Class 4: Mitochondria and Peroxisomes Transport and Related Diseases
Mitochondria
Mitochondria are organelles found in eukaryotic cells and are essential for cellular energy production through oxidative phosphorylation.
Proteins destined for the mitochondria are translocated from the cytosol via specialized import machinery.
Mitochondrial dysfunction is significantly implicated in Parkinson's Disease, contributing to the disease's pathology and progression.
Parkinson's Disease symptoms include:
Motor impairment: Tremors, rigidity, bradykinesia (slowness of movement), and postural instability.
Cognitive and psychiatric symptoms: Depression, anxiety, cognitive decline, and dementia in later stages.
Autonomic dysfunction: Constipation, orthostatic hypotension, and urinary problems.
Parkinson's Disease involves:
Mitochondrial dysfunction: Impaired mitochondrial respiration and increased oxidative stress.
α-synuclein aggregation in dopaminergic neurons: Formation of toxic aggregates that disrupt neuronal function.
It is normally in the cell, but in the disease state it changes in terms of its location and function in the cell
Selective loss of dopamine neurons in the substantia nigra: Leads to dopamine deficiency and motor symptoms.
Lewy body formation: Intracellular inclusions containing aggregated α-synuclein.
α-Synuclein
α-Synuclein undergoes a conformational change upon lipid binding, shifting to a highly α-helical conformation (71% α-helix when bound to lipid), which is crucial for its interaction with membranes.
Positively charged amino acids are clustered on one face of the α-helix, with nonpolar amino acids on the opposite face, facilitating membrane binding and interaction with other proteins.
We want to know if the mutated protein interacts with TOM20
Proximity Ligation Assay (PLA)
A technique used to detect protein interaction and quantify protein proximity in cells.
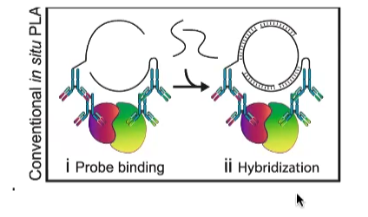
Antibodies are conjugated to oligonucleotide sequences. These antibodies bind to the proteins of interest.
In proximity (less than 40nm), connector oligonucleotides hybridize with the probes, forming a closed circle template in the presence of a ligase. This occurs when the proteins are in close proximity.
Addition of polymerase and fluorescent-labeled oligonucleotides results in rolling circle amplification of the closed circle template, allowing for signal detection.
α-Synuclein and TOM20 Interaction
There are some drawbacks to this method. Size of the protein may limit the outcomes of this assay. Additionally, immunoprecipitation can also be used to verify the outcomes, with or without specific identifications of proteins.
Co-precipitation is also an option.
α-Synuclein interacts with TOM20 in dopamine neurons, reducing import to mitochondria, thus disrupting mitochondrial function.
Ndufs3 is a nuclear-encoded, mitochondrially targeted protein and a subunit of mitochondrial complex I, essential for electron transport.
TH catalyzes Dopamine synthesis and is crucial for dopamine production in neurons.
Model system: Rats exposed to rotenone (inhibitor of complex I) to mimic mitochondrial dysfunction in Parkinson's disease.
Observed in postmortem brain tissues of patients, confirming the relevance of these interactions in human disease.
One of the ways that we can differentiate between an issue with transcription and expression of this protein is to determine is mRNA for the protein was generated, which we can do through a number of methods (cDNA for example) to assess the presence of mRNA transcripts in brain tissues. This allows us to confirm whether the observed decline in dopamine levels is a result of reduced transcription or other cellular processes affecting protein expression.
What is the issue: The mRNA is generated for the Ndufs3, but the protein does not make it into the the mitochondrial membrane due to impaired transport mechanisms (alpha synuclein is a mutated protein, can’t interact normally with TOM20 to enter the mitochondrial membrane), leading to mitochondrial dysfunction.
Overcoming Mitochondrial Dysfunction
TOM20 overexpression restores mitochondrial dysfunction and prevents cell death in vitro, suggesting a potential therapeutic strategy.
DJ-1 and Mitochondrial Localization
Mutations in the cytosolic DJ-1 protein in Parkinsons patients also mediate mitochondrial localization of DJ-1 and are associated with autosomal recessive Parkinson's, indicating a role for DJ-1 in mitochondrial maintenance.
HSP60 is a chaperon protein that is in the intermembrane space of the mitochondria and assist in the proper folding of proteins once they have penetrated the mitochondrial structure.
Due to the mutation of DJ-1, the hypothesis was that there is a change in the mitochondrial targeting sequence and the protein possesses a lower affinity to the HSP60 chaperon. Therefore less protein enters the mitochondria, leading to impaired mitochondrial function and increased susceptibility to oxidative stress which can contribute to neuronal degeneration in Parkinson’s disease.
Therefore they generated a mutation library with alanine (every residue location was replaced with alanine to see which positions were essential for the protein to be transported into the mitochondria.
Alanine Scanning
Alanine scanning is performed by site-directed mutagenesis to identify functional regions of the protein. Replacing amino acids with alanine can reveal which residues are critical for protein function and localization.
DJ-1 Mutations and Mitochondrial Localization
Cytosolic vs. mitochondrial mutations affect colocalization with TOM20, suggesting different mechanisms of action.
Pearson correlation coefficient is used to quantify colocalization with TOM20, providing a quantitative measure of protein association.
DJ-1 Structure and Mitochondrial Localization
Mutations buried in the DJ-1 structure are responsible for mitochondrial localization, indicating that conformational changes can affect protein targeting.
Folding instability of DJ-1 pathogenic mutants determines their mitochondrial localization, linking protein stability to cellular location.
Folding Stability and Mitochondrial Localization
Correlation between DJ-1 mutant instability and mitochondrial localization, highlighting the importance of protein folding for proper function.
DJ-1 and N-terminal MTS
DJ-1 has a cryptic N-terminal MTS that is essential for mitochondrial localization, suggesting a regulatory mechanism for mitochondrial import.
Mutations in region A of the E18K mutant inhibit mitochondrial localization, pinpointing specific regions involved in targeting.
DJ-1 and Structural Change
Structural change in DJ-1 allows the cryptic N-terminal signal to be exposed, revealing a mechanism for regulated mitochondrial targeting.
Restricted unfolding and subsequent mitochondrial translocation is essential for the genuine function of DJ-1, emphasizing the importance of controlled protein dynamics.
Summary of Mitochondrial Transport
Most mitochondrial proteins are translocated from the cytosol via specialized protein import complexes.
Proteins are transported in an unfolded state across both outer and inner membranes to facilitate entry into the mitochondrial matrix.
ATP hydrolysis and membrane potential across the inner membrane drive translocation, providing the energy needed for protein import.
Cytosolic hsp70 chaperones maintain precursor protein unfolded, and matrix chaperones pull the polypeptide into the mitochondria, ensuring proper folding and preventing aggregation.
A signal sequence directs protein translocation.
N-terminal signal is cleaved off after import by signal peptidases.
Internal signals can be retained for proteins residing in the inner membrane space.
A second hydrophobic signal can direct transport to the inner membrane, crucial for the insertion of proteins into the inner mitochondrial membrane.
Transport into Peroxisomes
Peroxisomes are organelles found in eukaryotic cells and are involved in various metabolic processes.
Peroxisome Characteristics
All eukaryotic cells have peroxisomes, highlighting their fundamental role.
Peroxisomes contain oxidative enzymes, essential for their metabolic functions.
Like mitochondria, they are a major site for oxygen utilization.
They use molecular oxygen to remove hydrogen from organic substrates and produce Hydrogen Peroxide:
Detoxification of harmful molecules in the kidney and liver.
β-oxidation of fatty acids, breaking down fatty acids into smaller molecules.
Peroxisome Structure and Targeting
Peroxisomes are surrounded by a single membrane, distinguishing them from other organelles with double membranes.
They do not contain DNA or ribosomes; thus, they rely on protein import from the cytosol.
Peroxisomal targeting signal (PTS) directs proteins to peroxisomes, ensuring proper localization.
Peroxisomal Import
Dynamic pore dimensions enable oligomeric cargo transport, allowing large protein complexes to enter the peroxisome.
Zellweger Syndrome
Zellweger syndrome results in peroxisomal deficiency, leading to severe developmental abnormalities.
It is a rare congenital disorder characterized by the reduction or absence of functional peroxisomes, impairing essential metabolic functions.
Caused by mutations in different PEX genes, which encode proteins involved in peroxisome biogenesis and protein import.
Results in abnormalities in the brain, kidney, and liver, due to impaired peroxisomal functions.
Peroxisome-localized Translation
Peroxisome-localized translation of peroxisomal membrane proteins occurs in yeasts, suggesting a mechanism for localized protein production.
mRNA of peroxisomal membrane proteins colocalizes with the peroxisome, supporting local translation.
BiolD Proximity-Dependent Biotinylation
Used to detect mRNA found in proximity to peroxisomes, providing insights into localized translation.
Biotin ligase is attached to a peroxisome membrane protein, enabling the biotinylation of nearby molecules.
AviTag is fused to a large protein ribosomal subunit, allowing for the identification of ribosomes near peroxisomes.
Biotin is added to the reaction mixture, facilitating the isolation and subsequent analysis of peroxisome-associated mRNAs.