Lec 1: Drug Receptors and Pharmacodynamics: Study Notes
Receptors and Pharmacodynamics: Key Concepts
Pharmacodynamics describes the actions of a drug on the body and how drug concentrations influence the magnitude of the response.
Therapeutic and toxic effects arise from interactions with receptors; receptors confer selectivity of drug action.
The molecular size, shape, and electrical charge of a drug determine whether it will bind to a receptor and with what affinity.
The affinity of a receptor to a specific molecule will determine the concentration needed for this molecule to produce a response. The interaction between a drug and a receptor is specific.
Receptors are the primary targets for drug action; understanding receptor–drug interactions explains dose–response relationships and therapeutic windows.
Agonist: an agent that activates a receptor to produce a biologic effect.
ENHANCES CELLULAR ACTIVITY
Antagonist: an agent that prevents the action of an agonist on a receptor, without producing an effect on its own.
Antagonists:
• Antagonists do not activate a signal generation.
• Antagonists do NOT produce a reverse signal of the agonist.
• Antagonists occupy the receptor and BLOCK the ability of an agonist to activate the receptor.
aka ZERO INTRINSIC ACTIVITY
Signal transduction (conceptual)
Drug binding to a receptor
generates signal transduction and elicits a biological response.
Second messengers or effector molecules
translate receptor binding into cellular responses
Receptors states:
exist in at least two states, inactive (R) and active (R*), in reversible equilibrium: R ⇄ R*.
Agonists shift the equilibrium toward R* to produce a biological effect.
Antagonists occupy the receptor but do not shift to R*; they block activation.
Partial agonists shift toward R* but yield a smaller fraction of receptors in the active state than a full agonist.
The magnitude of the biological effect is proportional to the fraction of receptors in the active state, R*.
Dose–response relationships and key metrics
The effect of a drug depends on its concentration at the receptor site, influenced by dose and pharmacokinetics (absorption, distribution, metabolism, elimination).
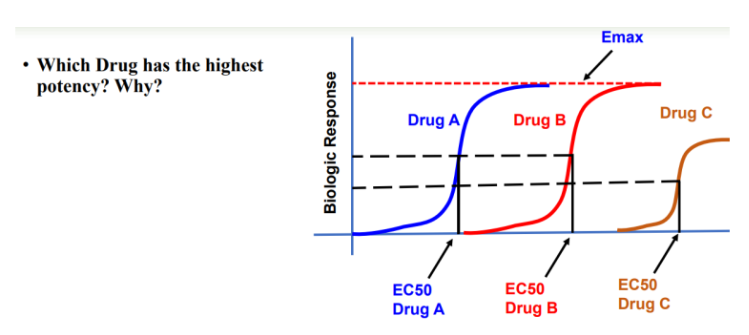
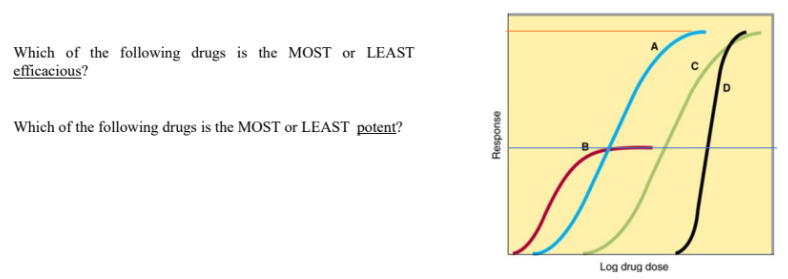
Potency: usually represented by EC50; the concentration that produces 50% of the maximal response.
EC50= Concentration of drug that produces 50% of the maximal response = DETERMINES POTENCY
indicates the concentration needed to achieve half-maximal effect; lower EC50 means higher potency.
EC50 is used to describe:
a) the potency of the drug
b) The efficacy of the drug
c) The toxicity of the drug
d) The affinity of the drug
EC50 is:
a) the concentration of drug that exerts 50% of the intrinsic activity
b) the concentration of the drug that produces 70% of the intrinsic activity
c) the concentration of the drug that produces 25 % of intrinsic activity
d) the concentration of the drug that produces no intrinsic activity
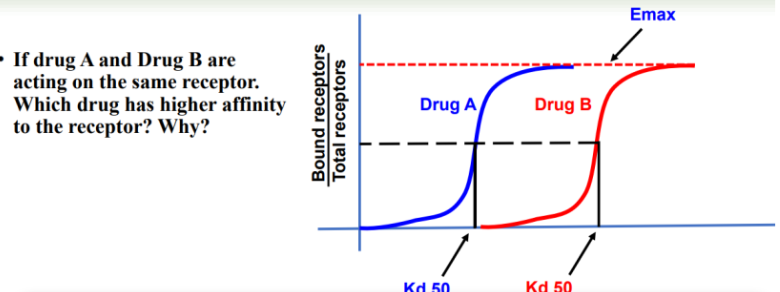
Kd = Concentration of the drug that binds to 50% of the available receptors
Used to determine the affinity of a drug for its receptor
Kd 50 is used to describe:
a) the potency of the drug
b) The efficacy of the drug
c) The toxicity of the drug
d) The affinity of the drug
Kd:
Kd = dissociation constant of D-R complex; it quantifies the affinity of the drug (D) for the receptor (R), where a lower Kd indicates a higher affinity, higher Kd indicates weaker interaction.
Higher affinity (lower Kd) generally yields greater receptor occupancy at a given concentration.
Receptor–drug complex: [DR], free receptor [R], free drug [D], total receptors = [R] + [DR].
Dose–response relationships are often plotted as a hyperbola or logarithmically; curves can be used to determine potencies and compare efficacies.
Fraction of receptors bound at a given drug concentration (occupancy) for a simple 1:1 interaction
Intrinsic activity (IA) refers to the ability of a bound ligand to activate the receptor.
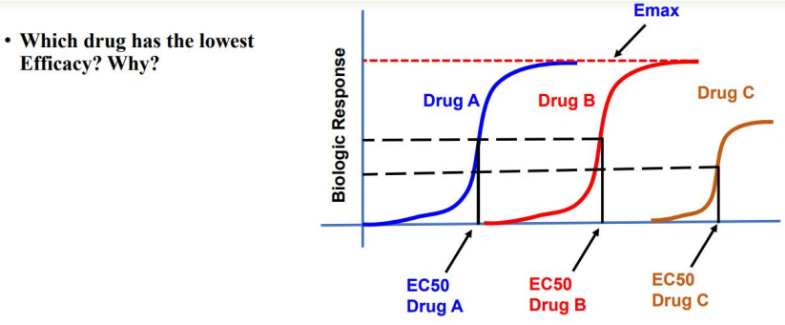
Efficacy: represented by Emax; the maximal effect achievable by the drug.
Efficacy (Emax) is the maximal response achievable with a drug; it reflects the ability to activate receptors and produce a cellular response.
As receptor occupancy increases, the pharmacologic effect rises until all receptors are occupied (Emax).
Full agonist: binds to receptor and produces a maximal biologic response equivalent to the endogenous ligand.
Example: Phenylephrine as a full agonist at α1-adrenoceptors, producing the same Emax as norepinephrine (NE) in causing vasoconstriction.
ALL FULL AGONISTS FOR A RECEPTOR POPULATION SHOULD PRODUCE THE SAME EMAX
Partial agonist: binds and activates but yields a maximal response less than a full agonist even when all receptors are occupied.
Partial agonists may have higher, lower, or equal affinity compared to full agonists.
EVEN IF ALL RECEPTORS ARE OCCUPIED, PARTIAL AGONISTS CANNOT PRODUCE THE SAME EMAX AS A FULL AGONIST
Spare receptors: a subset of receptors exist in excess; full maximal effect can be achieved without occupying all receptors.
Inverse agonist: produces the opposite pharmacologic effect of an agonist when binding to the receptor.
Competitive and non-competitive antagonism; allosteric modulation
Antagonists bind receptors without activating them; they block the action of agonists.
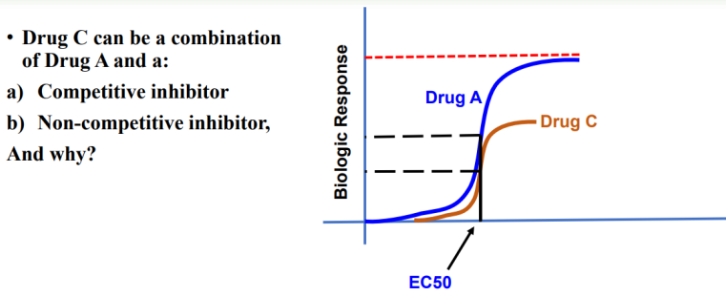
Competitive antagonists (reversible): bind the same site as the agonist; can be overcome by increasing agonist concentration; shift the dose–response curve to the right without changing the maximum effect (Emax).
can shift agonist dose response curve TO THE RIGHT (INCREASED EC50 WITHOUT AFFECTING EMAX)
BIND SAME RECEPTOR
Non-competitive antagonists: bind to a site other than the agonist binding site (allosteric or irreversible binding); reduce the maximum effect (Emax) and cannot be overcome by simply increasing the agonist concentration; may be allosteric activators or inhibitors.
can shift EMAX DOWNWARDS, with NO SHIFT OF EC50 VALUES
BIND DIFFERENT RECEPTOR
Allosteric site: a binding site distinct from the active site where allosteric modulators (activators or inhibitors) bind to modulate receptor activity.
Allosteric modulator: binds at a site other than the active site and modulates receptor activity as an activator/inhibitor
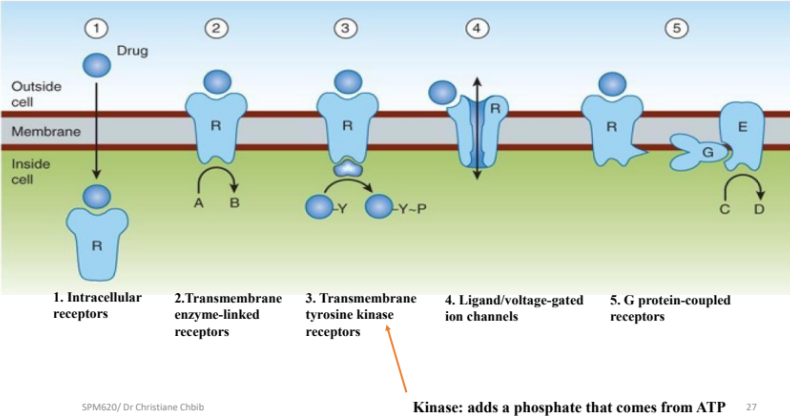
Receptors can be classified into four major transmembrane/ intracellular classes, plus ligand-gated/voltage-gated ion channels:
1) Intracellular receptors (cytoplasm or nucleus); ligands are lipophilic (e.g., steroids, thyroid hormone).
Intracellular ligands: steroids, thyroid hormones; ligands diffuse through the membrane to reach intracellular receptors because they are lipid soluble
2) Transmembrane enzyme-linked receptors with extracellular ligand-binding domain and cytosolic catalytic domain (often tyrosine kinase).
3) Transmembrane tyrosine kinase receptors (a subset of enzyme-linked receptors).
most abundant
4) G protein-coupled receptors (GPCRs); activate heterotrimeric G proteins.
GPCRs couple to heterotrimeric G proteins composed of α, β, and γ subunits.
The Gα subunits (Gs, Gi, Gq) regulate downstream effectors:
Gs activates adenylyl cyclase (AC) → increases cAMP → activates PKA.
Gi inhibits adenylyl cyclase → decreases cAMP.
Gq activates phospholipase C (PLC) → cleaves PIP2 into IP3 and DAG; IP3 increases Ca2+, DAG activates PKC.
When a G-protein coupled receptor (GPCR) binds to its ligand, the following sequence of events occurs:
The ligand binds to the extracellular side of the GPCR.
This induces a conformational change in the GPCR.
The GPCR acts as a guanine nucleotide exchange factor (GEF) for the G-protein.
The Gα subunit of the heterotrimeric G-protein exchanges GDP for GTP.
This exchange causes the Gα subunit to dissociate from the βγ subunits and the receptor.
The active Gα-GTP and the βγ dimer can then go on to activate downstream signaling pathways.
Once a G-protein coupled receptor (GPCR) has bound to its ligand, which of the following steps
allows the α subunit to dissociate from the receptor and trigger downstream cascades?
a. Exchanging the G-protein’s bound GDP for a GTP
b. Exchanging the G-protein bound ADP for an ATP
c. Exchanging the G-protein association with a magnesium ion for a calcium ion
d. Degradation of the GPCR’s C-termina tail that is bound too the α subunit
5) Ion channels: two primary types
Ligand-gated ion channels open in response to ligand binding.
Voltage-gated ion channels open in response to changes in membrane potential.
Receptor regulation: desensitization, tachyphylaxis, down/up-regulation, and tolerance
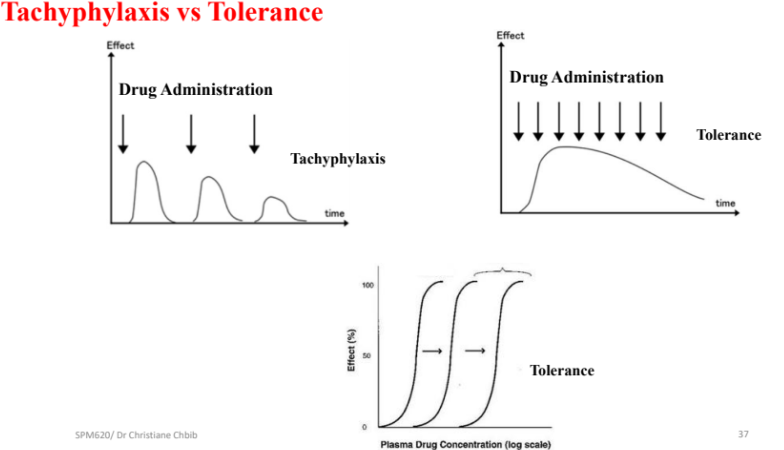
Tachyphylaxis: diminished response to repeated agonist exposure; an acute reduction in response.
Desensitization: receptor becomes less responsive after repeated stimulation.
Down-regulation: receptors are sequestered away from the cell surface and become unavailable; may be recycled (up-regulation) or degraded (down-regulation).
Up-regulation: increase in receptor number on the cell surface via recycling of receptors, restoring sensitivity.
Antagonist-induced up-regulation: repeated antagonist exposure can increase receptor reserves on the surface.
During this recovery phase, unresponsive receptors are said to be “refractory.”
• Repeated exposure of a receptor to an antagonist may result in up-regulation of receptors, in which receptor reserves are inserted into the membrane, increasing the total number of receptors available
Tolerance: a gradual decrease in response requiring higher doses to achieve the same effect; develops over a longer period and can sometimes be overcome by increasing dose, unlike tachyphylaxis.
Practical examples and exam-style prompts (based on the transcript content)