Endoplasmic Reticulum (ER)
The ER characteristics:
• Serves as the cell’s biosynthetic powerhouse and a highway for the transport of molecular materials
• Is a network of membraneous tubules and flattened sacs that extend throughout the cytoplasm
• Is contiguous with the outer nuclear membrane
• Is categorised as rough and smooth ER based on its microscopic appearence
ER functions
• Protein synthesis
• Lipid motabolism
• Calcium storage
•Drug detoxification
Section 1: Structure and Electron Microscopy of the ER (15 minutes)
Highlight the structural differences and specific features of rough and smooth ER.
Intracellular Location: Discuss where both types of ER are predominantly located
within the cell.Electron Microscopy: Show and interpret electron micrographs of the ER.
Interactive Review: Engage with the audience using questions about the micrographs.
Section 2: Functions of the Smooth ER (15 minutes)
What is the role of the smooth ER in lipid and steroid hormone synthesis?
How is the smooth ER is involved in metabolising carbohydrates?
How does the smooth ER detoxifies drugs and poisons?
What is the role of the smooth ER in muscle cell contraction and
signalling?
• Function Discussion: Use specific examples to illustrate each function.
Smooth ER
• SER lacks ribosomes and has more of a tube apperence
• It is involved in lipid and steroid hormone synthesis, crucial for cell membrane formation and signalling
• SER plays a role in detoxyfying metabolic by-products and xenobiotics
• SER regulates intracellular Ca2+ levels, which is important for muscle contractions and other signalling pathways
Section 3: Functions of the Rough ER (15 minutes)
• Protein Synthesis: Describe how the rough ER is involved in the synthesis of
membrane-bound and secretory proteins.
• Post-Translational Modification: Outline the initial modifications that occur to
proteins in the rough ER.
• Protein Quality Control: Explain how the rough ER is involved in quality control of
synthesized proteins.
• Case Studies: Present real-world examples to highlight the functions of the rough ER.
Break (10 minutes)
Protein manufacturing in RER
• Ribosomes translate into polypeptide chains that are co-translationally translocated into RER
• Inside the RER, proteins are folded with help of chaperones and undergo modifications such as glycosylation and disulphide bond formation
• Properly folded proteins are packed into vesicles for transport to the golgi apparatus
Rough ER:
• Characterised by by the presence of ribosomes on its cytoplasmic surface
• Is the site of synthesis for secretory, membrane-bound, and organelle-targeted proteins.
• Newly synthesised proteins enter the RER lumen where they undergo folding and post-translational modifications
Section 4: Protein Translocation (20 minutes)
• Co-translational vs. Post-translational Translocation: Define and differentiate between the two processes.
• Protein Types: Discuss which proteins undergo which type of translocation.
• Mechanisms and Animation: Use visual aids to depict the translocation mechanisms.
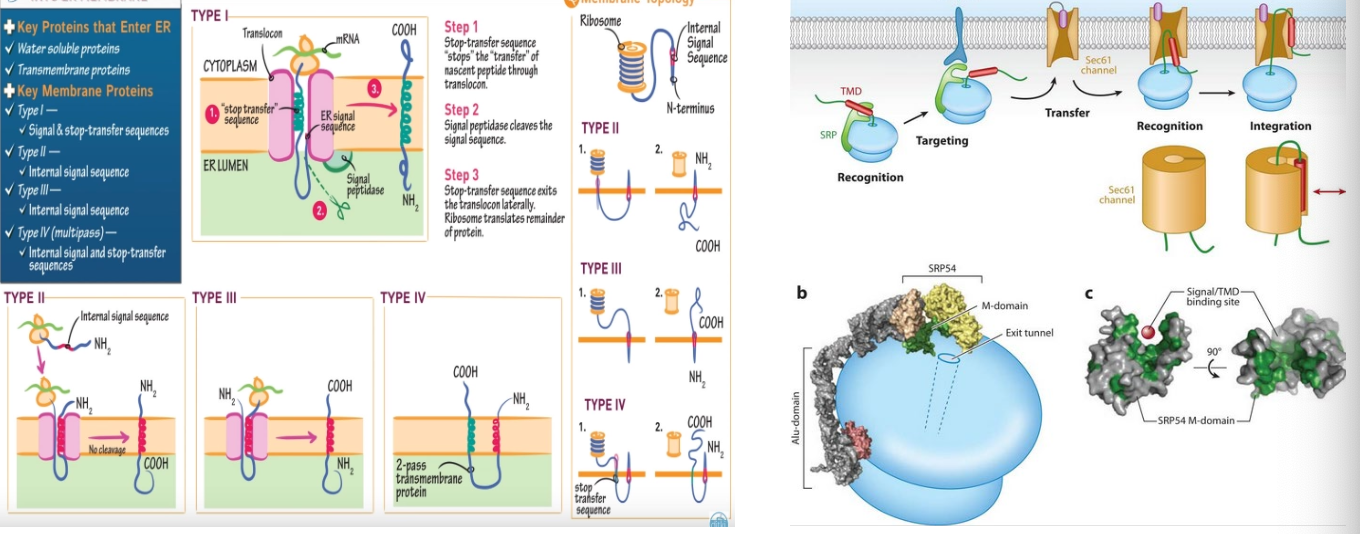
Protein insertion into the ER:
Type 1:
defining characteristics-
•single transmembrane domain
•has an N-terminus in the ER lumen, and a C-terminus in the cytosol
•contains a cleavable N-terminal signal sequence
insertion process-
begins with ribosome translation of the signal sequence
the signal recognition particles (SRP) pauses translation and directs the ribosomes to the ER
the signal peptide is inserted into the translocon and cleaved off as translation resumes
the hydrophobic stop-transfer sequence halts translocation, anchoring the protein in the membrane
the C-terminal continues to by synthesises into the cytosol.
example proteins -
glycophorin & CD4
Type 2:
defining characteristics -
•single transmembrane domain
•there is the N-terminus in the cytosol, and C-terminus in the ER lumen
•contains a signal-anchor sequence that is not cleaved
insertion process -
Begins with ribosome translation of the internal signal-anchor sequence
SRP directs the ribosomes to the ER, where the signal-anchor sequence initiates insertion
The orientation is dictated by a positive charge flanking the signal-anchor sequence
The polypeptide chain then grows into the ER lumen, forming the C-terminus
example proteins -
asialoglycoprotein receptor
some types of G protein-coupled receptors
Type 3:
defining characteristics -
•single transmembrane domain
•there is the N-terminus in the ER lumen, and C-terminus in the cytosol
•single-anchor sequence remains as a transmembrane segment
insertion process -
Translation begins with an internal signal-anchor sequence
the SRP binds to the signal-anchor and guides the complex to the ER
The orientation is generally N-lumenal, which is often influenced by the distribution of positive charges
The protein continues to elongate, inserting the C-terminal into the cytosol
example proteins -
cytochrome P450 enzymes
flippase proteins
Type 4:
defining characteristics -
•multiple transmembrane domanes
•complex orientation with both termini on the same or opposite sides of the ER
membrane
•contains several signal-anchor and stop-transfer sequences
insertion process -
Translation begins with a signal-anchor sequence that is not cleaved
Multiple signal-anchor and stop-transfer sequences guide the ribosome in inserting the protein
The protein loops in and out of the translocon, inserting multiple domains into the membrane
example proteins -
G-protein coupled receptors with multiple transmembrane domains
Ion channels like the potassium channel\
Section 5: Protein Trafficking Pathways (20 minutes)
• Translocation Overview: Describe how proteins are trafficked to lysosomes,
endosomes, and the plasma membrane.
• Vesicle Transport: Explain the role of vesicles in protein trafficking.
• Sorting Signals: Discuss the signals that direct proteins to their correct destinations.
• Interactive Activity: Map protein pathways with audience participation.
Proteins that are synthesised in the ER are destined for various locations - lysosomes, endosomes, or plasma membrane
The transport to these locations are highly regulated and it is critical for proper cellular functions to occur
The vesicles are the primary mode of transport for proteins from the ER to their destinations
Bud from the ER or golgi apparatus, vesicles ferry encapsulated proteins through the cytoplasm
Proteins that contain specific amino acid sequences that act as postal codes, by directing them to correct cellular addresses
these signals are recognised by adaptproteins which madiate the sorting of proteins into vesicles
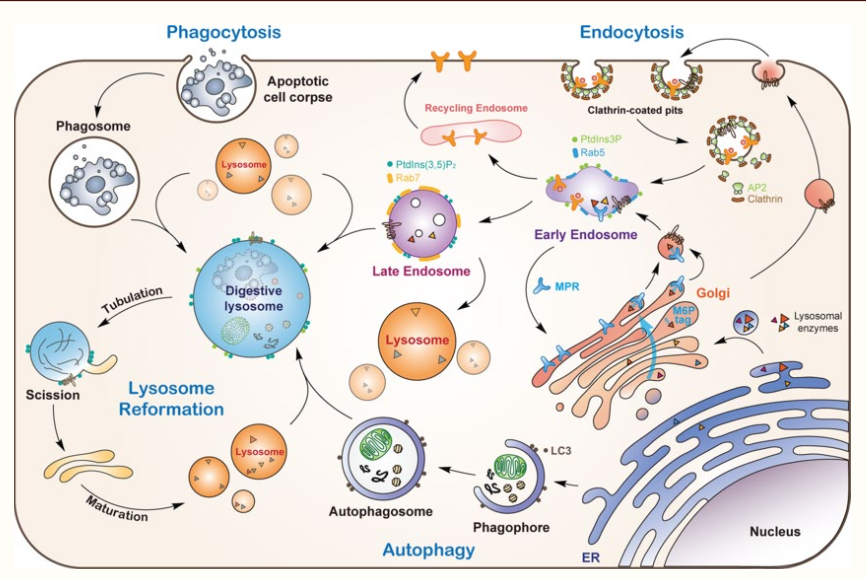
Secretory pathways:
The post-translational pathway is when proteins are synthesised in the ER, processed in the golgi and then transported out of the cell
Secretory proteins are packed into vesicles that bud from the golgi and migrate toward the plasma membrane
constitutive secretion is a continuous non-selective process where secretory vesicles fuse with the plasma membrane to release their contents
It operates constantly in all cells, delivering proteins like extracellular matrix components
The import mchanism is when the proteins destined for the ER have a signal sequence that directs them to the ER membrane
The signal recognition particle (SRP) binds to this sequence and pauses translation
The SRP-ribosome complex docks on the ER membrane, and the protein is threaded into the ER lumen through a translocon channel
Once inside the ER, proteins must fold into their three-dimensional shapes to become functional.
The ER provides an optimized environment for protein folding, with a unique set of enzymes and conditions.
Molecular chaperones, such as BiP , assist in proper protein folding and prevent aggregation.
Foldases, like protein disulphide isomerase (PDI), facilitate the formation of disulfide bonds between cysteines.
Proteins in the ER are modified through processes like glycosylation, which attaches sugar molecules to specific amino acids.
Other modifications include the formation of disulfide bonds and proper folding to achieve mature protein conformation.
Significance in Cell Physiology:
Importance of the ER in maintaining cellular health and
its role in various diseases.
Section 6: Protein Secretion (15 minutes)
• Secretory Pathways: Outline the process of protein secretion from the ER to the
outside of the cell.
• Constitutive vs. Regulated Secretion: Explain the two main pathways of protein
secretion.
• Real Cell Examples: Provide examples of proteins that follow each pathway.
Section 7: Protein Import and Processing in the ER (20 minutes)
• Import Mechanism: Describe how proteins are imported into the ER.
• Protein Folding: Discuss the role of the ER in protein folding and assembly.
• Chaperones and Foldases: Explain the assistance proteins receive in the ER.
• Post-translational Modifications: Outline the types of modifications that proteins
undergo in the ER.
Section 8: Quality Control in the ER (15 minutes)
What is the ER-associated degradation process for misfolded proteins.
What is the unfolded protein response and its significance.
How do defects in these processes lead to disease?
Persistent ER stress and an overwhelmed UPR can contribute to the development of diseases like neurodegeneration, diabetes, and cancer.
Therapeutic strategies targeting ER stress pathways are being explored to treat these conditions.
Section 9: Clinical Correlations (20 minutes)
What is the cellular pathogenesis associated with faulty protein folding?
Misfolded proteins can aggregate, leading to cellular stress and activation of the UPR, which may result in apoptosis if homeostasis cannot be restored.
Misfolding can occur due to genetic mutations, environmental factors, or a combination of both, leading to loss of function or toxic gain of function.
What are the current and emerging treatments?
Therapeutic Strategies:
Pharmacological Chaperones: Small molecules that stabilize the native state of proteins, improving their folding and trafficking.
Proteostasis Regulators: Compounds that modulate the UPR pathways, chaperone levels, and proteasomal degradation to alleviate stress.
Gene Therapy: Strategies to replace defective genes or introduce correct copies to restore normal protein function.
Related Diseases: Identify conditions like Cystic Fibrosis and Alpha-1 Antitrypsin
Deficiency.
Background on Alpha-1 Antitrypsin (AAT):
AAT is a protease inhibitor produced in the liver, functioning primarily to protect the lungs by inhibiting neutrophil elastase.
It is synthesized as a single polypeptide chain that folds into a stable tertiary structure within the ER of hepatocytes.
Pathogenesis of AAT Deficiency:
Caused by mutations in the SERPINA1 gene, leading to the production of a misfolded variant of AAT called Z-AAT.
Misfolded Z-AAT accumulates in the ER of hepatocytes, forming insoluble polymers that cause liver cell damage and ER stress.
Disease Manifestation:
Liver damage due to ER stress and apoptosis of hepatocytes.
Reduced levels of functional AAT in the blood lead to unchecked neutrophil elastase activity, resulting in lung tissue damage and emphysema.
Therapeutic Interventions:
Augmentation Therapy: Infusion of purified AAT to restore its protective levels in the lungs.
Small Molecule Correctors: Compounds that assist in proper folding and prevent polymerization of Z-AAT.
Gene Therapy: Approaches to correct the underlying genetic defect, providing a source of functional AAT.
Case History: Patient with Alpha-1 Antitrypsin
Deficiency
Patient Details:
•Age: 42 years old
•Sex: Male
Medical History: Non-smoker, moderate alcohol use, no significant past medical
history
Family History: Father died at 55 from liver cirrhosis, mother has chronic obstructive pulmonary disease (COPD)
Presenting Complaints:
Shortness of breath with exertion for the past 6 months
Chronic productive cough
Recent episodes of wheezing
Diagnosis
Clinical Workup:
Physical Examination: Reduced breath sounds, wheezing upon auscultation, no jaundice or other signs of liver failure
Pulmonary Function Tests (PFTs): Showed reduced FEV1/FVC ratio indicative of obstructive lung disease
Liver Function Tests (LFTs): Mildly elevated AST and ALT, normal bilirubin levels
Imaging: Chest X-ray revealed hyperinflated lungs; CT scan confirmed the presence of emphysema
Genetic Testing: Performed due to the combination of early-onset emphysema and family history; confirmed homozygosity for the Z allele of the SERPINA1 gene
Diagnosis:
Alpha-1 Antitrypsin Deficiency: The patient's symptoms, imaging, and genetic profile are consistent with this diagnosis, with manifestations including early-onset pulmonary emphysema and liver involvement.Treatment and Management
Immediate Management:
Smoking cessation counselling (despite patient being a non-smoker, counseling is standard due to the risk of exacerbating lung disease)
Bronchodilators to manage wheezing and improve breathability
Vaccinations to prevent respiratory infections (influenza and pneumococcal vaccines)
Ongoing Treatment:
Augmentation Therapy: Initiation of intravenous AAT replacement therapy to increase circulating levels of functional AAT and slow the progression of lung disease
Lifestyle Modifications: Patient education on avoiding environmental pollutants and maintaining a healthy weight to reduce stress on the lungs and liverTreatment and Management
Long-Term Management:
Regular monitoring of pulmonary function to assess progression of lung disease
Annual liver function tests and consideration of liver imaging to monitor for signs of liver disease progression
Patient education on recognising signs of liver disease (jaundice, ascites, easy bruising)
Consideration for liver transplantation in the event of liver failure
Special Considerations:
Referral to a support group for individuals with Alpha-1 Antitrypsin Deficiency
Genetic counselling for the patient and family members given the hereditary nature of the condition
Monitoring for potential side effects of augmentation therapy, such as transfusion reactionsFollow-Up and Prognosis
Follow-Up Care:
The patient will have follow-up visits with a pulmonologist and hepatologist every 3 months initially, then annually or as clinically indicated.
Monitoring will include PFTs, LFTs, imaging studies, and assessment of symptoms.
Prognosis:
With early diagnosis and appropriate management, the patient's lung function decline can be slowed, and liver disease can be monitored and managed. The prognosis is variable and depends on the level of lung and liver involvement and the patient's adherence to the treatment regimen.
Conclusion (10 minutes)
• Recap of Key Points: Summarize the major takeaways from the lecture.
• ER’s Role: Emphasize the ER's role in health and disease.
Recap of Key Points:
• The ER is essential for protein synthesis, folding, and trafficking, ensuring
proteins reach their destinations functional and intact.
• Misfolded proteins and ER stress can lead to diseases like Cystic Fibrosis and
Alpha-1 Antitrypsin Deficiency.
• The UPR and ERAD are critical for managing ER stress and maintaining cellular
homeostasis.
• Therapeutic interventions, including pharmacological chaperones, proteostasis
regulators, and gene therapy, offer hope for treating protein misfolding
diseases.