Lecture 24 - Immunodeficiency
primary (congenital/inherited) immunodeficiency
innate immune deficiencies
leukocyte adhesion deficiency
defects in integrins or other molecules involved in cell extravasation out of blood vessels
Pathophysiology: Mutations in integrins or related adhesion/cytoskeletal components disrupt selectin-glycoprotein rolling, integrin–ICAM firm adhesion, and transmigration (extravasation), impairing neutrophil slowing and squeezing between endothelial cells
Clinical impact: Neutrophils (and monocyte-derived macrophages) fail to reach infection sites, limiting inflammatory cell recruitment; macrophages may still arrive, but neutrophil absence is prominent
chronic granulomatous disease
Molecular defect: Mutations in components of the phagocyte oxidase (NADPH oxidase/“phox”) complex reduce reactive oxygen species (ROS) generation in phagolysosomes.
Reactive nitrogen species interplay: Nitric oxide (NO) production remains intact, but ROS deficiency prevents formation of combined reactive nitrogen species (e.g., peroxynitrite, ONOO−), reducing microbicidal capacity.
Susceptibility: Increased risk for bacterial and fungal infections, including opportunists such as Aspergillus fumigatus, due to impaired intracellular killing after phagocytosis
B and/or T cell deficiencies
severe combined immunodeficiency (SCID)
individuals that lack both T and B cells
Definition: Severe combined immunodeficiency; mutations in recombination/repair genes lead to absent T and B cells and loss of adaptive immunity
Clinical consequence: Profound susceptibility to bacterial, viral, fungal, and other infections; innate immunity is insufficient to compensate
agammaglobulinemia
functional T cells but defects in B cell development
Concept: Absence of immunoglobulins due to failed B-cell development while T cells may remain functional
Infection profile: Greater susceptibility to bacteria, fungi, and helminths (processes requiring antibodies for neutralization, opsonization, degranulation); viral infections are less impacted relative to bacterial/fungal when T cells are intact, though neutralizing antibodies also aid in viral control
defects in T cell development and maturation
defects in T and/or B cell activation
defects in genes specific to T cell development also inhibit B cell-mediated immunity
TCR complex signaling
hyper IgM syndrome — defects in B cell instrinic activation of isotype switching (AID required)
Absence of T cells or defective CD40–CD40L interaction prevents isotype switching and germinal center reactions
AID deficiency blocks somatic hypermutation and class switch recombination, locking antibody production at IgM
Downstream signaling defects (e.g., NEMO in NF-κB pathway) can similarly impair switching
outcome: elevated IgM with paucity of switched isotypes (IgG, IgA, IgE), leading to impaired responses to many pathogens
defects in genes involved in T cell-mediated activation of B cells
CD40/CD40L (also important for Th1 activation of macrophages)
defects in CD8+ T cell induction of apoptosis
Perforin mutations: Even with T cells present, perforin defects abrogate cytotoxic granule-mediated apoptosis of infected cells, impairing CTL responses
Macrophage activation: T helper cells are required for activating macrophages to kill intracellular microbes; T-cell deficiencies compromise macrophage control of infections
includes mutations of genes required for VDJ recombination
RAG1/2 - recombination activating genes
Artemis complex - component of recombination machinery, associated with RAD50 complex
DNA ligase 4 - required for sealing DNA breaks via phosphodiester bond formation
DNA-PKcs - crucial for DNA repair during recombination
T cell-development and -independent activation revisit
T cell-independent activation require multivalent binding
Signal 1: BCR binding antigen.
Signal 2: Innate co-receptors (e.g., PRRs or complement receptors) provide costimulation.
Signal 3: T-cell help via CD40–CD40L and cytokines leading to germinal center reactions.
does not allow for somatic hypermutation and affinity maturation, isotype switching, and memory formation
Downstream processes: Cyclin D1/myc-driven proliferation; AID-mediated somatic hypermutation and isotype switching; differentiation into plasma cells and memory B cells
T cell–independent activation: B cells can be activated without T-cell help but do not undergo somatic hypermutation, isotype switching, or memory formation; output is predominantly IgM
treatments for primary immunodeficiencies
hematopoietic stem cell transplant
addresses root causes by reconstituting hematopoiesis; bone marrow transplant for T- and B-cell developmental defects
intravenous Ig
gene therapy
gene editing
CRISPR used to correct specific mutations in DNA associated with immunodeficiencies
passive immunity
administration of antibodies (e.g., IVIG) to compensate for humoral defects; feasible even when T-cell transfer is not practical
secondary (acquired) immunodeficiency treatments
chemotherapy for cancer by myelosuppression affecting precursors for all blood cells, reducing leukocyte development, resulting in immunodeficiency
bone marrow cancers (metastasis & leukemia) occupy/reduce marrow niches and diminish leukocyte development capacity
protein-calorie malnutrition energy-intensive lymphocyte development (recombination, selection) falters without adequate nutrition; traditional admonitions to “eat well” reflect genuine immunologic needs
splenectomy
Phagocytosis impact: Loss of a major macrophage reservoir diminishes blood-borne pathogen clearance
Filtering role: Spleen filters blood, catching pathogens; removal impairs pathogen capture
B-cell maturation: Spleen houses B-cell maturation; removal reduces capacity for efficient antibody responses
immunosuppressive for graft rejection and inflammatory diseases
primarily used for organ/cell transplantation or autoimmune disease
historically small molecule inhibitors
glucocorticoids/corticosteroids
nature and entry: Steroidal lipids traverse membranes easily; bind intracellular glucocorticoid receptor (GR), the receptor for cortisol, the stress hormone
nuclear actions: GR translocates to nucleus; binds glucocorticoid response elements (GREs) to activate/suppress target genes; can also bind NF-κB (and other transcription factors) to inhibit its DNA binding, suppressing pro-inflammatory gene expression
outcome: Broad repression of NF-κB–target genes and inflammatory cytokines; metabolic effects also noted
mimics of the endogenous hormone cortisol “stress hormone”
affects metabolism and inflammation
bind to glucocorticoid receptor (GR) — directly bind to promoters in DNA to activate expression of anti-inflammatory genes
bind to other transcription factor complexes to inhibit activity
tran-repression
NF-kB transcription factor is a targe
activates expression of regulators of mRNA stability
decreases protein expression of cytokines like TNF-a
calcineurin inhibitors such as cyclosporin & tacrolimus
biologics
anti-TNF agents: mAbs bind TNF-α, preventing receptor engagement and NF-κB activation
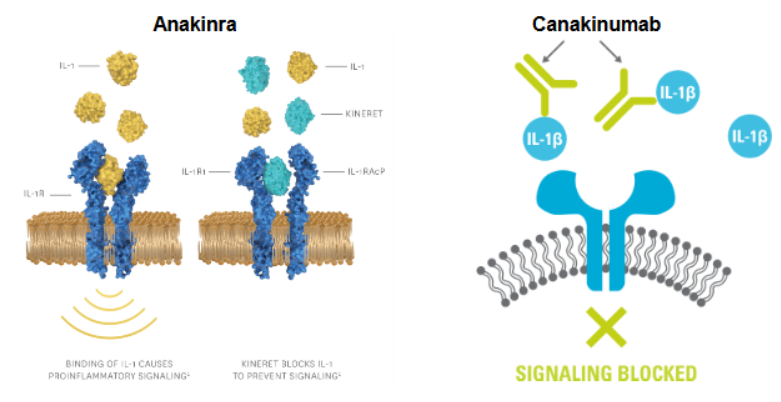
IL-1 pathway blockers:
anakinra (Kineret): mimics IL-1 structure, binds IL-1 receptor to block activation (receptor antagonism)
Anti–IL-1β monoclonal antibodies: Neutralize IL-1β directly.
IL-6 pathway blockers:
target IL-6 receptor (membrane-bound or soluble forms); biomolecules bind and neutralize receptor signaling regardless of receptor form, preventing downstream activation
biopharmaceutical drug products synthesized in or extracted from a living organism such as insulin, vaccines, and engineered mAbs that are structurally resembling biological components and administered by injection
functions of Ab — neutralization, agglutination, ADCC, degranulation and opsonization
infliximab
targeted suppression of specific pathways
block TNF-a signaling by engineered mAbs that bind TNF-a
block IL-1B signaling with anakinra (an antagonist that binds to IL-1 receptor) & canakinumab (a mAb that binds to IL-1B)
block IL-6 signaling with tocilizumab (an antagonist that binds to IL-6 receptor)
JAK inhibitors
block all JAK-STAT signaling downstream of multiple receptors
Mechanism: Many cytokines signal via JAK-STAT; inhibitors block intracellular kinases, preventing transcriptional responses even when cytokine and receptor are intact
Positioning: Acts downstream of receptor engagement; complements extracellular neutralization strategies
Signaling cascade summary: cytokines released bind receptors on target cells (macrophages, B cells, T cells, etc.)
activation triggers NF-κB or JAK-STAT pathways
drugs act by blocking ligand binding, receptor function, or intracellular signal transduction to reduce inflammation and immune activation
inhibit TCR signaling and downstream T cell activation by:
calcineurin inhibitors such as cyclosporin, tacrolimus and glucocorticoids
inhibit downstream activation of IL-2 signaling with mTOR inhibitors and cell cycle inhibitors
receptor activation to nucleus: TCR/PRRs activate intracellular cascades leading to NF-κB and other transcription factors, initiating expression of inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6)
drug action points: Suppress NF-κB activity or downstream gene expression; block cytokine–receptor interactions; inhibit JAK-STAT signaling
revisit to NF-kB regulated pro-inflammatory cytokines
IL-1B, TNF-a, IL-6, CXCL8, IL-12
inflammation: redness, swelling, pain, fever
secondary (acquired) immunodeficiency
human immunodeficiency virus infection — HIV/AIDS
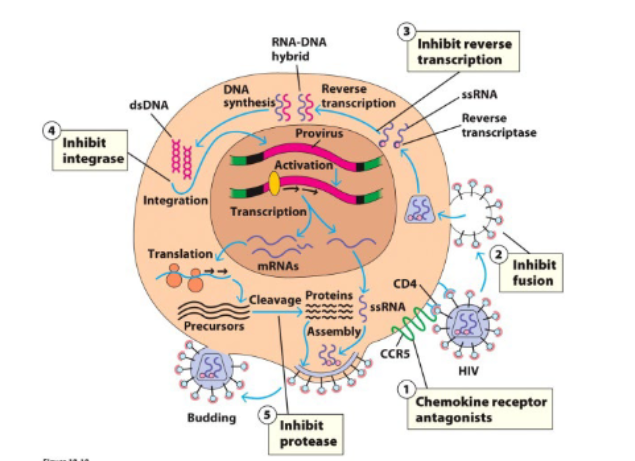
a retrovirus with RNA genome and reverse transcriptase enzyme which synthesizes a DNA intermediate
RNA-dependent DNA polymerases have no proofreading = high rate of mutation
HIV specifically infects immune cells
entry into cells depends on binding of virus to CD4+ and a chemokine receptor (CCR5 or CXC4)
viral replication depends on activation of host NF-kB TF
cells must be activated by cytokines and/or PAMP-PRR signaling
mutations in CCR5 cause resistance to HIV
CCR5-Δ32
32 bp deletion that causes frameshift mutations & causes premature stop codons
makes receptor non-functional, HIV cannot bind
“Berlin patient” in 2008
HIV positive & had acute myeloid leukemia
treated with hematopoetic stem cell transplant
used stem cells from a donor homozygous for Δ32 mutation
patient now has no more detectable virus
process repeated for “London patient” in 2019
in 2018, a scientist in China genetically edited human embryos with CRISPR to mutate CCR5
not ethical!
many current anti-retroviral drugs are very effective against HIV and mutations in CCR5 can cause more susceptibility to other infections
Anti-retroviral drugs
elite controllers
a subset of infected people are able to suppress HIV replications for decades
“long-term nonprogressors”
multiple molecular mechanisms:
specific MHC allotypes are better able to present HIV peptides and stimulate strong cytotoxic T cell responses
specific alleles of the NK cell receptors KIR also promote NK cell activation and control HIV
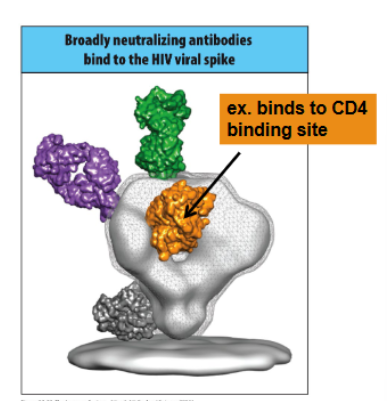
broadly neutralizing Ab formed from multiple rounds of somatic hypermutation bind conserved and required portions of the HIV envelope glycoprotein