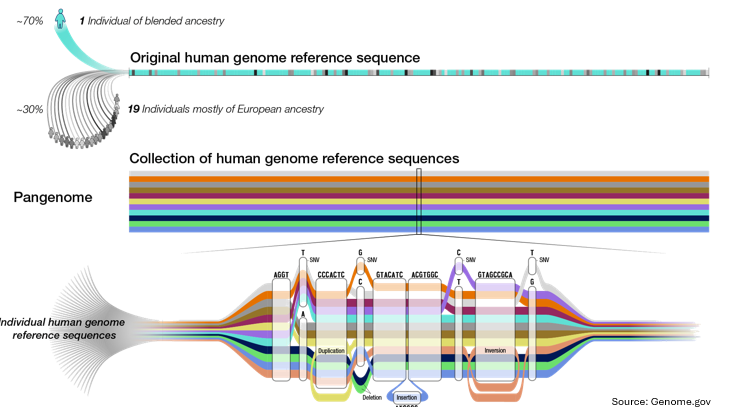
From Single Reference to Pangenome:
The original human genome reference sequence consisted of from one individual of blended ancestry and from 19 individuals (mostly European).
A Pangenome is a collection of diverse reference sequences that capture better human variation, including SNVs (Single Nucleotide Variants), indels (insertions/deletions), duplications, and inversions.
Gene Annotation and Functional Identification
Gene Annotation Procedures: Software programs use models to detect Open Reading Frames (ORFs) and annotate DNA sequences.
computer programs have been developed to find protein coding genes within genomes
Key Genomic Markers:
TATA box (consensus: )
CAAT box
Translation initiation sites
Splice sites (Exons and Introns)
Stop codons and the Start codon (ATG)
Poly-A addition sites (consensus: )
Transcriptomics Support: Sequencing RNA (the transcribed DNA) is critical for identifying protein-coding genes and the exact location of exons.
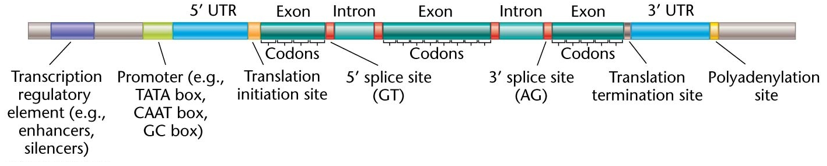
Protein Prediction: Once coding genes are identified, knowing the genetic code allows for predicting the amino acid sequence starting with Methionine (Met) with a few specific codons to encode for a stop signal.
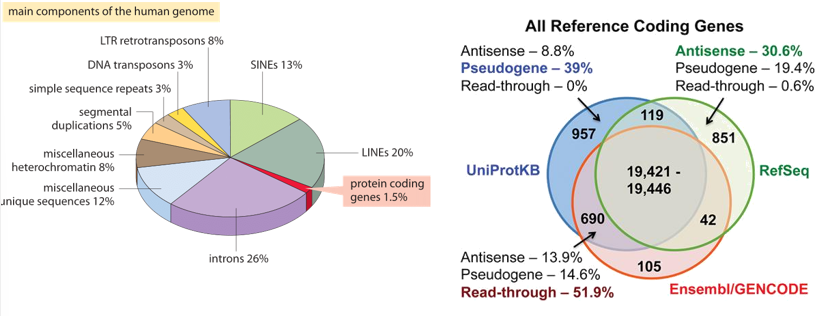
Composition of the Human Genome
Human Gene Paradox: Humans have fewer protein-coding genes than expected. There are approximately to protein-coding genes.
Non-Coding Dominance: Most of the human genome is non-coding.
Protein-coding genes (UniProtKB): of the genome.
Introns:
LINES (Long Interspersed Nuclear Elements):
SINES (Short Interspersed Nuclear Elements):
LTR retrotransposons:
DNA transposons:
Simple sequence repeats:
Segmental duplications:
Miscellaneous heterochromatin:
Miscellaneous unique sequences:
Gene Definitions by Database (Examples):
RefSeq: coding genes with pseudogenes.
Ensembl/GENCODE: coding genes with pseudogenes.
Bioinformatics Tools and File Formats
Genome Browsers: Tools like the UCSC Genome Browser (Human Feb. 2009 GRCh37/hg19 assembly) allow researchers to visualize:
Gene locations (e.g., SOD1, SCAF4).
Transcription levels (RNA-seq).
Epigenetic marks (e.g., H3K27AC).
Conservation across species (e.g., Lamprey, Zebrafish, Chicken, Dog, Mouse).
SNPs and RepeatMasker regions.
File Formats:
FASTA/FASTQ: Sequencing data (text-based).
Wig / bigWig: Genomic continuous data.
SAM / BAM: Mapping locations (BAM is binary).
VCF: Genetic variants.
BED: Genomic intervals.
MAF: Multiple alignments.
Searching and Mapping:
BLAST: A heuristic tool for fast "fuzzy" searches to identify similar sequences in massive databases.
NGS (next generation sequencing) Mapping: allows for experiments producing millions of short reads. Need to know for each of these where they best match the reference genome. development of fast, fuzzy algorithms to map short DNA sequences using indexes, some algorithms trade accuracy for more speed. Some need specialized for reads coming from spliced RNA or modified DNA (methylation)
identification of conserved sequences
comparison of species
can be done using DNA sequences directly or translated protein sequences
multiple alignment software is used (CLUSTAL omega)
allows identification of functional regions in proteins as theyre likely to be conserved between species
allows study of how protein sequences encoded by conserved genes have changed during evolution
multiple alignment algorithms
algorithm - sequence of steps to perform a function
needleman-wunsch: finds the best global alignment
smith-waterman: finds the best local alignment
both guarantee finding the best alignment but need to compute the whole matrix
Functional and Evolutionary Genomics
Functional Annotation Projects:
ENCODE: Large-scale collaboration to decrypt the function of "junk DNA," focusing on Transcription Factor (TF) binding sites, gene expression, epiginetic modifications and 3D contacts.
Gene ontology: Hierarchical classification of known genes on multiple dimensions (biological process, molecular function, protein domains, gene expression cellular context, etc)
NIH Roadmap: Integrative analysis of 111 reference human epigenomes across various cell types.
Transcription Factor Binding Site (TFBS):
Proteins bind specific DNA motifs often represented by Position Weight Matrices (PWMs).
JASPAR is a key database for experimentally validated transcription factor motifs.
Hidden Markov Models (HMMs) are used to predict TF binding sites by scanning genomes with algorithms.
anything in the genome can be defined as a genomic interval, using bioinformatics tools such as bedtools you can manipulate and combine these
Comparative Genomics:
Human vs. Chimpanzee: DNA is similar in coding regions with similar orthologues, but whole-genome similarity is due to TE (Transposable Element) insertions and indels. Approximately of genes differ by at least one amino acid.
Specialized Genomic Applications
Ancient DNA (aDNA): Studying shattered, chemically degraded DNA (e.g., Neanderthals, Mammoths).
algorithms to correct sequence obtained from fragments of DNA molecules damaged by time
useful to refine human history, epidemics, domestication of animals, ancient crops, etc.
comparison of genomic sequences between species allow for the inference of their evolutionary relationship
Svante P: Awarded the Nobel Prize in 2022 for his work on ancient genomes.
Record holder: Mammoth DNA fragments dating back million years.
Epidemiology: Real-time analysis of viral evolution (e.g., COVID-19, Nextstrain.org) and tracking antibiotic resistance. rapid diagnostic, sequencing and large scale efforts allows for almost real time analysis of viral evolution.
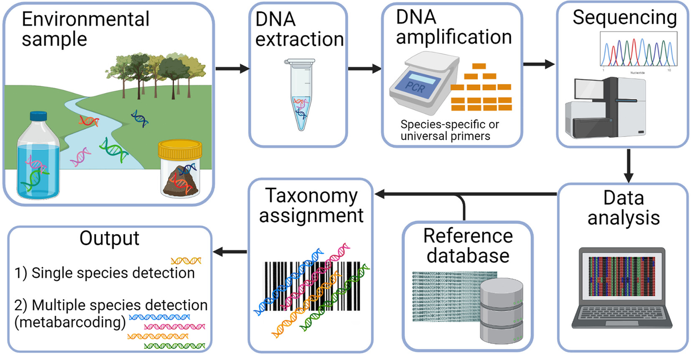
Metagenomics and eDNA: Identifying bacteria/organisms in soil, gut, or water samples without needing to grow them in a lab.
Medical Genomics:
GWAS (Genome-wide association study): Statistical framework analyzing massive datasets to link phenotypes (like height or diabetes) to specific genomic loci, often visualized in a Manhattan plot. analyses massive amounts of genomic data and identify likely genomic loci that are linked to phenotypes of interest
Cancer Genomics: Tracking somatic mutations and inherited risks across thousands of samples using databases like Xena. understand how cancer develops and how best to treat it.
Summary of Principles
Genomics and bioinformatics have a symbiotic evolutionary history.
Bioinformatics is the essential processing layer for genomic information.
Massive current scales permit a functional understanding of genomes once thought to be "junk."
The search for genetic causes of human disease is at an foundational, ever-expanding stage, reliant on open-source software and generous international funding.