2 inborn of carbohydrate 1
These are inborn errors that affect the catabolism and anabolism of carbohydrates.
Galactosemia
Galactose is an aldohexose and is a constituent of lactose of milk. Inability to metabolize galactose results in galactosemia.
Classic galactosemia is caused by defect in the enzyme galactose-1- phosphate uridyl transferase ; GAL-1-PUT (which catalyzes the rate limiting step of galatose metabolism pathway).
Biochemical Implications and Clinical Features
Galactose-1-phosphate will accumulate in liver due to the block in the enzyme (GAL-1-PUT). This will inhibit galactokinase as well as glycogen phosphorylase, resulting in hypoglycemia.
Bilirubin conjugation is reduced; so unconjugated bilirubin level is increased in blood.
There is enlargement of liver, jaundice and severe mental retardation.
Free galactose accumulates, leading to galactosemia. It is partly excreted in urine (galactosuria).
Galactose is reduced to galactitol. The accumulation of galactitol in the lens results in cataract due to its osmotic effect.
This is called congenital cataract and is a very important feature of galactosemia
A variant of galactosemia occurs due to the deficiency of galactokinase. But here the symptoms are milder.
This is because galactose-1-phosphate is not formed and hence toxic effects of this compound (such as liver insufficiency, hypoglycemia) are not manifested.
Cataract is the only manifestations of galactose kinase deficiency.
Diagnosis And Management
Collection of fetal cells by amniocentesis may be useful in prenatal diagnosis.
Screening is also performed by measuring GAL- 1-PUT activity
Management is by elimination of galactose from the diet.
Elimination of implicating substance; measuring enzyme activity, amnioncentesis)
Hereditary Fructose Intolerance (Hfi)
HFI is caused by a defect in Aldolase B, hence fructose-1- phosphate cannot be metabolized and will accumulate.
Biochemical Implications and Clinical Features
Patients are asymptomatic unless they ingest fructose or sucrose.
Accumulation of fructose-1-phosphate will inhibit glycogen phosphorylase. This leads to hypoglycemia.
Vomiting and loss of appetite are seen.
The infants often fail to thrive.
Hepatomegaly and jaundice occur. If liver damage progresses, death will occur.
DIAGNOSIS: Genetic testing and Aldolase B assay.
MANAGEMENT: Withdrawal of fructose and sucrose from the diet will immediately relieve the symptoms.
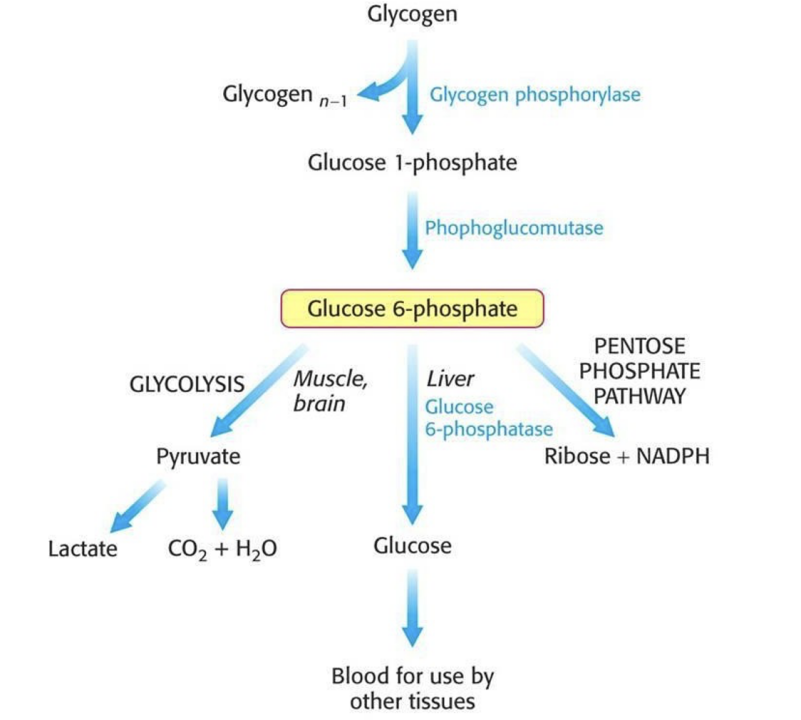
Glycogen Storage Diseases (GSD)
Enzyme deficiencies that leads to impaired synthesis or degradation of glycogen are also considered disorders of carbohydrates metabolism.
The two organs most commonly affected are the liver and the skeletal muscle
Glycogen storage diseases that affect the liver typically cause hepatomegaly and hypoglycemia, relating to impaired mobilization of glucose for release to the blood during fasting
Those that affect skeletal muscle cause exercise intolerance, progressive weakness and cramping resulting from low glucose levels, hence there is inability to increase glucose entry into glycolysis during exercise