Biochem
The beginning
biochemistry
dictionary definition: study of the chemicals and reactions in living organisms
working definition: study of how macromolecules carry out chemical reactions and how these prcesses are regulated in a living organism
went over the syllabus, class structure, and Moodle
encourage to read the book but…. watching the in-detail videos will be fine
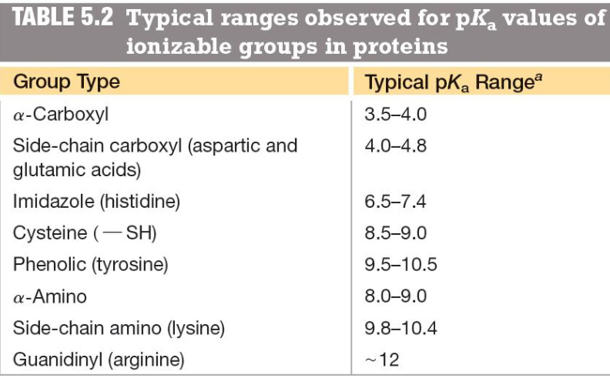
learn the 20 naturally occurring AA
Functional groups
biochemistry is important bc it provides a basis for understanding many issues in biology, medicine, and other life sciences
biochemistry is also important to the chemists and physicists because it provides many important, interesting and fundable topics for applications of the basic physical sciences
biochem = metabolic pathways, chloroplast, mitochondria
combines chem, physics, genetics, life sciences, and medicine
biochem: study of the chemicals and reactions in living organisms
working def= study of how macromolecules carry out chemical reactions and how these processes are regulated in a living organism
three characteristics of living organisms:
structurally complex: living organisms have intricate internal structures, which are a consequence of assembly of diverse molecules
extract, transform and utilized energy from their environments
autotrophs: utilized radiant energy and inorganic compounds to make organic compounds
heterotrophs: use chemical organic nutrients as energy source
capacity for precise self-replication and self-assembly
all three of these characteristics involve biochemical processes and require combinations of reactions that are catalyzed y enzymes or occur spontaneously
general primer on organic compounds and names of functional groups
remember the number of valence electrons elements have
C,N,O,H,P
remember names i.e.
organic compounds
alcohol, thiols, aldehydes, ketones. carboxylic acids, amines (primary, secondary, and tertiary)
functional groups
hydroxyl, acyl, carbonyl, carboxylate, amino, phosphoryl, phosphate, sulfhydryl
btw ammonium is protonated
linkages
esters, ether, amide, phosphate ester, phosphoanhydride, carbon-carbon, primary, secondary, tertiary, and quaternary
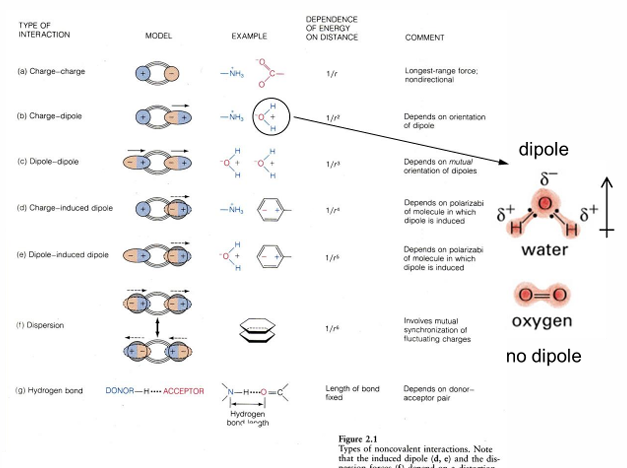
The big 4; all of these classes exhibit specific covalent structure as well as opportunity for additional important intermolecular associations through non-covalent interactions
proteins: AA, polypeptides, proteins
nucleic acid: mononucleotides, oligo- and polynucleotides, in DNA and RNA series
carbs: sugars, disaccharides, polymers of sugars
lipids: fatty acid, derivatives of glycerophospholipids and other compounds containing fatty acids, cholesterol
Noncovalent interactions
the chemistry of living organisms is based on carbon
carbon has four outer shell electron and has the ability to participate in four covalent bonds
this allows carbon to form an extraordinary number of complex linear and branched molecular structures; the formation of polymer structure is particularly important
carbon is relatively neutral in terms of its electronegativity
molecules contain carbon without added electronegative functional groups will be generally hydrophobic
however, atom, such as O, N, S, P add important properties to organic molecules
molecules containing these other atoms gain electronegative and polar characteristics
there is greater chemical and functional diversity
there are greater chances for H bonding
there are greater chances for ionic bonding
still chances for hydrophobic interactions
chemical composition of cells

the reason why O is 60-70% bc water constitutes 60-90% of cell mass
exceptions to this water content are seeds and spores which contain much less water and are revived by hydration
life on earth is described as a carbon-carbon based phenomenon, but H2O is the solvent of life
the physical properties and functional activity of macromolecules are intimately connected to their association with h20
therefore, understanding of water structure and interactions is absolutely critical for understanding the structure and function of biological molecules
review of O, H and H2O molecular structure
water us an example of a molecule with covalent bonding in which atoms share outer shell electrons to completely fill each atom’s outer shell
covalent bond strength is related to difference in atom electronegativities and also distance between outer shell electrons and nucleus and nuclear charge
in water the difference in O and H electronegativities establish strong covalent bond
other bonding is ionic bonding in which electrons are transferred from one atom to another to fill the outer shells and create oppositely charged ions
metals in biological systems will be cations and will be associated with anions (Cl-, CH3O-) through ionic bonding
the electronegative scale was derived by Linus Pauling for electronegativity; this quantifies the atom’s strength to attract electrons
structure of H2O
the bond angle is 104.5 degrees
normal sp3 would have a bond angle of 109 but the lone pairs experience electron repulsion
there is a dipole moment
an unequal charge or electron distribution
the arrow points toward the negative end of the dipole
how can you determine the dipole strength quantitatively
dipole moment of each covalent bond is the difference in their electronegativity values
for the moment of the whole molecule use vector addition
if permanent dipoles are designated by fancy upside down u and are expressed in D = Debye units, then:
H2O u=1.85 D
NH3 u= 1.47 D
CH3OH u= 1.71 D
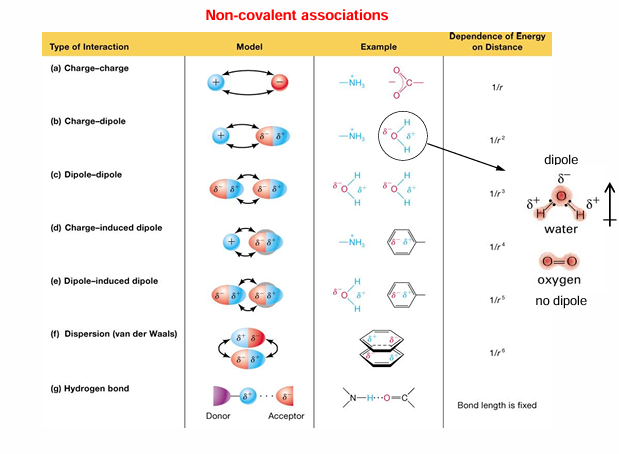
summary of covalent and non-covalent interactions
covalent bonds involve sharing of electron pairs between atoms in outer electron shells
non-covalent interactions:
charge-charge interactions transfer electron with resultant charge transfer; this results in electrostatic interaction
H-bond interactions: the arrangement in H2O; there are many more examples of H donor/acceptor pairs
van der Waals interaction: occurs between non-polar molecular surfaces; derives from transient changes in electron distribution in non-polar molecules; transient diploes have weak interactions
hydrophobic interactions: derives from entropy increase when water is released from non-polar surfaces
strengths of non-covalent vs covalent interactions:
covalent bonds > charge-charge interactions » H-bond interaction > hydrophobic interactions > van der Waals interaction
although hydrophobic and van der Waals interactions are very weak, there are many of them present in biomolecules that lead to the formation and stabilization of their 3-D structure
electrostatic (charge-charge) interactions
electrostatic interactions happen between two charged particles
these interactions con also be called ionic interactions, ion pairing or salt bridging
these interactions can be the strongest of the non-covalent interactions
attraction for interactions between opposite charges; repulsion for interaction between like charges
electrostatic interactions involving the virtual charges of permanent dipoles, although these charges do not have the full value of actual ions
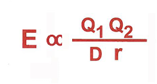
e= energy of attraction
Q1 and Q2 = electric charges of each particle
D = dielectric between charges particles
r = distance between charged particles
the strength of attraction
proportional to the strength of the charged particles
inversely proportional to the distance between the particles
the polar solvent H2O weakens electrostatic interactions by shielding the charged particles with water molecules
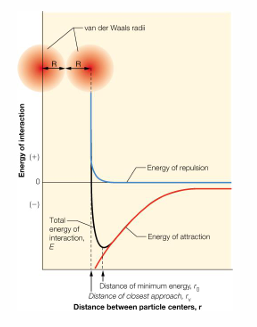
van der Waals interaction
occur between non-polar molecular surfaces
weak attractive force develops from the interaction of induced transient dipoles; these are very weak interactions, but if they occur over a large area they are a significant contributor to overall stability
a repulsive force occurs when two molecules are too close; this is attributed to repulsive part of the van der Waals force field
a property of atoms that describes their size is called the van der Waals radius; this radius comes from the measurements of atoms that are in close contact; there are tables summarizes these values
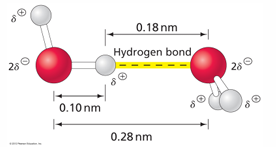
hydrogen bonds found in biomolecular interactions
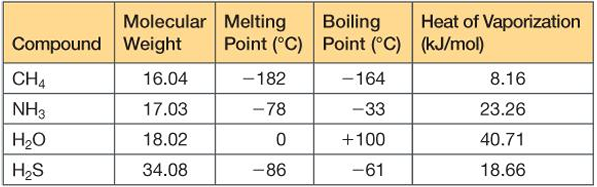
Properties of water
3-D structure of H20
tetrahedral structure (a trigonal pyramid)
the oxygen is buried in the center of the tetrahedron and the four corners are occupied by the 2 H and 2 lone pairs from the O
the structure of water is very conducive to hydrogen bonding in which the hydrogen atom interacts with a electronegative atom of another molecule
H2O can form intermolecular H bonds so one H2O can be associated with four other H2O through H bonding
properties of water vs other H- containing compounds
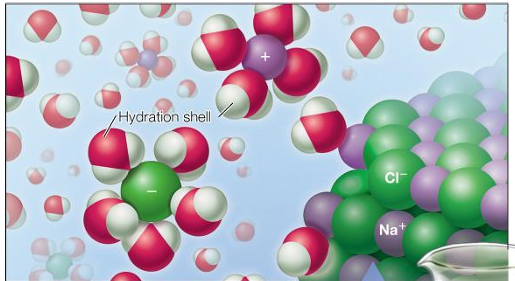
water readily dissolved polar molecules
charged ions are hydrated by water because it is polar
water readily dissolves salts (charged ions) because it is polar
ions are transported hydrated
this affects their size and screens their charge, unless the water is removed
biological compounds are soluble in water because they form hydrogen bonds
these molecules are called hydrophilic or water loving
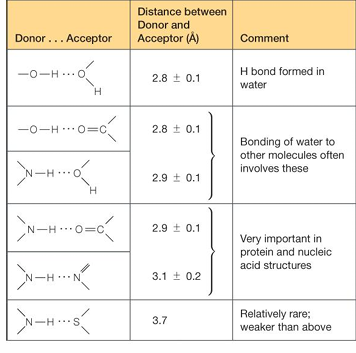
critical features for H-bond strength
defined length
2.8-3.0 A
this distance is the total heavy atom to heavy atom ie O to O bond
linear positioning
linearity maintains H-bond strength
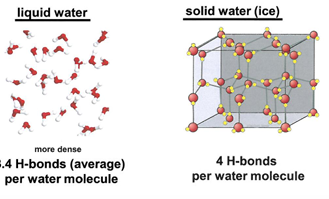
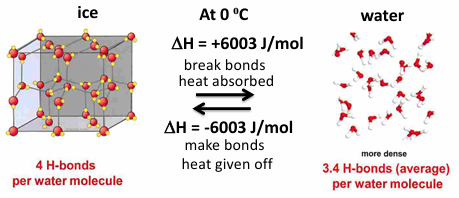
how does H2O H-bond in liquid water in ice
in liquid water H-bonding is not complete due to irregularity of liquid water structure
in ice a regular crystal structure is present, H-bonding is complete, density is less than that of liquid water
at 0 C water density is 1.00 g/ml and ice density is .92 g/ml
this explains why your plumbing pipes may break when it freezes- water expands when it freezes
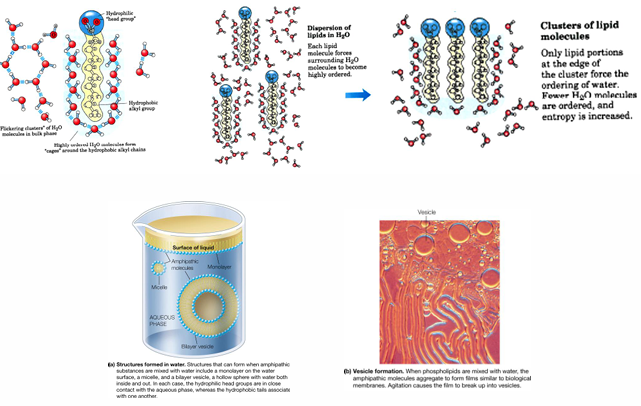
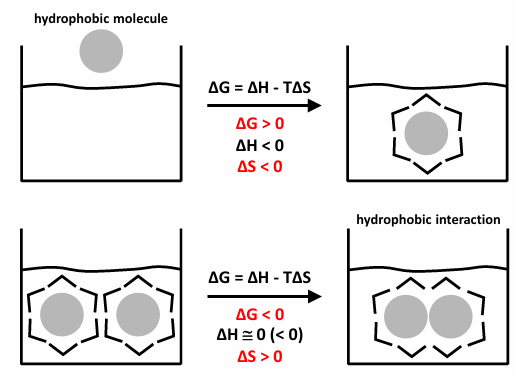
hydrophobic interactions
non-polar molecular surfaces give rise to the situation in which water is constrained in specific orientations (water is ‘‘ice-like” to create a hole to accommodate the non-polar group)
thermodynamically unfavorable
if two non-polar surfaces associate, the water is released (the “ice-like” structure is partially broken down to release water molecules to the bulk solvent)
thermodynamically favorable
an example is lipids (amphipathic molecules - “two-faced”)
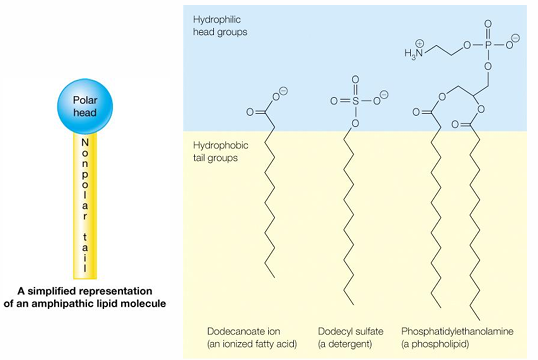
interactions of amphipathic molecules (lipids) with water
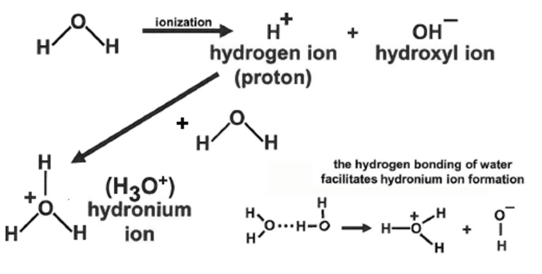
ionization of water
how can h2o ionization be described mathematically
describe in terms of chemical equilibrium
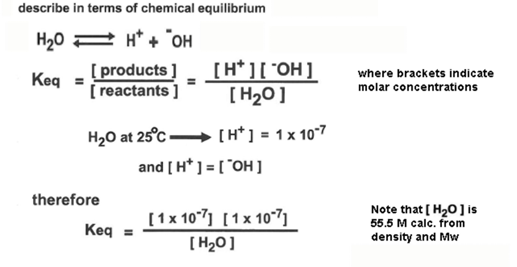
the quantity [h20] is the denominator is constant, and it can be combined with Keq to give a value, named Kw = Keq *55.5 M = 1 × 10 ^-14
another way to express this relationship is pH + pOH = 14
therefore, the equilibrium equation for the dissociation of water can be written as
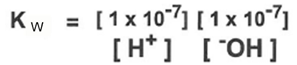
the equilibrium of H and OH ions determine the acidity of the water, which is described by the parameter pH
where pH = -log[H+]
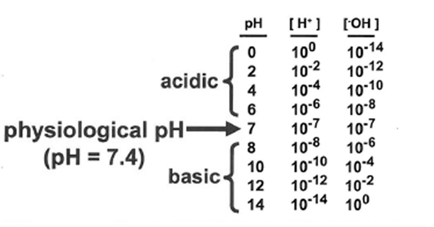
Chemical properties of acids and bases
definitions
acid: a proton (h+) donor
base: a proton acceptor
this is the Lowry-Bronsted definition, which i most useful i thinking about the structure and interactions of biological molecule
strong acid: acid that completely dissociates
weak acid: acid that practically dissociates
a term Ka (acid dissociation constant) will characterize the dissociation
if the Ka value is large then
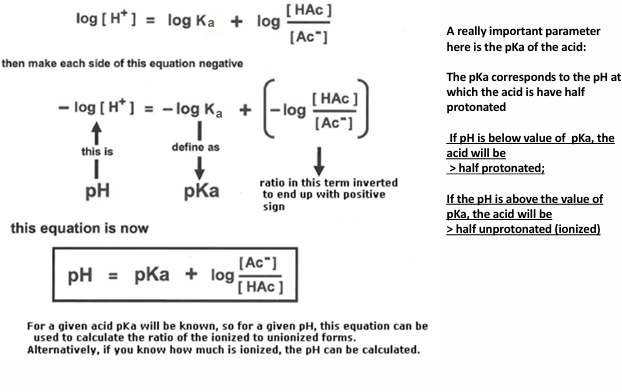
was eventually rewritten
= pH= pKa + log_10 (A/HA)
a term Ka (acid dissociation constant) will characterize the dissociation
bases can also be defind as strong or weak based on their association strength
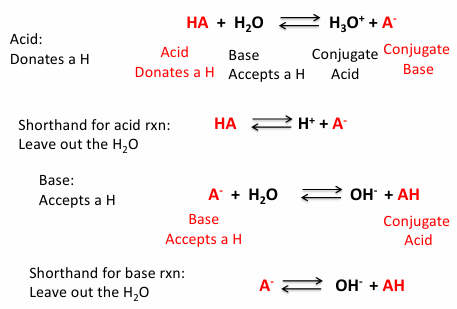
strong vs weak acids
a strong acid dissociates almost completely in water
HCl is strong acid →/← H+ and Cl-
Ka - acid dissociation constant
1) write the expression of the acid as dissociation
2) Ka = [product]/ [ reactants]
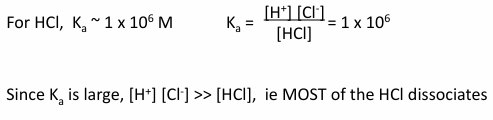
a weak acid does not dissociate much in water
acetic acid, H3COOH is a weak acid:
CH3COOH →/← H+ and CH3COO-
Ka - acid dissociation constant = 1.74 × 10-5 M
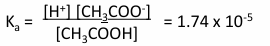
since Ka is small, [H+][CH3COO-] « [CH3COOH]
ie most of the CH3COOH does not dissociate
strong acids: Ka >1
weak acids: Ka < 1
why is it important to understand weak acids and bases
many biological molecules or components of biological molecules are weak acids i.e. amino acids, DNA, RNA
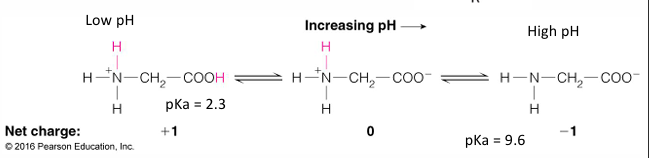
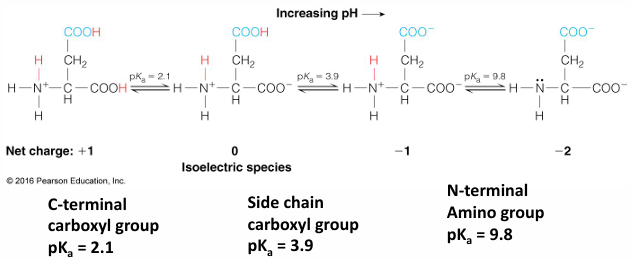
all AA have an amino group (weak base) and a carboxylate group (weak acid)
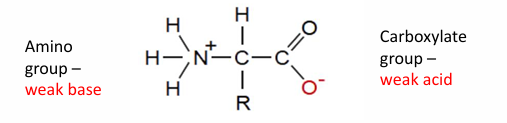
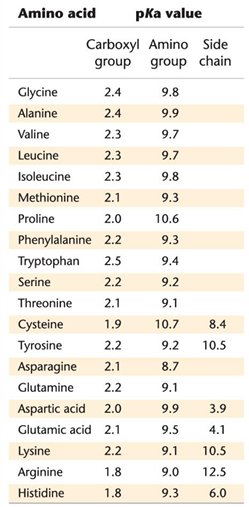
the o- has a pKa around 2.2 and the N+ has a pKa around 9
the stronger acid is the on with the lower pKa
this is what it would look like a pH 7
aka a znitterion ion
depending on the acid depends on where it would be protonated of deprotonated
if the pH is 2.2 then the N would be deprontated
if pH 0 then O will be protonated
at 2.2 is where the titration value levels off
then at pH 9 the N would be titrated
the state of the weak acids (ionized vs protonated) of biological molecules depends on the pH, which regulates their function and how they interact with other molecules
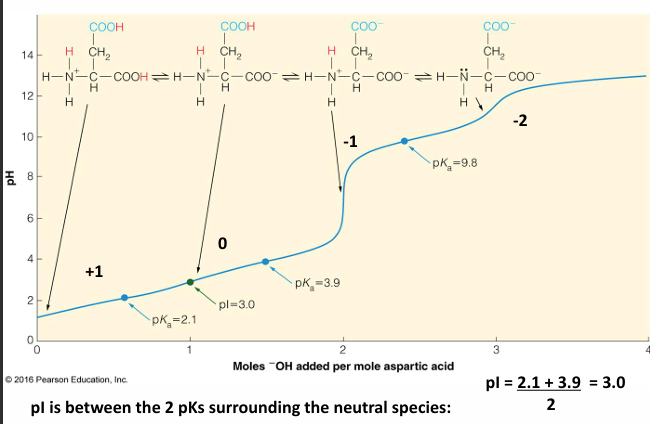
titration
a titration curve describes pH change of solution as base is added to an acid
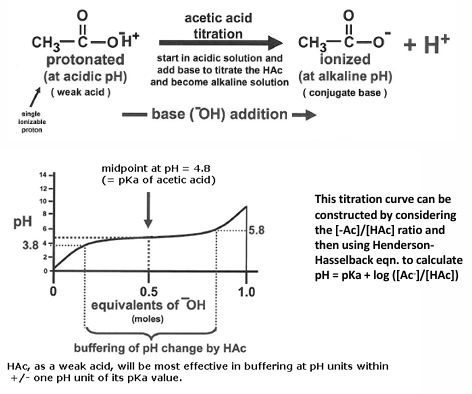
buffers are weak acids or bases that maintain the pH of a solution
ampholyte- molecules with both acidic and basic pKa’s
for example: the amino acid glycine: COOH - acidic and NH2 is basic
zwitterion: containing both positive and negative charge, with a net charge of 0
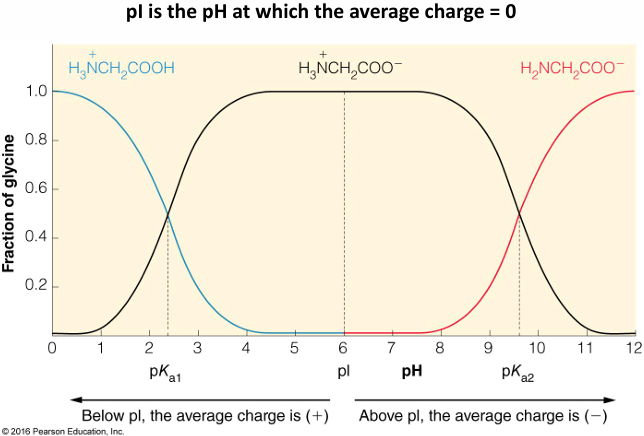
isoelectric point: the pH in which the average charge sums to 0. this happens at only one pH
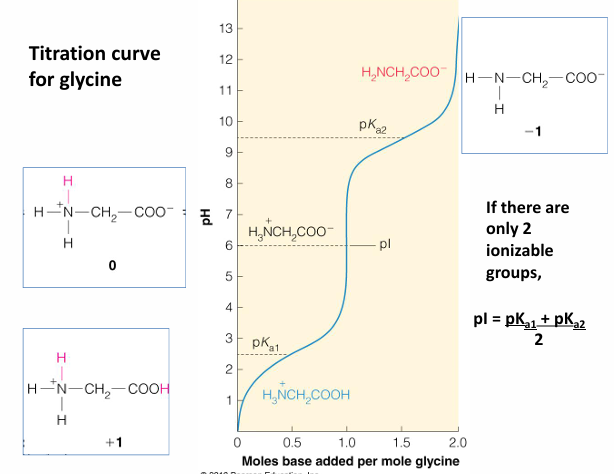
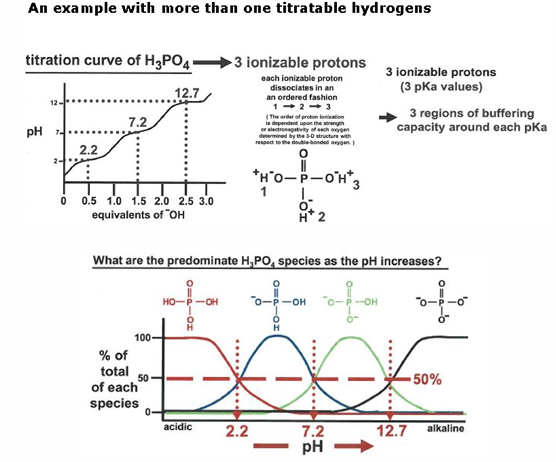
what is the pH if there are more than 2 ionizable groups
example: an aspartic acid amino acid
Henderson-Hasselbalch Equation
to derive this equation, start with the logarithms of terms
pH and the pK_a the concentrations are the same
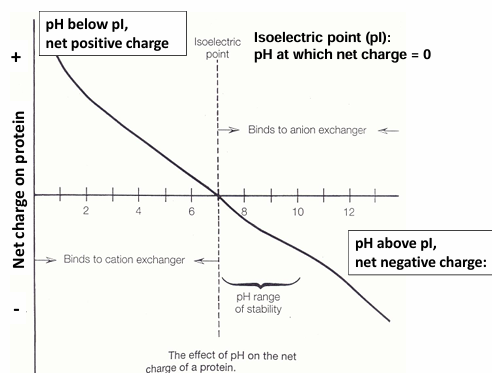
proteins contains many titratable groups: ubiquitin at 8 different pH’s
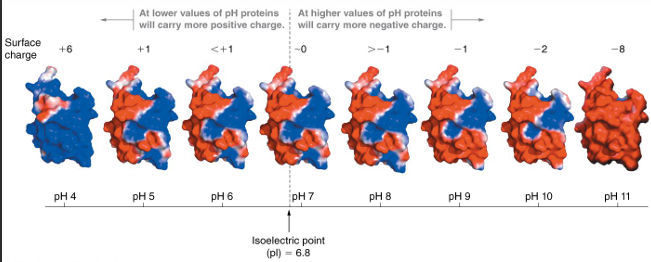
with many ionizable groups, it is difficult to calculate the pH
only the surface residues affect the pH of the folded ; can measure
charge sitribution changes at different pH
even at the pH, the surface is charged: net average charge = 0
solubility in water depends partly on surface changes
DNA is very electronegative
molecules repel each other so they will not aggregate: soluble
repulsion: DNA molecules with many negative charges, strongly repel one another in solution
proteins binding DNA are typically positively charged
attraction: if DNA is mixed witha positively charged protein, these molecules have a strong tendency to associate
pH of proteins affect solubility: example Beta-lactamase
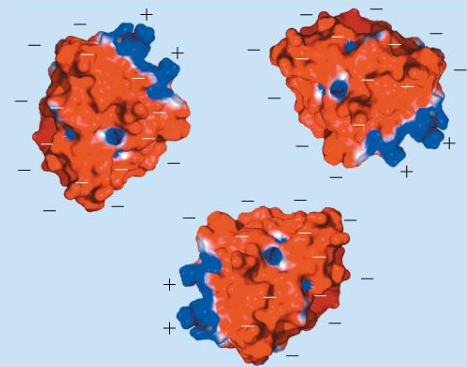
pH > pI
negative surface charges (red) repel → soluble
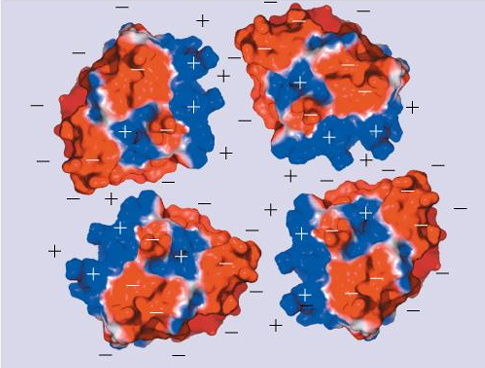
pH of proteins affect solubility: example beta-lactamase
pH = pI
negative and positive surface charges attract → promotes aggregation, protein is less soluble
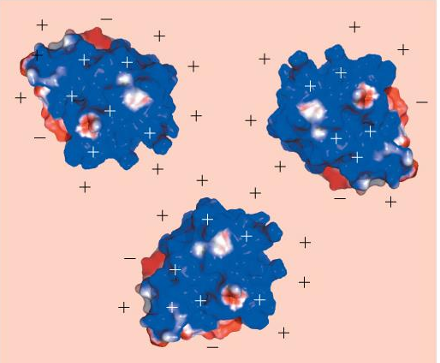
pH of proteins affect solubility: example - beta-lactamase
pH < pI
positive surface charges (blue) repel → soluble
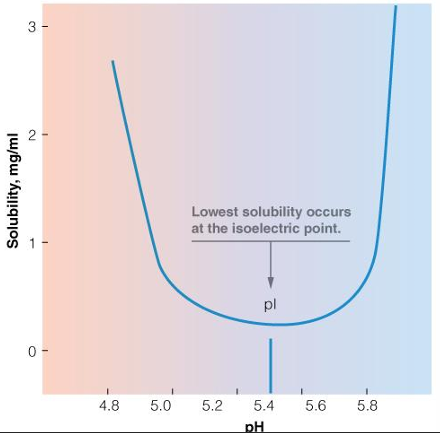
protein solubility as a function of pH
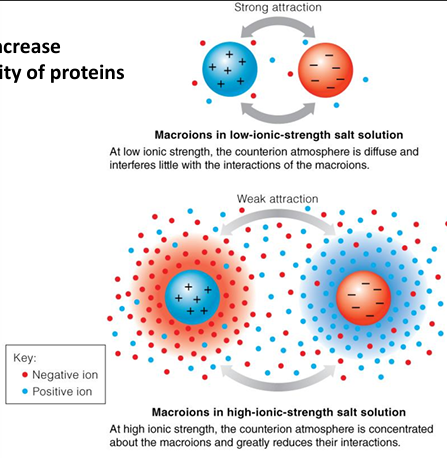
salts increase solubility of proteins
strong attraction
macroions in low-ionic strength salt solution
at low ionic strength, the counterion atmosphere is diffuse and interferes little with the interactions of the macroions
weak attraction
macroions in high-ionic strength salt solution
at high ionic strength, the counterion atmosphere is concentrated about the macroions and greatly reduces their interactions
the influence of ionic strength
Quick reminder
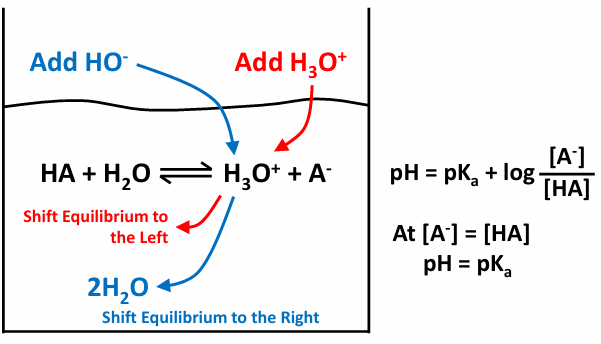
HA + H20 →/← H30+ + A-
pKa pH=pKa + log [A-]/[HA]
buffer is a weak acid
needs to keep the pH at 7.4
chemical properties of buffers
a buffer is a weak acid or weak base that is added to a solution in order to maintain a constant pH
without a stable pH, acidic/basic groups on biomolecules (like proteins) will contain different chages and can lead to protein cinformational changes, alter interactions, or promote aggregation
FINISH THIS PART
and not all good buffers are actually good
bicarbonate: the blood buffer
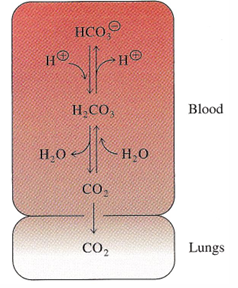
this basically removes protons when exhaling CO2
how buffers work
start off with the equasion
to get to the right pH so pH=pKa we can either add protons or create more acid/base
to add more H+ we could add HCl
will dissociate easily
or we could add a buffer change the pH
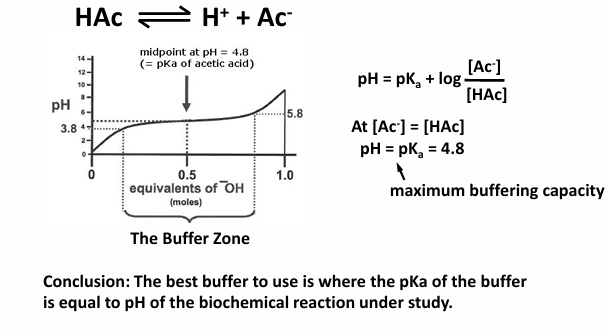
choosing the best buffer
consider acetic acid (a weak acid)
conclusion: the best buffer to use is where the pKa of the buffer is equal to pH of the biochemical reaction under study
making buffer system
experimental: to make the buffer soluctio, you need to know the pKa of the buffer and the pH of the reaction solution inorder to calculte the proportion if the weak acid and its conjugate base
problem solving: for an given buffer problem, you will need to isolate the unknown variable from the henerson-hasselbalch equation
example: what is the pH of a solution containing 150 mM potassium acetate and .3 M acetic acid?

buuuut problems can be more complicated, so keep consistent with units and what is being asked
molar, millimolar, picomolar
example problem 1
from 450 mM acetate buffer at pH 4.45, determine the concentrations of the acid and conjugate base. The pKa of acetic acid is 4.75
on paper
thermodynamics, equilibrium, and kinetics
Thermodynamics
thermodynamics describe the energy and entropy transformations in biochemical and chemical reactions
two types of systems of biochemical or chemical reactions
closed system: energy and materials are retained within the system during any reaction transformation
open system: energy and materials can be exchanged with the surrounding environment during the reaction or transformation
a cell or living organism is an open system because there can be import of thermal energy, nutrients, chemical signals, as well as export of reaction products and energy a heat
three laws of thermodynamics
first law of thermodynamics (energy is conserved)
energy can be converted from one form to another, but the total energy remains the same during a reaction
mathematical description
delta E = Q + W
delta e = change in internal energy of the system
Q = heat transferred into/out of the system
W = work done by/on the system
biological systems operate at constant pressure and volume; therefore, the first law can be described in terms of the enthalpy of the system
delta H = Q + W
reactions will generally be more favorable if there is a decrease in enthalpy, but a decrease in enthalpy is not the only driving force in determining if a reaction will proceed
in a closed system energy is conserved
delt H = enthalpy
delta H = H(final) - H(initial)
heat envolve inna reaction, for example from breaking bonds (at a constant pressure)
enthalpy is a state function - does not matter the path, only the initial and final states
calorimeter measures heat of a reaction
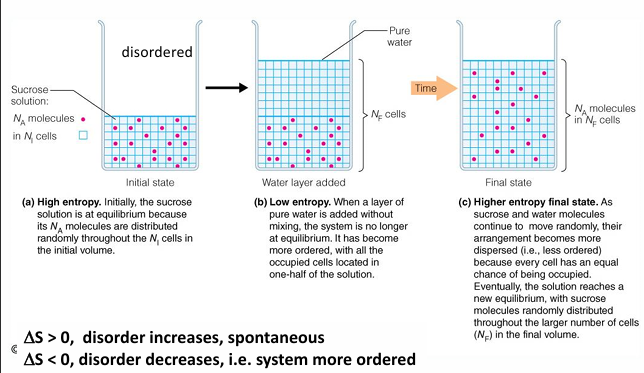
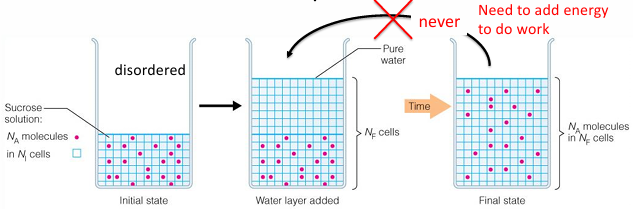
second law: systems tend to go to disorder
the entropy of an isolated system will tend to increase; this means that systems tend to become more random or disordered
delta S = the change in entropy (a measure of disorder)
S becomes larger as disorder increases
S becomes smaller as disorder decreases
liquid water would have a higher S than solid water
When a reaction is not at equilibrium, the reaction is driven to move towards equilibrium
Third law: entropy is at absolute zero temperature
the entropy of a system goes to 0 as the temperature goes to absolute 0
zeroth law: if two systems are in thermal equilibrium with a third system, they must be in equilibrium with each other
this law supports the existence of a temperature scale
free energy (delta G) determines the overall direction of a reaction

since a reaction will go to a lower free energy if it can, this equation means that
a decrease in enthalpy during the reaction and/or an increase in entropy during the reaction will contribute to a favorable negative delta G change
example

thermodynamic of the hydrophobic interaction
chemical equilibria
a chemical reaction can be described by the equation

the relationship between rate constants and equilibrium constants
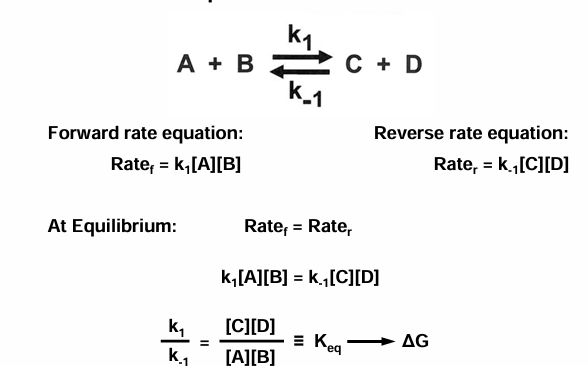
if you add stuff then the rate would stay the same, but the concentration would change
the value of G for the transition state
although delta G can be used to determine whether a reaction is spontaneous or not the concept of free energy can be applied to a explain reaction rates
the activation free energy, which is seen as the delta G needed to reach the transition state will determine the rate at which reaction occurs
activation free energy is designated by G+-
the transition state is the activated form of the reaction(s) in which there is a partial chemical reaction and represents a molecular intermediate in the chemical reaction
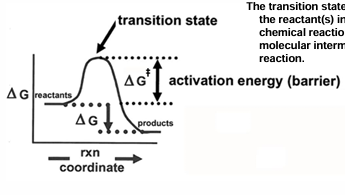
forward and reverse rate constants in both exergonic and endergonic reactions are connected to free energies of activation
for exergonic reaction
the forward rate k1, is proportional to e^- (delta G+-/RT) and the resverse rate k-1 is proportional to e^-((delta G+- + delta G)/RT))
so, the forward rate has a smaller activation energy to breach, and it is faster
for the endergonic reaction
the opposite situation occurs, and the reverse reaction will be faster
the free energy also depends on the concentrations of the reactants and products
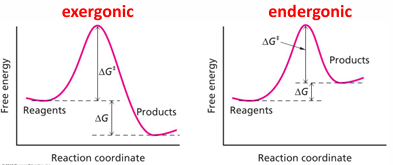
delta G at standard conditions
the delta G 0 means standard condition
everything is in 1 M at 25 degrees C
products over reactions = product quotient

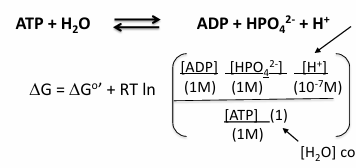
standard delta G biochemical standard free energy
add a condition to standard free energy: pH = 7
most biochemical reactions occur near pH 7 in cells
at pH 7 [H+] = 10^-7 M
if a reaction includes [H+]. the reference concentration is not 1M but 10^-7 M
the reaction quotient (Q) is unitless, so units are removed by dividing each compound by its standard concentration
where, R = 8.314 J mol-1 K-1 and T = temp in Kelvin (C+273)
the [H2O] constant set = 1 (defined)
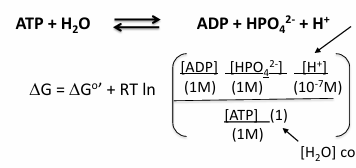
at equilibrium (delta G = 0)
aA +bB →← cC + dD

Example from Textbook
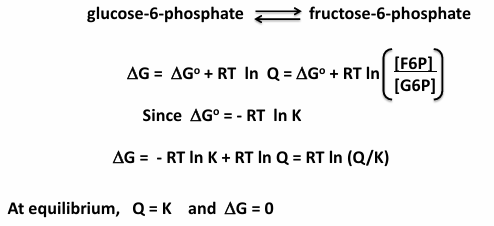
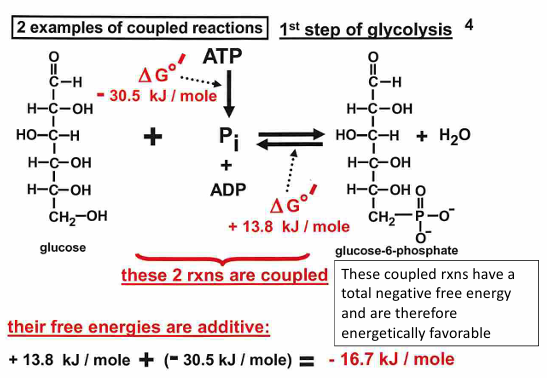
2nd step of glycolysis:
glucose-6-phosphate→← fructose-6-phosphate
delta G0 = 1.7 kJ/mol
at standard conditions, the reaction is endergonic
at equilibrium the reaction favors G6P at 25 C

during glycolysis, formation of F6P is favored because the reaction is not at equilibrium
if [F6P] is low because its used up rapidly in the next step of glycolysis:
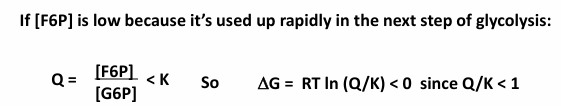
the reaction is now exergonic in the forward direction, favoring formation of F6P
fits LeChatlier’s Principle
interplay between Q and K
if Q<K, delta G<0 exergonic, which favors forward reaction
if Q=K, delta G=0 equilibrium, forward and reverse reactions occur at the same rate (though there may be more reactants or products depending on the K)
if Q>K, delta G>0 endergonic, which favors the reverse reaction
reactions that are part of pathways are typically not a equilibrium
the direction of the reaction depends on Q (not K)
example problem 1
the phosphate transfer potentials for glucose-1-phospahte and glucose-6-phosphate are 21kJ/mol and 14J/mol respectively. (a) calculate the equilibrium constant for this reaction at 25 C? (b) if a mixture were prepared containing 1 M glucose-6-phospahte and 1 × 10^-3 M of glucose-1-phosphate, what would the thermodynamically favored direction for the reaction be?
example problem 2
consider the following chemical reaction A+B→← C+D where delta G0’ is 4.1 kJ/mol
if this reaction were to occure at 37 C with the following steady state concentrations of A at 1 mM, B at 1.0 nM, C at 0.5 uM, and D at 2.0 nM, calculate the delta G and determine of the reaction is endergonic or exergonic.
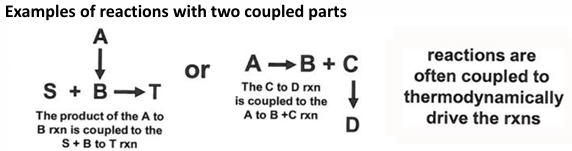
Biochemical reaction coupling
coupling of biochemical reactions
a thermodynamically unfavorable reaction can be performed by coupling it to a thermodynamically favorable reaction
there are other schemes involving two reactions and schemes involving more than 2, or even many, reactions; in these cases, the total free energy change is the sum of all of the free energy change for all connected reactions
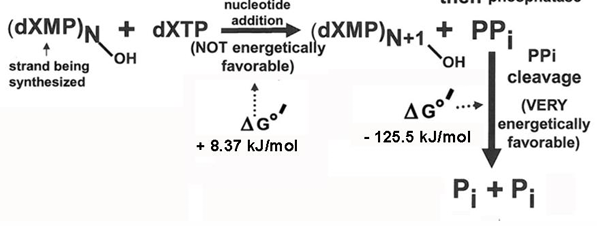
second example of coupled reaction DNA polymerase reaction
so, the total delta G0’ change is +8.4 kJ mol -1 + (-125.5kJ mol-1) = -117.1 kJ mol-1
therefore, addition of nucleotide to chain is favorable, but only if addition is followed by the hydrolysis of the PPi product to Pi
three important points about thermodynamic reactions
biochemical reactions can be coupled
the total free energy change is the sum of the separate parts; this process can aid in a reaction that would otherwise be unfavorable
some reactions proceed even with delta G0’>0 because concentration of product is kept very low, this results in delta G0’<0, which is favorable
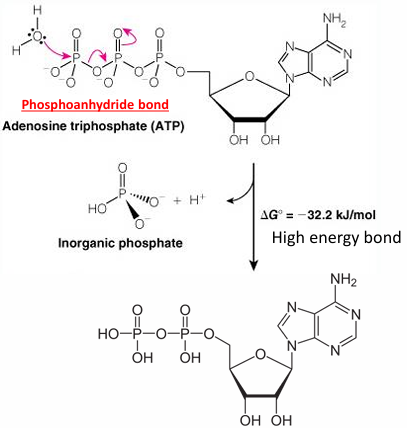
ATP: the universal currency of cellular energy
phosphoryl group transfer - energy transducers in cells
has three phosphoryl groups
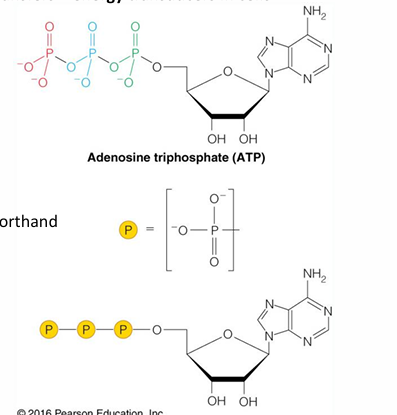
hydrolysis of ATP - phosphoryl transfer to water
nucleophilic attack by the lone pair of water
phosphate: HPO4 2-
Hydrolysis of ADP to AMP
high energy phosphoanhydride
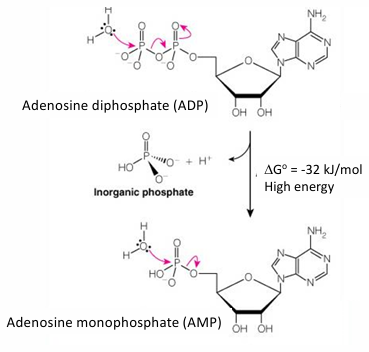
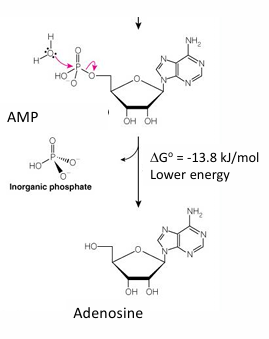
Hydrolysis of AMP to Adenosine

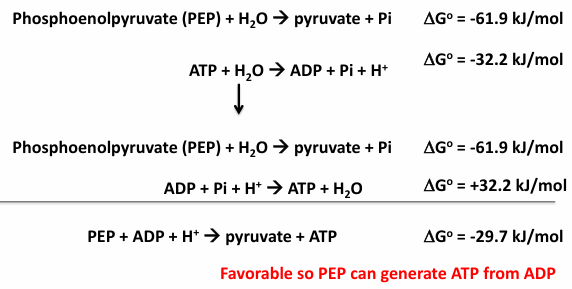
phosphoryl group transfer potential
higher enrgy phosphoryl groups can phosphorylate (transfer the phosphoryl group) to make lower energy phosphoryl bonds
this is the thermodynamic coupling reactions
biochemical redox reactions
oxidation-reduction reactions
energy is transferred in many biological reactions by the flow of electrons
there is exchange of electron(s) in these reactions; this one will be from A to B:
a
remember OIL RIG
oil = oxidation is loss
rig = reduction is gain
two “half reactions” can be written for the substances in this equation
s
A is reducing B (reducing agents are initially reduced)
A is the electron donor, or reductant or reducing agent which becomes oxidized
B is oxidizing A (oxidizing agents are initially oxidized)
B is the electron acceptor, or oxidant or oxidizing agent which becomes reduced
how do we know which direction a redox reaction will go?
half reactions have compared to the reaction 2H+ +2e- → H2 (at pH = 0) using an electrochemical cell
standard reduction potential is measured - it indicated the tendency of the oxidized state to accept electrons
half reactions that appear at the top of the table will be oxidized, they are matched with a half reaction from lower down in the table

Potential difference is measured in galvanic cell
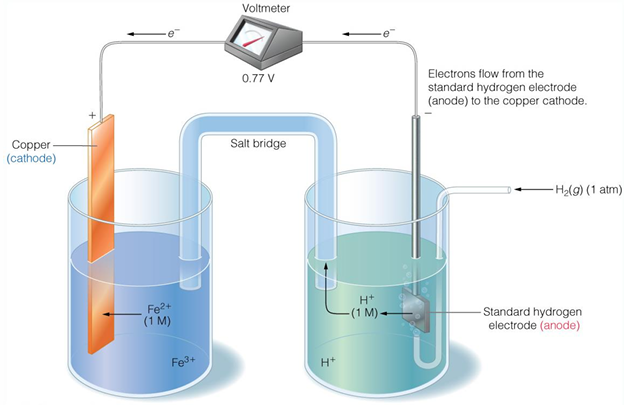
Determination of electron flow in redox reactions

Example: reduction of O2 by NADH
O2 is a stronger oxidizing agent than NAD+ since it is lower in the table (ie O2 more readily reduced that NAD+)
NADH is a stronger reducing agent that H2O since it is higher in the table (ie NADH is more readily oxidized than H2O)

Thermodynamics of redox reactions
the standard reduction potentials in the table are a measure of the tendency of the electron movement. they can be used calculated the free energy change in the reaction
the definition of the standard free energy in the redox reaction is where n is the number of e- transferred, F is Faraday constant (= 96.48 kJ V-1 mol -1)

Calculating the free energy of oxidation/reduction reactions

example problem 1
using the standard reduction potentials (E0’) for ubiquinone (Q) and FAD of 0.04 V and -0.22 V respectively, (a) calculate the free energy for the oxidation of FADH2 by Q (b) is this redox reaction able to generate enough free energy to drive the synthesis of ATP from ADP and Pi under standard conditions?
more highly reduced organic carbon molecules have larger free energies of oxidation
in general, more reduced substances, when they are oxidized, produced a larger free energy change
for instance, in the series of 1-carbon molecule, free energies of oxidation are highest for methane
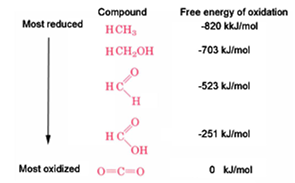
how can there be so many different reduction potentials for organic compounds?
there are different oxidation states for each carbon atom depending on the carbon partners
the important question for judging the C oxidation state is what part of the eight electrons that carbon can potentially possesses does it actually have
C trumps H for owning shared electrons
C vs C share electrons equally
O and N trump C for owning shared electrons
as a consequence of these comparisons, the oxidation state of a C atom can be anywhere 8 to 0
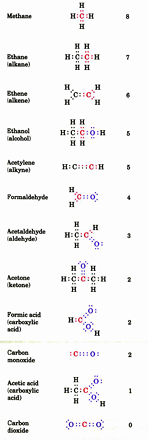
example problem 2
determine the oxidation state for each carbon atom in the following compounds
structure and properties of amino acids
key issues for biological macromolecules
macromolecules usually have specific 3-D shapes
occupies volume and has a general shape
at detailed level, has irregular surfaces with crevices and protrusions
conformational flexibility
their structures can be altered by co-factors, allosteric effectors, or upon interaction with other macromolecules
dynamics
spontaneous and constant change in local and possibly in global shape, even though a preferred conformations is present
these fluctuations occur because of thermal motion
function is intimately connected to the structure of the molecule
biological macromolecule classes
proteins: polypeptides polymers of amino acids
nucleic acids: polymers of nucleotides
carbohydrates: polymers of sugars
lipids: synthesized from acetate (lipids more modest in molecular weight compared to other classes of macromolecules)
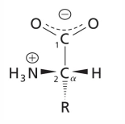
amino acid structure
of the 20 common naturally occurring AA used in protein synthesis, all are based on the same underlying chemical structure arrangement
carboxyl group and amino group are both attached to a central carbon atom (the alpha C)
in 19 of the of the AA, an “R” group is attached to the alpha carbon
proline is connected to the amine, making a five-atom ring
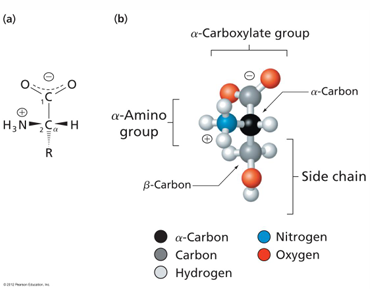
3-D structure of AA
a-carbon: the carbon next to the carboxylic acid
side chain: one of the 20 possible side groups
at pH 7, the AA is mostly zwitterionic
the amine group is deprotonated at pH 9-10
the carboxyl group is deprotonated at pH 2-3
AA stereochemistry
the alpha carbon is chiral because it is attached to 4 different substituents
the H is a dashed bond
the R group is a wedged bond
stereoisomers: molecules with the same chemical formula but different spatial configurations
emil fischer realized amino acids were chiral - rotated light
stereoisomers of alpha amino acids
all common AA are found in the L enantiomer in living systems
to judge the configuration, arrange the AA with CO2- group up (and away) and the R group down (and away), then
if the NH3+ group is on the left (levo or L), the AA has the L configuration
of the NH3+ group is on the right (dextro or D), the AA has the D configuration
the R,S system - based on priority of substituents
SH>OH>COOH>CHO>CH2OH>CH3>H
a chiral center has four different functional groups
identify the function group with the lowest priority
view the chiral center down the bond from the chiral center to the lowest priority atom
assign priorities to the three other functional groups connected to the chiral center, using the above ranking
if the priorities of these other groups go in clockwise direction, the chirality is R
if the priorities of the other groups go in a counterclockwise direction, then the chirality is S
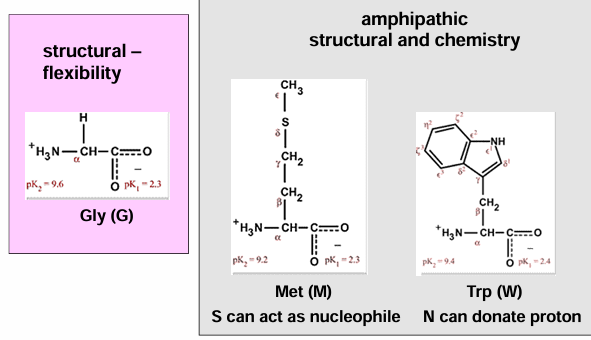
AA side groups
the AA side chains impart unique structural and chemical properties for each individual AA
these side groups establish 4 basic categories of AA based upon the physical characteristics of the side chain groups
hydrophobic AA (gly, ala, val, ile, met, pro) → non-polar side chains
polar AA (ser, thr, cys, gln, asn) → electronegative atoms in the side chain
charged AA (asp, glu, his, lys, arg) → positively or negatively charged side chains
aromatic AA (phe, tyr, trp) → ring side chains with electron resonance
side chains determine the physical and chemical character of each AA and the proteins they assemble
hydrophobic AA - structural
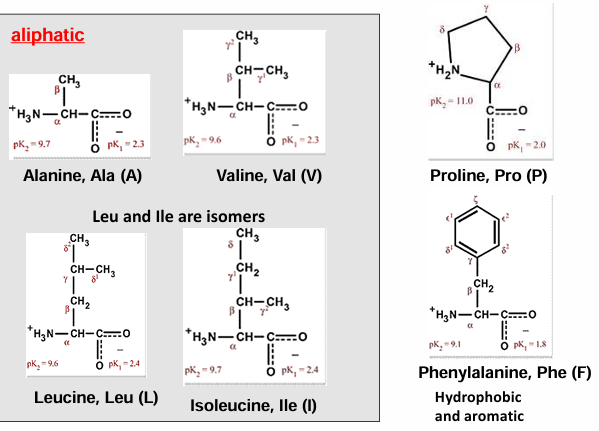
proline: the one cyclic AA
proline is frequently found in proteins in a cis peptide conformation, in addition to the standard cis conformation
aliphatic, nonreactive, the peptide bond preceding proline is more likely to be trans
1:4 ratio of cis to trans
normally it is 1:100
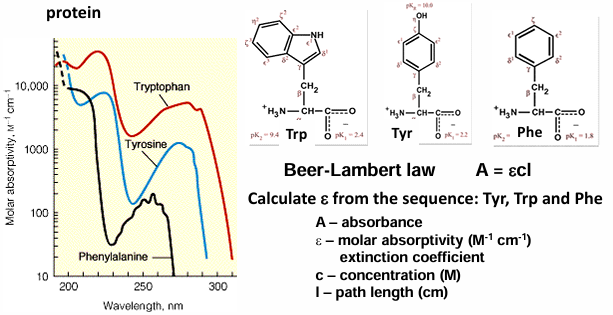
spectroscopic properties of AA
all AA absorb around 200 nm due to the peptide bond
the longer the aromatic system the larger the absorption
aromatic AA absorb around 280 nm
phe, tyr, trp
absorbance at 280 nm can be used to quantify the concentrations of protein
Beer-Lamert law
hydrophilic AA - basic
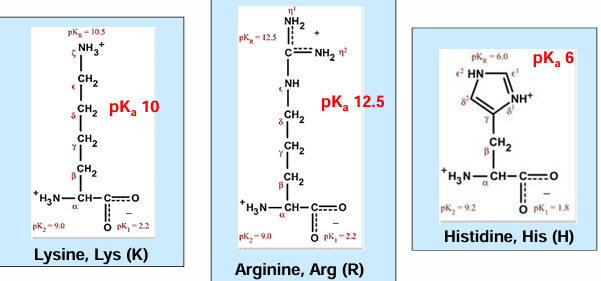
hydrophilic AA - acidic
gives up a proton to ionize and become negatively charged
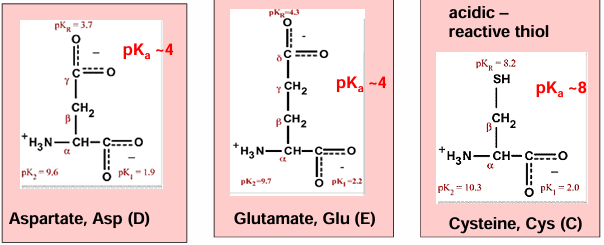
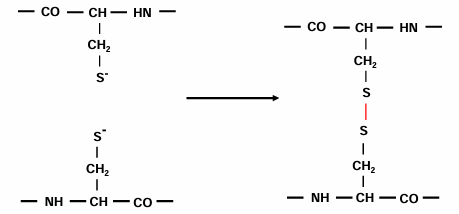
cystine can form disulfide bonds
fun this is a sugar
where do disulfide bonds form:
mostly not in the reducing environment of the cytoplasm
extracellular proteins or soluble domains of membrane proteins translocated to the endoplasmic reticulum
hydrophilic - hydrogen bonding
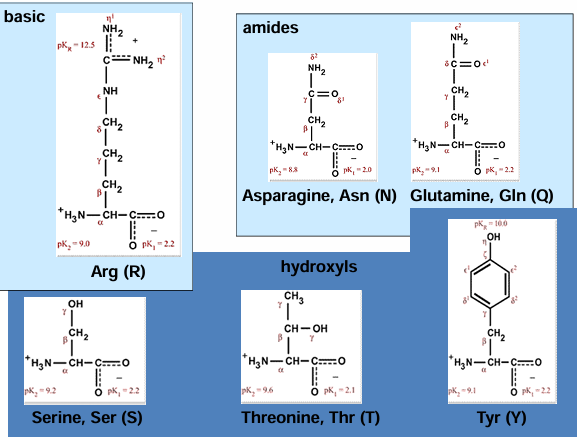
AA one letter codes

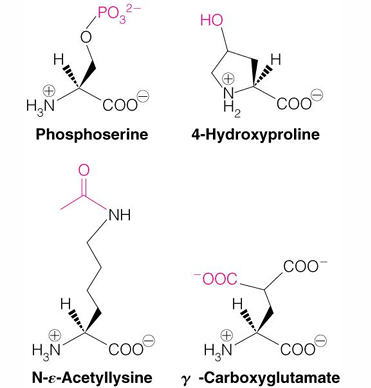
AA can be modified in a protein (post-translationally), increasing the number of possible side chains
has to have an enzyme to phosphorylate
acid/base properties of AA
the pH of the solvent determines the ionized state an charge of the AA and proteins
there are several types of questions that can be answered (and calculated) from the pKa values of the AA and side chain if present
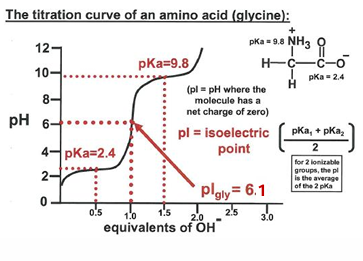
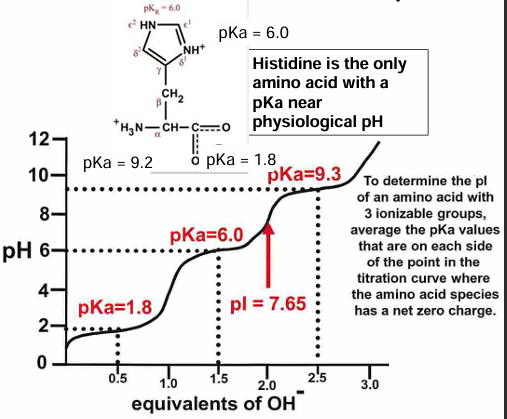
for glycine shown and what is their ratio at pH = 2.4, 6.1, 9.8?
what is the significance of 0.5 equivalents of OH- used to reach pH 2.4?
same question for the amount of OH- needed to reach 6.1 and 9.8
what is (are) the best buffering range for glycine and why?
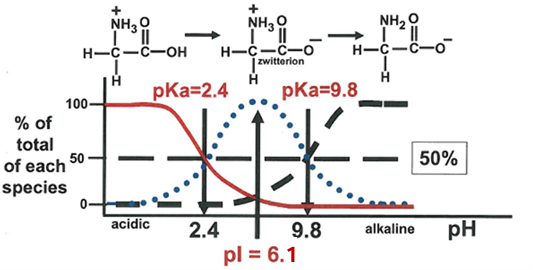
summary of predominant species of glycine present at different pH values
this plot shows the sequential glycine ionic states
the titration curve of an AA with side chain pKa (his)
structure and properties of peptides
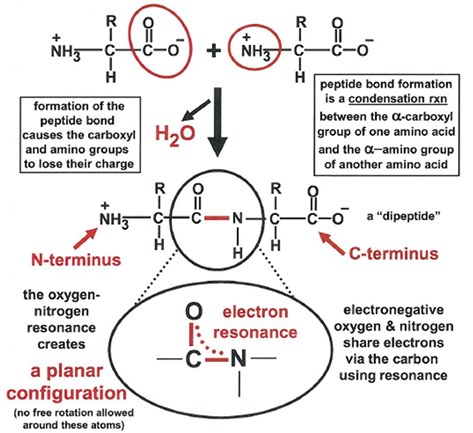
formation of peptide bond as condensation reaction between two AA
linkage of AA results in loss of one water molecule and joins carbonyl group to amino group to form a strong peptide bons
this usually happens during catalyzed protein synthesis in a different context, but the result is the same
there will be multiple peptide bonds formed in a linear fashion and this results in a polypeptide
a protein us folded into a 3D structure but a polypeptide is not
the identity of the AA in the polypeptide constitutes the primary structure of the protein
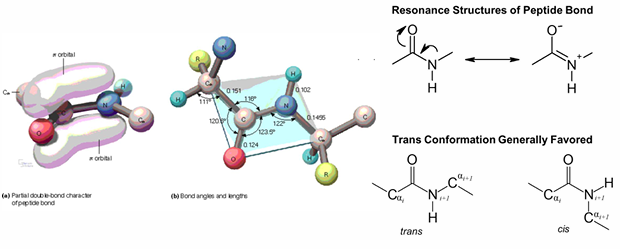
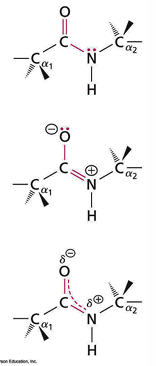
peptides are strong and planar due to resonance of an electron pair
almost always O and H have a trans configuration
consecutive alpha C and alpha C atoms in trans conformation
electronic dipole is present and points negative end towards the oxygen, positive end towards the nitrogen
3-D view of the peptide bond and cis/trans isomerization
peptide/protein nomenclature
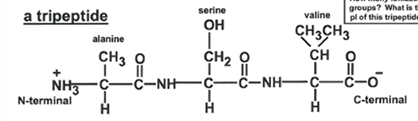
how to name a peptide/protein
→ read the peptide sequence
N-terminal to C-terminal
alanyl - seryl - valine
for naming AA lose the -ine or the -ate and replace it with -yl
the acceptions are gln, asn, and cys where the final -e is dropped and replaced with -yl
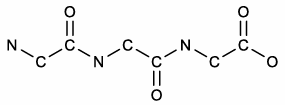
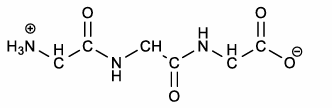
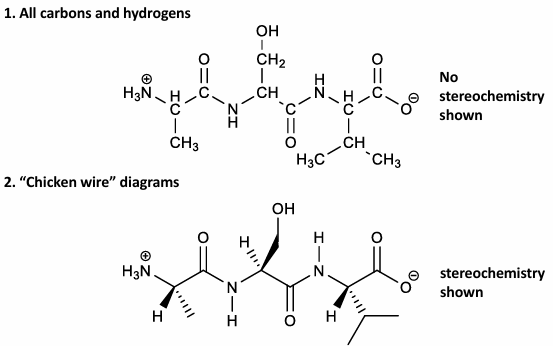
how to draw a peptide: ASV
draw the N-aC-C=O backbone
add H atoms to N and aC atoms
add each side chain
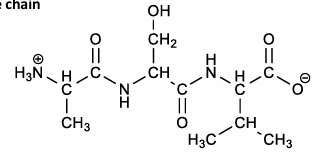
be sure to include the formal charges in the N-terminals, C-terminals, and the side chains
two drawings of peptide ASV
proline is more likely to form a cis peptide bond than most AA
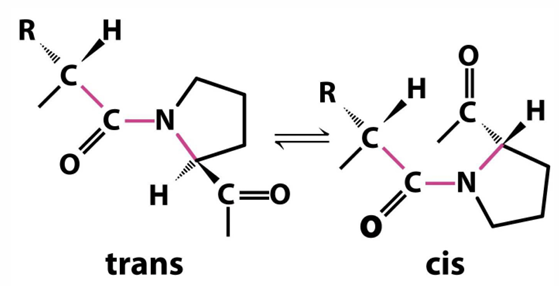
ratio of trans to cis Pro in proteins is about 4:1
titrations of peptides with multiple charged side chains
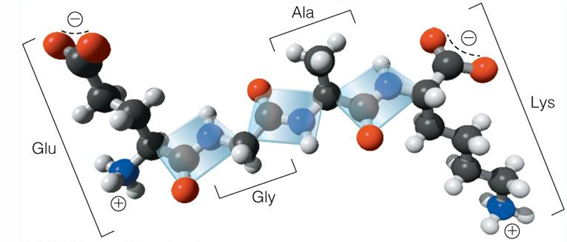
tetrapeptide: Glu-Gly-Ala-Lys (EGAK) - four titratable groups
titration of EGAK tetrapeptide
workflow: calculating pI
gather all the pKa values for the different ionizable groups in the molecule and rank them lowest to highest
draw the molecule at pH = 0, where all of the possible protons are on the molecule
determine the net charge of this structure
call this value n
go to your ranked list of pKa values
find the nth pKa and the next one in the list is nth +1 pKa
average these two pKa values to determine the pI
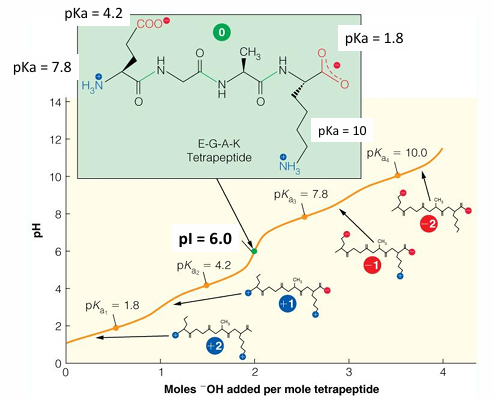
pI of EGAK tetrapeptide
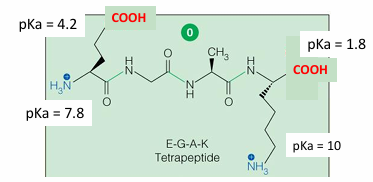
pKa’s ranked them from lowest to highest: 1.8, 4.2, 7.8, 10
draw the molecule at the pH = 0
determine the net charge at pH = 0 (2+)
find the nth and the nth +1 value (4.2 and 7.8)
average the two pKa values = 6
pKas of ionizable groups in proteins
titration curves of proteins: many titratable groups
protein primary and secondary structure
proteins have many and diverse functions in the cell
function as enzymes to catalyze biochemical reactions
store and transport other, molecules
serve as membrane channels for small specific ions and cofactors
can act as transporters for molecules, and molecular complexes
serve as structural components in interior of cells, in organelles and in tissues/organs
serve as mechanical motors for movement of cells and cellular components
serve as receptors for extracellular signals and intracellular communications
serve as regulators for cell regulation and gene expression
all processes - DNA replication, RNA transcription, translation initiation are subject to regulation
levels of protein structure
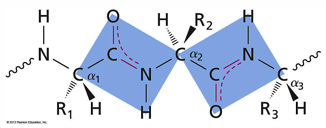
primary structure: sequence of AA covalently linked to form a polypeptide chain. it is not the folding of a protein, but the sequence that determines the fold
secondary structure: local folding held together by hydrogen bonds
tertiary structure: folding of the secondary structures elements into a compact, 3D structure. often this folding creates domains of folded proteins into a multiunit complex. these may be identical or different subunits
quaternary structure: association of 2 or more folded proteins into a multi-subunit complex. these may be identical or different subunits
non-covalent associations
protein secondary structure
the peptide bond exhibits resonance
the consequence of this is that all of the atoms associated with the amide are planar
the fact that amide bond is planar, reduces the number of free rotations in the protein backbone
there is rotation of the plane on the left of the alpha carbon atom
rotation of the plane on the right of the alpha carbon atom
rotation at the R2 group- alpha C bond
note that here that adjacent alpha atoms, and also the O and H atoms, across the peptide bond are in trans arrangements
torsion angled
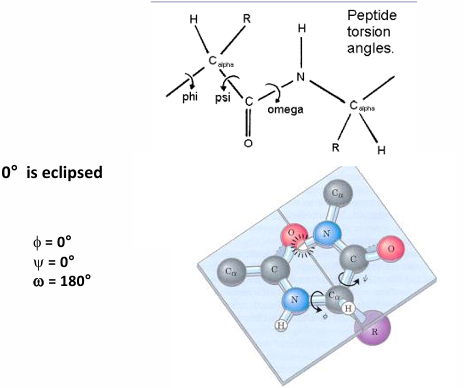
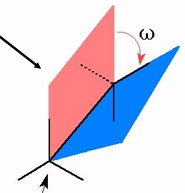
definition of a torsion angle: the angle between planes
the planes are defined by the largest substituents bound to each atom in the bond
torsions angles are defined by 4 atoms
in a + direction, back substituent rotated
alpha helix
for a helix, all c=o points up and all n-h point down
side chains point outwards, pointing towards the amino end of the helix
the side chains don’t generally form H-bons, but some side chains geometries favor helix formation (ie Ala)
right-handed helix is lower in energy than left-handed helixes because the side chains are near NH instead of near c=o
residues at N- and C- terminus are not H-bonded
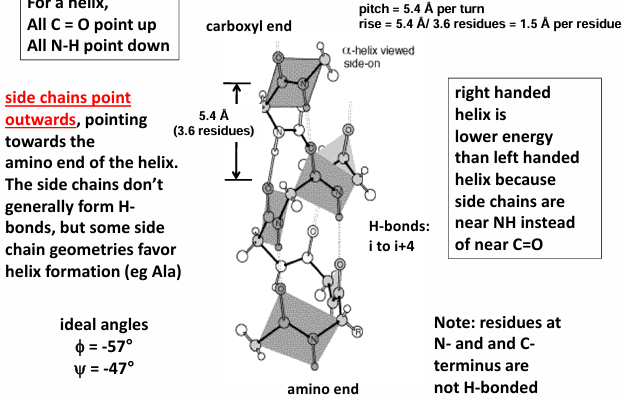
helical dipole moment
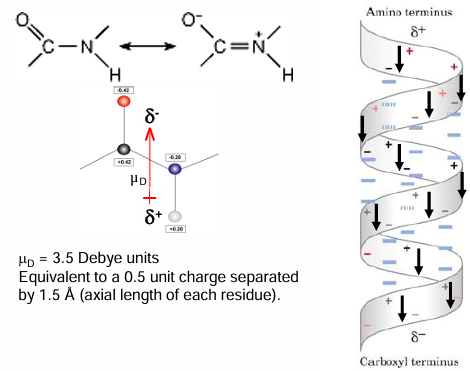
all the H bonds in an alpha-helix point in the same direction because the peptide bonds are aligned in the same orientation along the helical axis.
in addition, each peptide bond has a dipole moment arising from polarity of the NH and c=o groups that is aligned along helical axis
the overall effect is a net dipole for the alpha-helix that elicits a partial net positive charge on the amino end and a net negative charge on the carboxyl end
the total helix dipole = n x 3.5 Debye units, where n is the number of helical residues
overall, this translates into 0.5-to-0.7-unit charge at each end of the helix
as a result, various ligands can be expected to be attracted to the polarized ends
negative ligands like phosphate bind to the amino end, but positive ligands rarely bind to the carboxyl
other helical structure
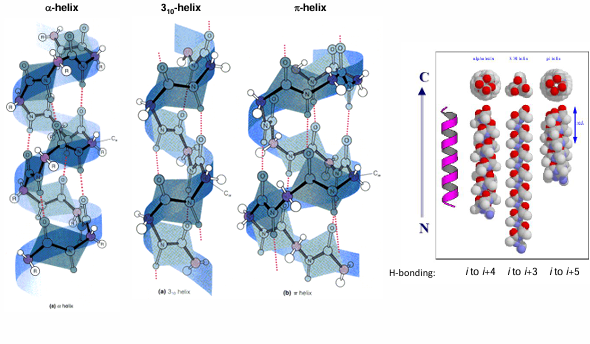
AA preference in alpha helix content
some AA are much likely to be found in alpha helix than others
Ala is most likely due to its small size, intermediate hydrophobicity and specific geometry
Pro and Gly are least likely due to cyclic structure in Pro which is incompatible with the helical structure and due to high flexibility of Gly
look out for free energy changes of different AA in alpha helixes compared to alanine
the sequence is important
ie if there are many glu/a
also if it is a short sequence there is smaller chance that a helix will be formed due to electrostatic repulsion
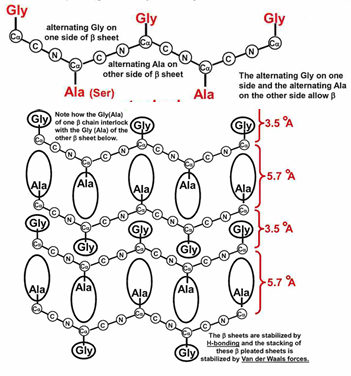
beta sheet
beta sheets are very common secondary structure feature
stabilized by the inter-strand H-bonding of beta strands to form beta sheets
these strands are usually part of the same polypeptide
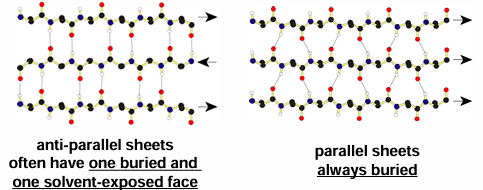
H bonds can occur again between carbonyl oxygens and amide hydrogens but exact patter and geometry of these a little different in the parallel and anti-parallel sheets
strands can be arranged in parallel or anti-parallel manner (where direction is defined by N to C sense in each strand)
in both of structures, R groups alternate above and below the surface defined by H bonding network
there is usually a natural twist to the sheet- even with just two strands
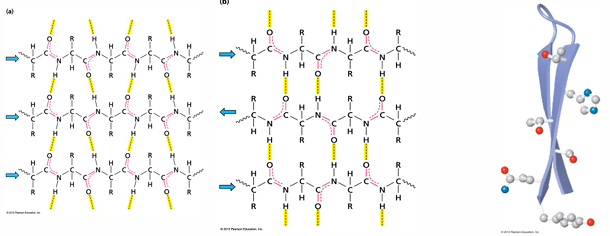
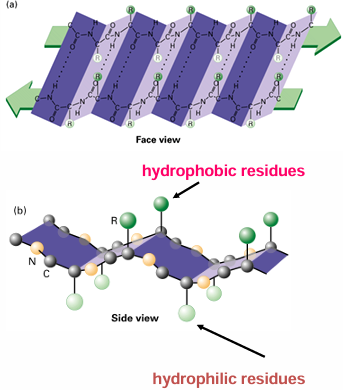
parallel beta sheets
the H-bonds are angled
strands orient in the same direction when paired
(N=>C, N=>C, N=>C)
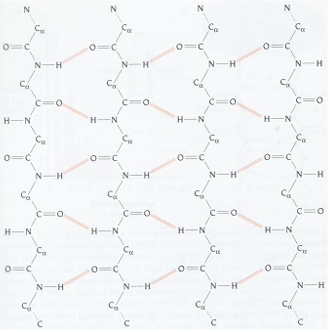
the pleat: side chains alternate pointing up and down with the direction of the pleat fold
parallel beta sheets have a right-handed twist
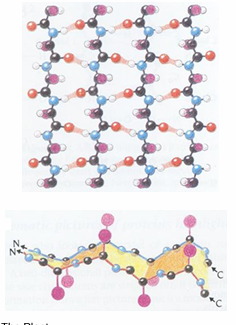
antiparallel beta sheets
the antiparallel beta sheets
H-bonds are linear and thus stronger than those in the parallel form
strands alternate in direction when paired (n=>c, c=>n, n=>c)
more common than the parallel form
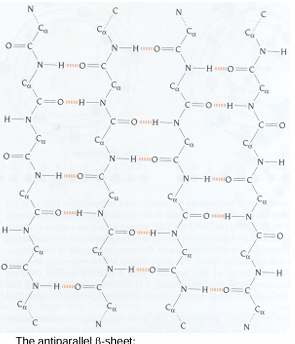
the pleat:
alpha carbon atoms are raised slightly above and below the plane
side chains alternate pointing up and down of the pleat folds
no twisting in the sheet
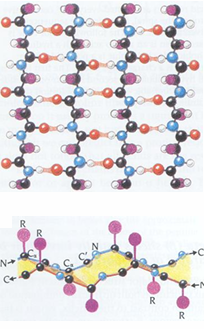
alpha helix vs beta sheets
beta-sheet
side chains alternate above and below the sheet
H-bonds perpendicular to strand extension
alpha helix
side chains extend perpendicular to helical axis
H-bonds parallel to helical axis
connectors between secondary structure elements
globular proteins change directions frequently in order to be compact
there will be non-structured loops
structured beta turns are often between antiparallel beta strands, 4 residues in the turn, 2 residues are not H-bonded
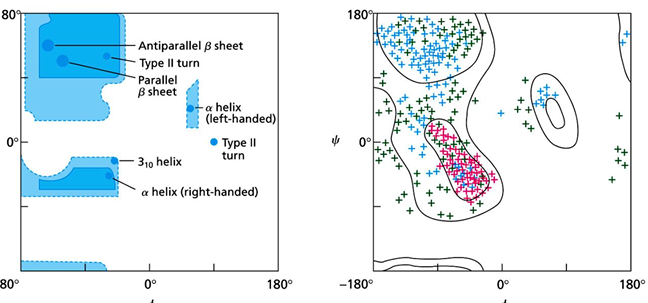
Visualizing dihedral angles of protein structures
a 2-d plot of phi vs theta values for each amino acid is called a Ramachandran Plot
each type of secondary structure will have fairly specific values of phi and theta
by looking at the location of the phi and theta for a particular AA you can conclude what secondary structure is present.
note that there are large areas in the theoretical and actual plots that are empty of data; these are restricted areas because of steric clashes would occur if an AA possessed those phi and theta values
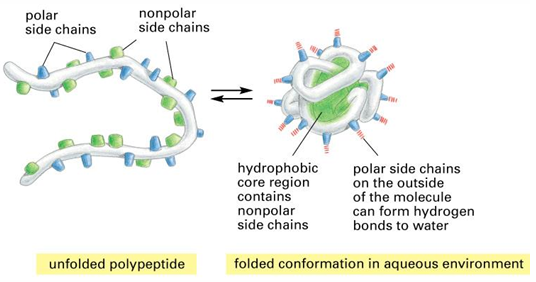
protein tertiary and quaterna637ry structure
tertiary structures
the driving force for folding water-soluble globular proteins is to pack the hydrophobic residues into the core of the interior and create hydrophobic core and hydrophilic surface
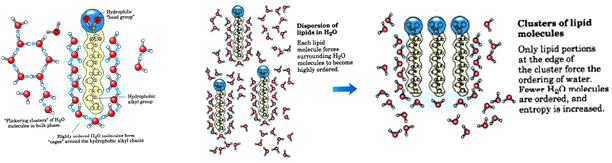
hydrophobic interactions
the non-polar molecular surfaces give rise to the situation in which water is constrained in specific orientations. this is very unfavorable entropically (ordered water at the non-polar surface ha low entropy)
if two non-polar surfaces associate, the water is released, and the result is a large increase in entropy - this is associated with a large free energy decrease
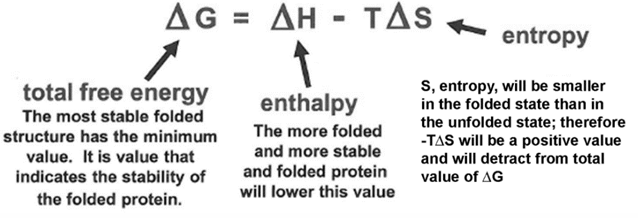
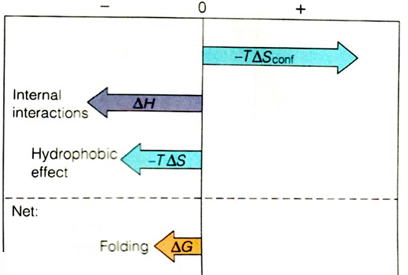
Overall free energy change for protein folding
the overall free energy change for protein folding delta G folding is negative, but usually surprisingly small, and it is the difference between large favorable and unfavorable conditions
it is sum of favorable internal interactions that are made in folded structure, “hydrophobic effect” interactions (contribute to favorableness of folding), but large unfavorable free energy change due to entropy of constraining the polypeptide chain
because delta G folding is so modest, the proteins cannot partially unfold - they would be too unstable; hence the highly cooperative, all-or-none behavior
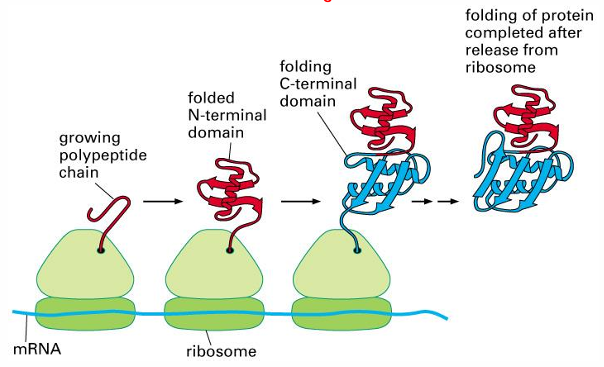
protein folding in vivo
Folding in vivo is assisted by molecular chaperones
many chaperones function by preventing aggregation of unfolded proteins, allowing them time to fold properly
E. coli GroEL is an example of a chaperone
unfolded proteins bind in hydrophobic cavity of GroEL, allowing them time to refold
chaperones like GroEL are heat shock proteins, preventing protein aggregation in response to heat
another example is HSP90- heat shock protein 90kDa
some chaperones assist with folding specific complexes
Misfolding Diseases
Alzheimer’s disease - amyloid beta or a-beta peptide
Parkinson’s disease - alpha-synuclein
spongiform encephalopathies (such as Creutzfeldt-Jakob disease and kuru) - prion protein
amyotrophic lateral sclerosis (ALS, or Lous Gehrig’s disease) - superoxide dismutase 1
Huntington’s disease - huntingtin with polyQ tracts
cataracts - gamma-crystallin
type ll diabetes - islet amyloid peptide (IAPP)
injection-localized amyloidosis - insulin
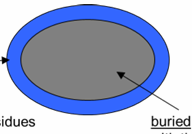
Average Composition of Buried and Accessible Regions
of a Globular Protein
accessible: made up of residues with their side chains 5% or more accessible to water
side chain composition
27% charged
34% polar
39% nonpolar
buried: made up of residues with their side chains 5% or less accessible to solvent
side chain composition:
4% charged
18% polar
78% nonpolar
Depictions of protein structures
cartoon model: follows the protein backbone
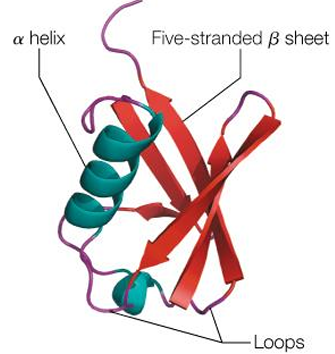
stick model: shows the location of all atoms including H atoms
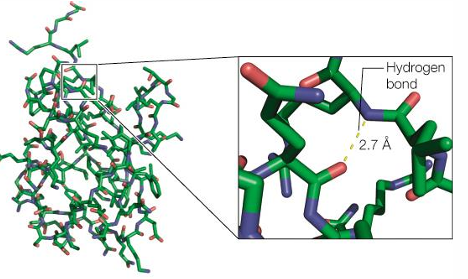
Hydration shell surrounding proteins in solution
ordered water molecules in the crystal structure of lysozyme
water molecules from hydrogen bonds with surface residues of proteins, and other large biomolecules. this helps solubilize the structure
other roles for water:
water also contribute to the hydrophobic effect, helping to fold proteins. “Structural waters” help keep the protein folded. water is often essential for catalysis
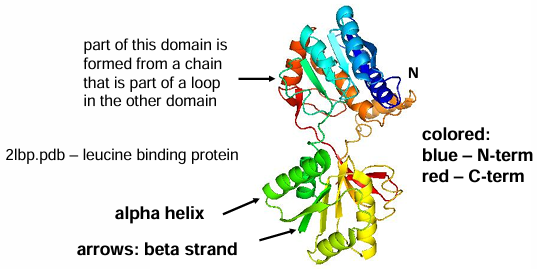
Protein Domains
domain: a compact unit pf protein structure that is usually capable of folding stably as an independent entity in solution. domains do not need to comprise a contiguous segment of peptide chain, although this is often the
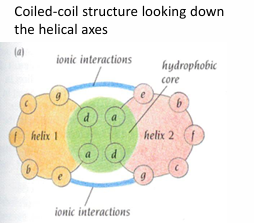
alpha-helix motifs: coiled coils
packing hydrophobic cores - interior of the protein
amphipathic - having both polar and nonpolar character and therefore a tendency to form interfaces between hydrophobic and hydrophilic
Helical wheel diagram of alpha-helix motif: coiled coils
looks down the helix axis
determines if there will be hydrophilic/hydrophobic regions
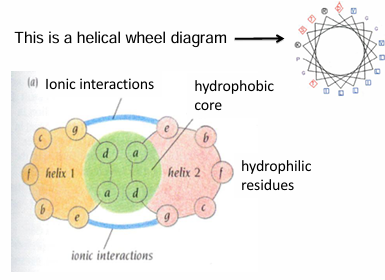
AA separated by 3-4 AA are on the same side of the helix
note: heptad repeat - it takes 7 AA and 2 turns of the helix for the position of the residues to repeat
Four-helix bundles – a proteins
topology - sequence order of secondary structure in the folded protein
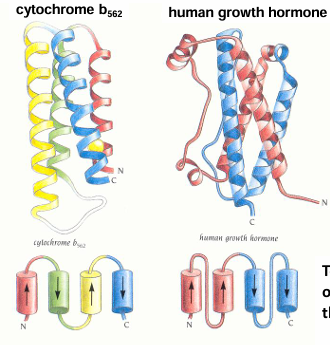
Amphipathic b-sheets
all the hydrophobic residues are on one side and all the hydrophilic residues are one the other side
beta-domain structures
anti-parallel beta sheets are more common that parallel beta sheets
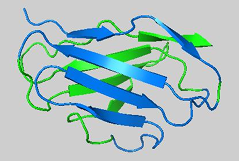
All beta-sheet proteins: immunoglobulin fold
an antibody is made up of 12 immunoglobulin domains
beta-sandwich: one immunoglobulin domain is composed of 2 layers of beta sheets
Triosephosphate Isomerase (TIM)
common alpha-beta motif
most common domain fold known- occurs in 10% of enzyme structures
varying number if beta-alpha-beta-alpha motifs are possible usually 4
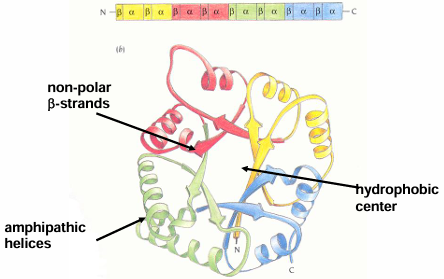
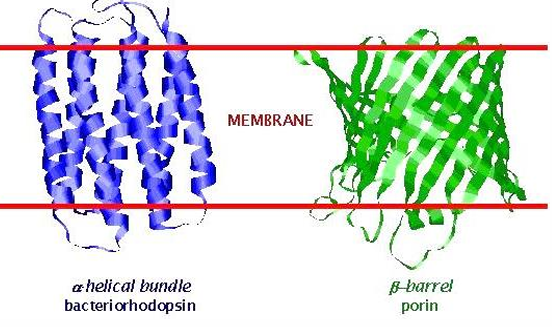
Membrane Spanning Proteins
integral membrane proteins display only two structural motifs: membrane spanning alpha-helical bundles and beta-barrels
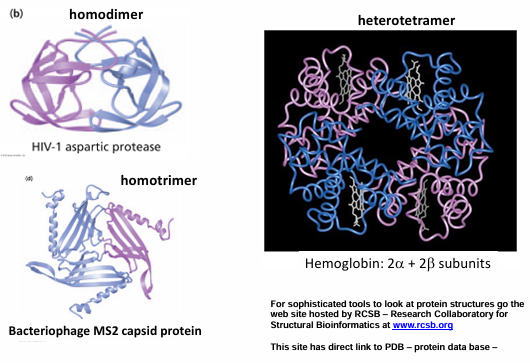
Quaternary Structure
the assembly of multiple protein chains into a functional unit
oligomers: protein assemblies of more than one polypeptide chain
monomers (or subunits) - the individual chains of an oligomer
possible oligomers
dimers (2), trimers (3), tetramers (4), pentamers (5), hexamers (6), etc
homodimer: composed of identical monomers
heterodimer: composed of different monomers; often the monomers resemble each other
examples of quaternary structure
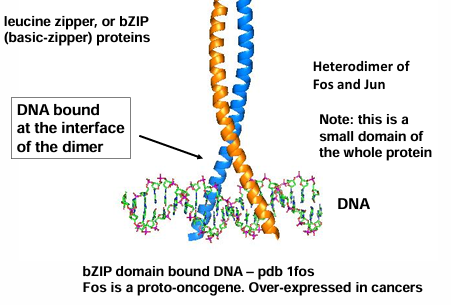
reasons for quaternary structure
ligand binding or active sites often occur at the interface of multimers
cellular recognition
regulation
multimers allow for allosteric (cooperative) regulation
allosteric effector: a small molecule that binds outside of the active site and modulates the activity of a protein
i.e. 2,3 BPG (2,3 biphosphoglycerate) stabilizes a conformation of hemoglobin that stimulates release of O2
allows for formation of large, complex, multi-functional “molecular machines”
myoglobin and hemoglobin
myoglobin and hemoglobin
compare O2 binding to myoglobin vs hemoglobin:
myoglobin stores O2 in the muscles
hemoglobin absorbs O2 in the lungs and delivers it to cells
binds O2 cooperatively
allostery
ligand binding equation: general equation to calculate the fraction of a protein bound to ligand
ligand binding: comparison of O2 binding to myoglobin vs hemoglobin
oxygen needs to be bound during transportations because it is a great oxidizers
we do not want oxygen stealing electrons from places it should not
hemoglobin transports O2 through the blood
oxygen diffuses to tissues or transported by myoglobin in muscles
CO2 carried back by hemoglobin or in plasma as HCO3-
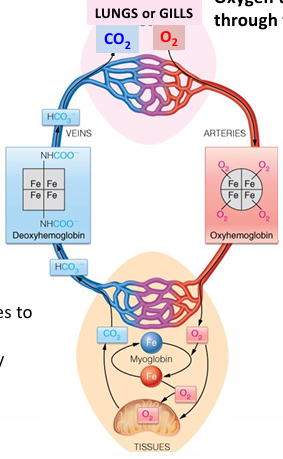
Oxygen is carried through the blood in red blood cells or erythrocytes
each red blood cell carried 300 million molecules of hemoglobin
hemoglobin carries 100x more O2 than would be soluble in blood plasma
hemoglobin and myoglobin are members of the globin family
all globins bind oxygen, but not all globins are used for O2 transport
apoprotein: protein without its prosthetic group
holoprotein: protein with a prosthetic group bound
How do these proteins bind to oxygen?
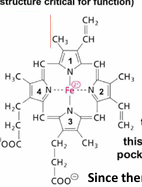
these proteins utilize a prosthetic group (tightly bound non-peptide structure critical for function)
the prosthetic group of hemoglobin and myoglobin is the heme group
composed of a protophorphyrin ring and Fe2+
the N of each pyrole ring coordinates/binds the Fe2+ atom
4 pyrole rings are covalently liked by mthly groups
this heme group binds in a hydrophobic pocket of myoglobin/hemoglobin proteins
since there is no H2O in the site Fe2+ remains as ferrous and not oxidized to ferric, Fe3+
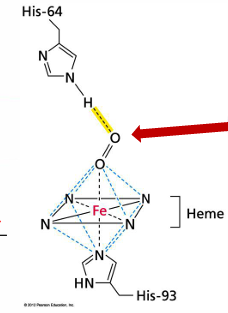
how does the heme prosthetic groups bind to oxygen
this oxygen is bound reversibly so that it can bind and then dissociate
this is critical for the myoglobin/hemoglobin proteins to function in supplying oxygen to body tissues
CO2 and CO also bind to heme group of hemoglobin
carbon monoxide binds very tightly, essentially irreversible
it is not displaced by O2
high levels of CO binding can cause suffocation
proximal: the histidine binds Fe directly
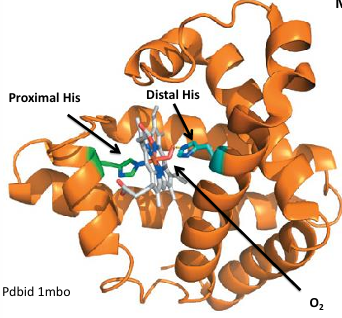
myoglobin structure
myoglobin is a monomer
single globin chain consists of 8 alpha helixes. the heme binds in a hydrophobic pocket
this shows O2 bound but how is O2 binding measured
derivation of binding curves

binding curve: bound vs free ligand
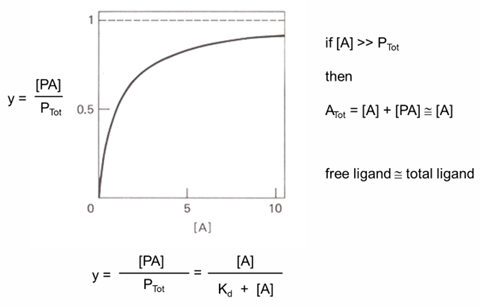
Kd determination
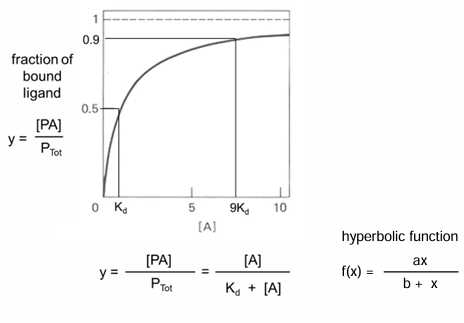
how to describe O2 binding to myoglobin (Mb)
hyperbolic binding curve
Yo2 = fraction of Mb bound by O2
Yo2 = (Mb bound by O2)/ (total concentration of Mb)
from equilibrium constant for:
MbO2 →← Mb + O2
Kd = ([Mb][O2])/ [MbO2]
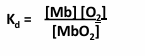
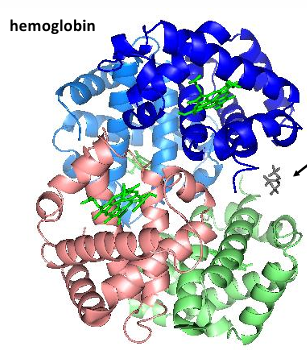
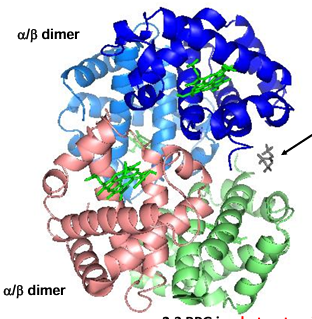
Hemoglobin structure
tetramer of 2 alpha chains and 2 beta chains
note the 4 heme groups - one each on each group
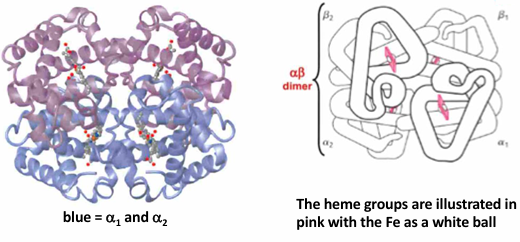
How similar are the structures of the myoglobin and hemoglobin chains
alpha globin = blue
beta globin = purple
myoglobin = green
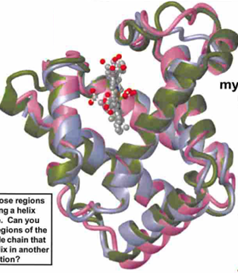
myoglobin and hemoglobin belong to the same → protein family: typically, members of a protein family exhibit → similar overall folded structures
identify those regions exhibiting in a helix structure. can you also find regions of the polypeptide chain that turn the helix in another direction
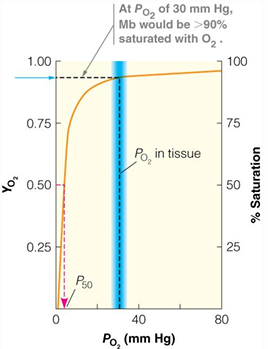
O2 binding curves for hemoglobin compared with myoglobin
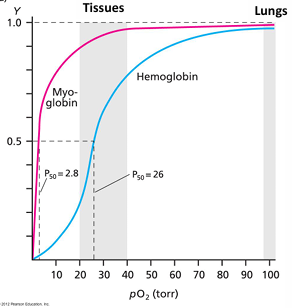
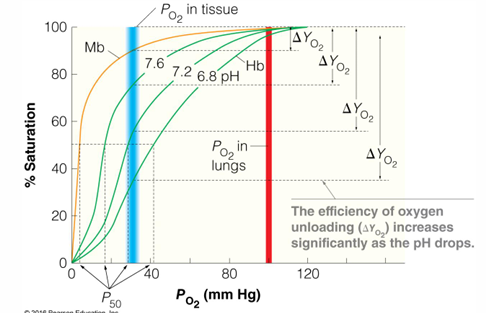
at 30 torr myoglobin about 90% bound and hemoglobin about 50% bound
at 100 torr in the lungs myoglobin is about 100% bound and hemoglobin is about 98% bound
sigmoidal binding curve of hemoglobin:
binding of O2 is cooperative
larger affinity changes over a smaller difference in pO2
allostery
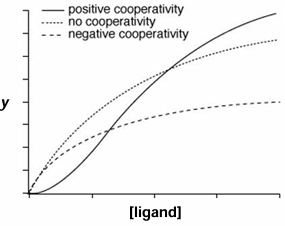
definition: the capability of one ligand to bind and effect the binding of a second ligand at a distant site
positive cooperativity: binding of the first ligand increases the affinity of subsequent ligands
negative cooperativity: binding of the first ligan decreases the affinity of subsequent ligands
hemoglobin is adapted to transport O2
hemoglobin increases the concentration of O2 in the blood
torr is a measure of Patial pressure
1 torr - 1 mm of Hg at 20 degrees C
760 torr - 1 atm
blood itself has about 100 torr of O2
hemoglobin increases the carrying capacity of blood about 80-fold
atrial blood ha about 100 torr of O2
venous blood has about 30 torr of O2
hemoglobin functions over 3-fold change in O2 concentration
the affinity for O2 changes from about 90% in the lungs to about 30% in the veins
myoglobin stores O2 in muscles
binds O2 more tightly than hemoglobin
not quick to release O2
the sigmoidal shape of the hemoglobin curve results from changes in quaternary structure that increases binding affinity
Monod, Wyman, Changeaux (MWC) model of allosteric regulation of hemoglobin
2 states of the hemoglobin tetramer exist:
a tense state (T) that does not bind O2 well
a relaxed state (R) that binds O2 tightly
in the T state, if O2 binds one of the monomers of the tetramer, it promotes the conformational change of the tetramer to the R state, increasing the O2 affinity of the other 3 sites of the tetramer
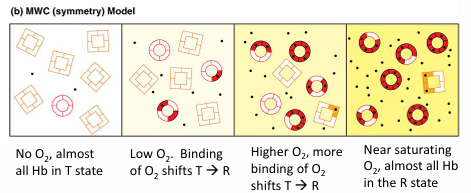
MWC model for cooperative oxygen binding to hemoglobin
T-state: low O2 affinity
R-state: high O2 affinity
increasing O2 binding increases the probability of Hb switching from low affinity T-state to high affinity R-state
O2 is homotropic effector of Hb since it affects its own binding by other O2 molecules
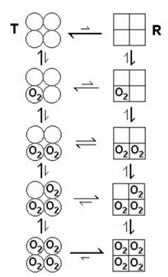
Allosteric effectors
allosteric effectors: a small molecule that binds outside of the active site and modulates the activity of a protein
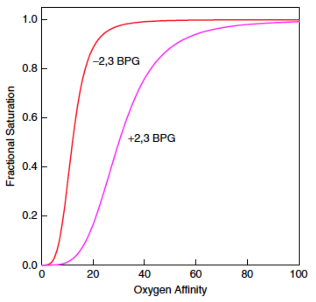
2,3 BPG is a heterotopic effector of Hb since it binds to a regulatory site distant from the active site
binding of 2,3 BPG stabilizes the T-state (i.e. 2,3 BPG binds better to the T-state than the R-state) as a result, 2,3 BPG stimulates the release of O2
example of allosteric regulation: 2,3 biphosphoglycerate induces oxygen release in peripheral tissues
Bohr effect - lower affinity of O2 at lower pH, releasing O2
CO2 accumulation in the blood lowers the pH: CO2 + H2O →← HCO3- + H+
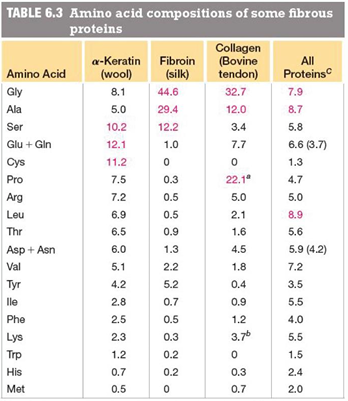
fibrous proteins
Fibrous proteins
structural proteins of limited sequence variations
properties of fibrous proteins
composed of long, extended chains
typically contain hydrophobic AA allowing chains to interact
limited sequence variation
insoluble in water
high tensile strength
examples
alpha- keratin: fingernails, hair, skin
intermediate filaments in cells for structure of nuclei and cytoplasm
beta- keratin: feathers and scales
collagen: bone matrix, tendons, skin
fibroin: silkworm silk, spider webs
fibrous proteins are composed of repeated, short sequence motifs
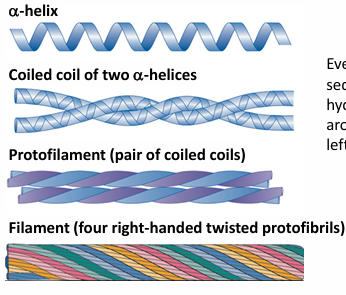
example #1 of fibrous protein: alpha keratin
dominant protein of hair, wool, anils, claws, quills, horns, hooves, outer skin layer
sequence: predominantly: ala, val, ile, met, phe
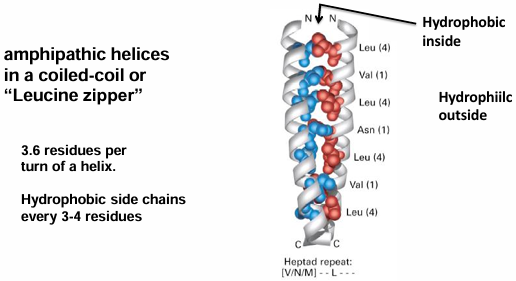
hydrophobic residues, form a coiled coil with leu and hydrophobic residues packing between helices, at the a and d positions
helix: 3.6 residues per turn
organization of alpha-keratin fibers
every 3-4 residues in the sequence are hydrophobic. the hydrophobic stripe spirals around the helix, resulting in a left-handed super-helical twist
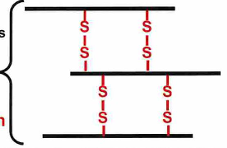
disulfide bonds between helices in alpha-keratin
a key feature of alpha keratin is the disulfide bond formation between helical chains
the more disulfide bonds crosslinking the harder the alpha keratin
the hardest alpha keratin is rhinoceros horn which has about 18% of the AA cross-linked with disulfide bonds
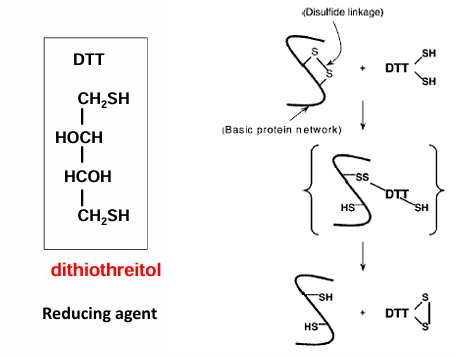
disulfide bonds in hair
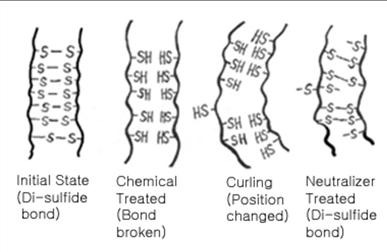
a permanent at a hair solon alters disulfide bonds
human hair possesses some disulfide bon crosslinking. when someone gets a perm at a hair salon, the mercaptan (reducing agent) compound initially breaks down these disulfide bonds by reducing the (cys is protonated again). this is why the 1st step in perms smell. then the hair is curled and these cys residues are oxidized to reform the disulfide bonds with the hair now newly curled. as long as these new disulfide bonds are maintained the hair remain curled with the perm
Example #2 of fibrous protein: collagen
dominant component of skin bone teeth tendons and cartilage
the most abundant protein in mammals
synthesized by fibroblast cells
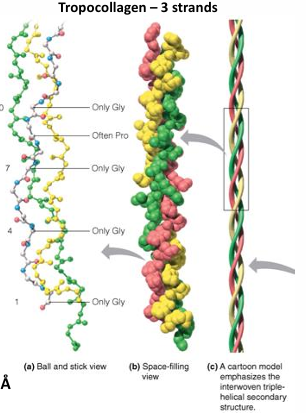
sequence predominantly: repeats of Gly-X-Y often gly-pro-y or gly-x-hyp
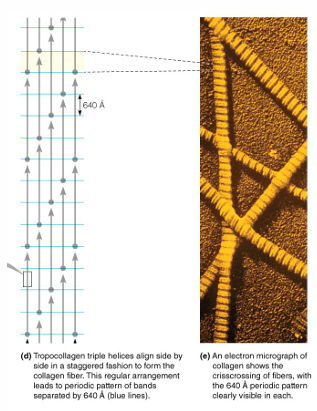
left-handed helices→ triple helices form a right- handed superhelix long and rigid: 3,000 A x 15 A
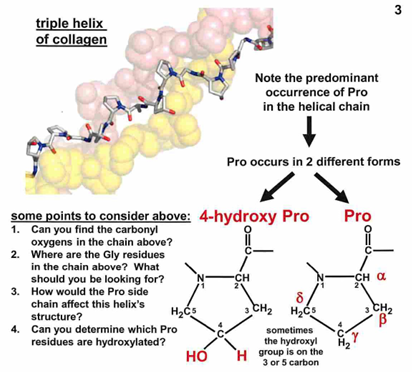
enzymes catalyze hydroxylation of Pro and Lys to form hydroxyproline and hydroxylysine.
cofactor: ascorbic acid (vitamin C)
scurvy: the deficiency of vC
common is sailors in the 15th century due to the lack of fresh fruit
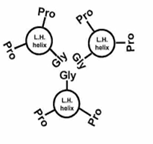
interactions within the triple helix gly-gly interactions:
3 AA complete one turn of a left-handed helix
the gly-pro-pro stabilizes the triple helix through interactions between gly residues, with pro rings located on the outside of the helix. gly is the only residue that can fit in the interior of the helix, which is very crowded.
H- bonds between amide H of gly of one chain and carbonyl O of residue X in another chain
also, H-bonds with OH of hydroxyproline
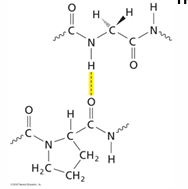
interactions between collagen chains forming rigid collagen fibers
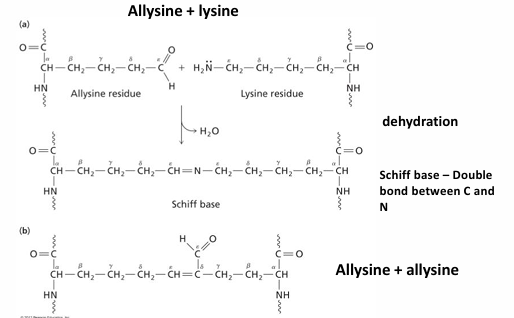
covalent cross-linked between triple helices by allysine: conversion of -CH2NH3+ to an aldehyde -CHO
Staggering of tropocollagen helices in collogen fibers increase the strength of collagen
Protein preservation of structural proteins
protein molecular data from ancient (>1 million years old) fossil material: pitfalls, possibilities and grand challenges
advances are resolution and sensitivity of analytical techniques have provided novel applications, including the analyses of fossil material. however, the recovery of original proteinaceous component from very old fossil samples from previously named limits in the literature is far from trivial. Here, we discuss the challenges to recovery of proteinaceous components from fossils, and the need for new sample preparation techniques, analytical methods, bioinformatics to optimize and fully utilize the great potential of information locked in the fossil record. we present evidence for survival of original components across geological time, and discuss the potential benefits of recovery, analyses, and interpretation of fossil material older than 1 Ma, both within and outside of the field of evolutionary biology
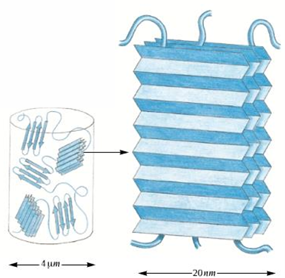
Example #3 of fibrous protein: silk
made by insects: silkworms caterpillar and spiders
sequence predominantly repeats of gly - ser - gly - ala - gly - ala)
antiparallel beta strands stabilize by H-bonds between the backbone NH and CO of different strands
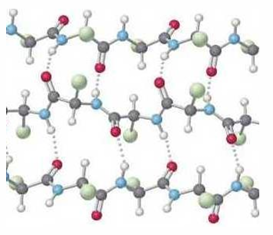
the alternating small side chains allow beta sheets to stack forming a fibroin
side view of a beta sheet shows the R-group are positioned on alternate sides of the sheet. form the angle, the backbone forms a zigzag or pleat appearance
silk fibroin - stacked pleated beta sheets
Silk is formed of crystalline sheets connected by amorphous regions, making silk elastic
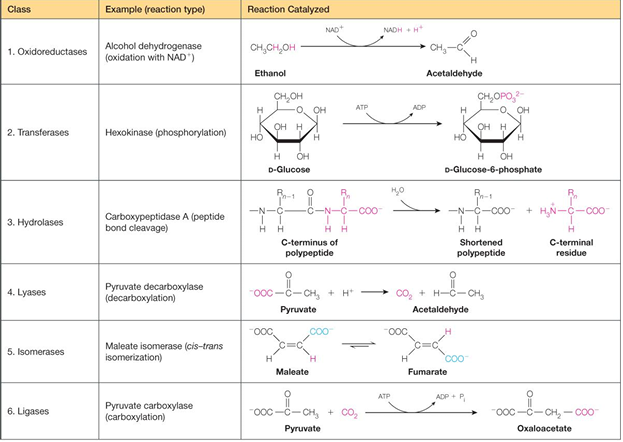
enzyme kinetics
general properties of enzyme
greatly accelerate the rate if a chemical reaction by factors off 10³ to 10^ 20
reactions can occur under physiological conditions (water, pH = 7, modest salt concentration) - these conditions would frequently no be conducive to reactions with otherwise require high temperature, high pressure, high or low pH, or organic solvents
note that enzymes will not induce a reaction that is unfavorable due to a positive free energy change
high specificity for the reaction substrate(s)
high reaction specificity - chemical reactions typically have a serious problem with unwanted contamination side reactions; this not the case in enzyme catalyzed reactions
enzyme is unchanged in the reaction, and can undergo rapid turnover
enzymes frequently are subject to regulation to alter substrate binding affinity or activity
largest class of enzymes are proteins, but another class are ribozymes and in evolution, RNA was the first enzyme
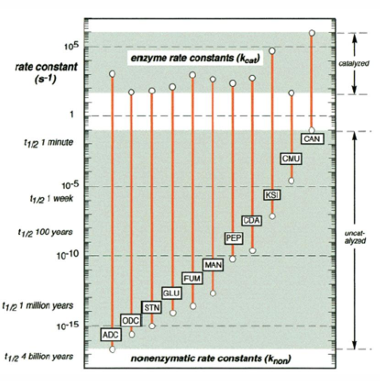
Raye enhancement by various enzymes
ADC = arginine decarboxylase
ODC = orotidine 5’-phosphase decarboxylase
STN = staphylococcal nuclease
GLU = sweet potato beta-amylase
FUM = fumarase
MAN = mandelate racemase
PEP = carboxypeptidase B
CDA E. = coli cytidine deaminase
KSI = ketosteroid isomerase
CMU = chorismate mutase
CAN = carbonic anhydrase
basics of an enzyme reaction
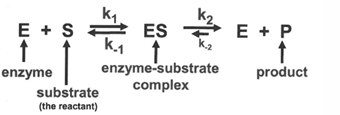
in the simplest case, consider a reaction in which an enzyme binds a substrate and catalyzes its reaction to form a product; the properties of this reaction an equation describing the kinetic steps will be developed for this case
important points about this reaction
1st step in the reaction, E + S →← ES is the slow or rate-limiting step in the reaction; the substrate must find the enzyme in the solvent; this rate us dependent on the concentration of the substrate and is limited by diffusion rate
the 1st is the reaction is reversible; for an enzyme-substrate interaction this is especially important; the binding of the substrate to the enzyme is very especially important; the binding of the substrate to the enzyme is very specific, but nevertheless a weak interaction - if there were strong forces, the discrimination of exactly the correct substrate would be as great so, there are forward and reverse rate constants for this association:
k1 = forward reaction rate constant → formation of ES complex
k-1 = reverse reaction rate constant → dissociation of ES complex
the second step of the reaction, ES «> E+ P is the rapid step; once the ES complex is formed, the reaction typically takes place very rapidly; therefore the ES complex does not remain long
the second step of the reaction is generally considered irreversible; forward and reverse rate constants, k2 and k-2 are written here nut frequently, the reverse reaction is so unfavorable that only k2 term is included in the overall reaction schemed
how the enzyme changes the reaction rate
the progress of the biochemical reaction is dependent on the free energy changes as the reaction proceeds
the rate of the uncatalyzed reaction is tied to the sizze of the activation free energy. the enzyme lowers the activation free energy needed to get to the transistion state and the consuences of this is to increase the foward rate constant
note that this does nt change the overall free energy og the reaction; also the activation free energy for the reverse reaction will affected, but the fractional change will be smaller)
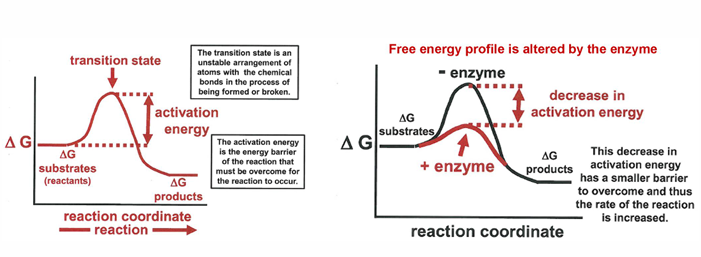
enzyme catalysis: stabilizing the transition state
enzyme kinetics
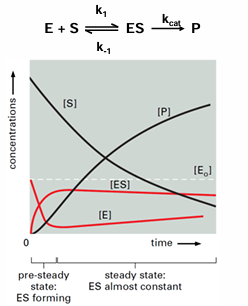
for any chemical reaction and in particular for the enzyme-catalyzed reaction described in the scheme: E + S →← ES →← E + P
the velocity of the reaction v = amount of product made in unit time (delta P / delta t) and at early times of the reaction before there is any reverse reaction the velocity will depend on the starting substrate concentration [S]
v0 = k[S] this is inital rate of reaction
if the reaction were going by itself (no enzyme needed), the following measurements could be made
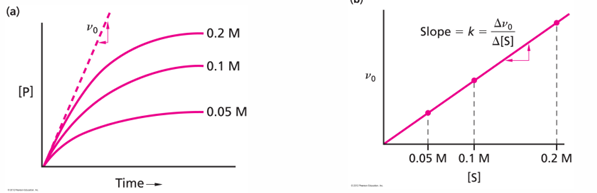
for a reaction that needs an enzyme, the initial velocity will depend on the amount of enzyme added (as long as there is a lot of substrates for all of the enzyme concentrations used)
plot of v0 vs [E] should be linear (right plot), again as long as there is enough substrate to keep all of the enzyme molecules completely occupied
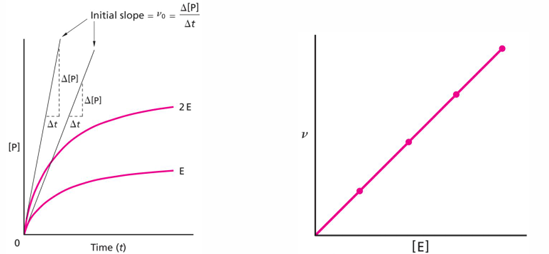
plots of initial velocity of reaction versus substrate concentration to determine enzyme properties
when the initial velocity measurement is made at constant enzyme concentration for different substrate concentrations S, the following plots show that the velocity increases rapidly at low [S] values, but at high [S] values reach a maximum value called Vmax.
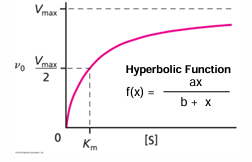
the [S] value at which the velocity is half the minimum is called Km (Michaelis constant)
the equation of the curve is hyperbolic and was derive by L. Michaelis and M. Menten

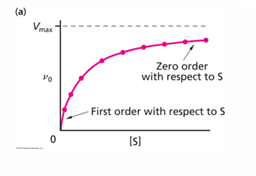
Michaelis-Menten kinetics are used to study enzymes at steady-state
pre-steady-state: time after mixing when intermediates build up
steady-state: the concentration of enzyme-bound intermediates is constant
assumptions
[Etot] « [S]
measure initial velocity of reaction before product accumulates and before substrate is depleted (where d[ES]/dt = 0)
the values of Vmax and Km give insights into the properties of the enzyme
at Vmax, the enzyme is working as fast as it can, which is determined by its catalyic constant Kcat and it concentration (rember [S] is saturating):
Vmax = Kcat[E]tot and rearranging Kcat = Vmax/ [E]total
Km is defined by the ratio of rate constants that dissociate (k-1 and k2) and associate the substrate (k1), but usually k2 is small compared to k-1
km = (k2 + k-1)/k1 = k-1/k1
when k2 is very small relative to k-1 the Km value is close to the dissociation constant between the substrate and the enzyme, but Km does not equal Kd
the strength of an enzyme can be characterized by two parameters
Kcat this is called the turnover number and indicated how fast the enzyme can work under conditions in which there is saturating amount of substrate; a strong enzyme will have a large Kcat value
a strong enzyme will have small value of Km
if [S]« Km the initial velocity can be written as v0 = Kcat/Km [E][S]
the rate constant in this condition can be taken as (kcat/Km) and the is the same as the second order rate constant for the reaction
kcat/Km is called the enzymatic rate constant (or catalytic efficiency constant) and is a measure of the overall efficiency of the enzyme (combination of binding strength and catalytic rate)
there is a limit to these constants called the diffusion-controlled limit, which is caused by the fact that the ES complex is formed by diffusion of S to E
this determines how small the Km value can be because the rate of the association reaction k1, cannot be larger than the rate of the diffusion of the substrate to the enzyme
therefore, because the reaction is limited by the diffusion-control process, the value of kcat/Km could not be larger than 10^8 - 109 M-1s-1, several enzymes have activates that are close to this range
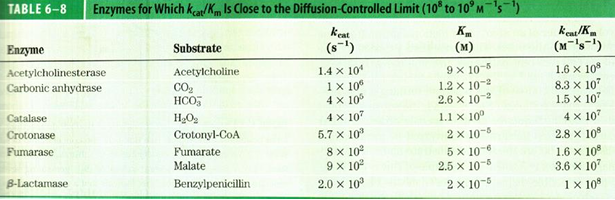
determination of Km and Vmax values
for the michaelis menten equation
v0 values in two regions could be used to determine kcat and Km
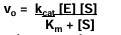
for reactions at low [S], Km»[S] and there is dependence on both the [E] and [S], so v0= (kcat/Km)[E][S] fits the limiting slope, and kcat/Km can be determined
at the high [S] values, because of all the E are saturated at high [S] v0= kcat[E], and kcat can be determined
so, with these two measures, it is possible to determine both kcat and Km
with modern curve fitting software you could also fit the curve according to the Michaelis Menten equation to determine kcat and Km
the meaning of Km and Vmax
Km = concentration of S when half Etot is bound
Vmax: maximum reaction rate occurs when all enzyme is bound Vmax = kcat[Etot]
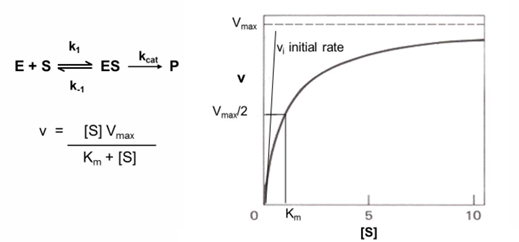
Michaelis-Menten equation transformed to a linear graph- lineweaver-Burk
inverting both sides of the Michaelis-Menten allows the derviation of an eqaution that will show a linear dependance of 1/v0 on 1/[S]
the y intercept is 1/Vmax and the x intercept is -1/Km
slope is Km/Vmax
this equation is useful when determining the effect of the effectors and inhibitors on kinetic parameters
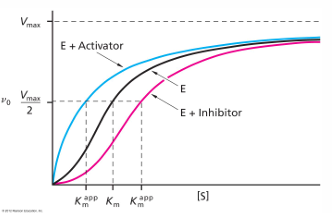
inhibitors vs activators: v0 vs [S] curves for allosteric enzymes
an enzyme that is allosteric will exhibit velocity curves that depend a lot on whether activator or inhibitor effects are present
the graph shows the apparent Km value will change, and the shape of the curve will change. with high [activator] the curve looks hyperbolic and has a low Km value, so even at low [S] there will be high v0
an inhibitor there is much higher apparent Km value- at the same [S], there is much smaller v0
allosteric activator: ligan that increases hr activity of the enzyme
allosteric inhibitor: ligand that decreases the activity of the enzyme
inital rate equations for 2 substrates (no inital product)
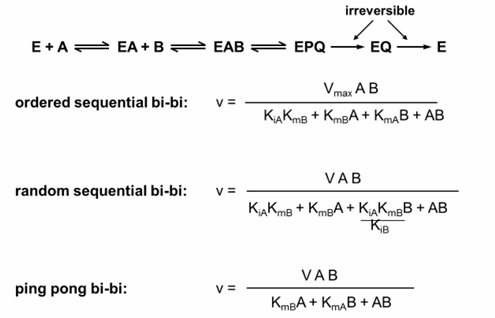
enzyme reactions with 2 substrates
the original Michaelis-Menten equation and the corresponding Lineweaver-Burk plot were developed for an enzyme reaction with a single substrate
most important enzyme reactions involve 2 substrates, and it is valuable to describe the steps in these schemes and acknowledge that the kinetic equations will be different
three reactions involving 2 substrates
the binding of 2 substrates, A and B in that order
the binding of 2 substrate A and B in any order
the sequential binding + dissociation of 2 substrates (ping-pong reaction); in this mechanism, the first substrate binds and dissociated from the enzyme leaving the enzyme altered and the second substrate binds and reacts - this last reaction is used frequently in protease reactions
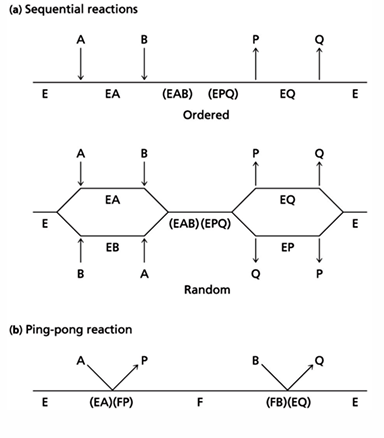
enzyme mechanism
enzyme mechanisms
there are several ways in which an enzyme can affect a reaction; these effects can be appreciated by considering the free energy state of the reactant, reaction intermediates and products if the reaction
the enzyme can increase the reaction rate by:
lowering the free energy of transition state
binding the substrate A and B; this raises their free energy, so it makes the activation free energy smaller
combination: bind A and B and also bind transition state to reduce its free energy
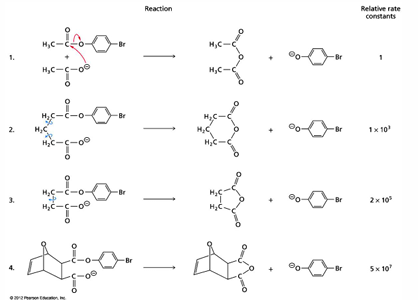
proximity effects on rate of reaction
an example of orgo literature shows how a reaction can be enormously accelerated by constraining the reactants. here the reaction increases by a factor over 10^7 compared to that seen for free reactants.
the same effect can be accomplished by an enzyme if it binds the reactants in the correct orientation
the first step reaction is bimolecular and is by far the slowest
the next version of the reaction constrains the reactants more and more through reduction of the conformational freedom they have
the last arrangement has a very favorable positioning of the nucleophile group (O-) for attack on the carbonyl C
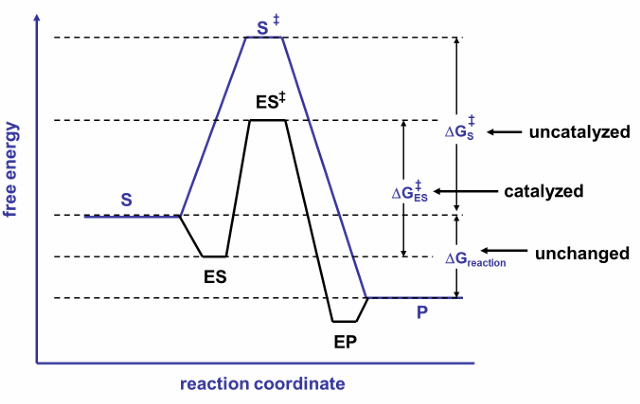
limit to binding strength (Km) values for enzymes
enzymes need to have low Km values to have good affinity to their substrates and good specificity to prevent inappropriate products
however, enzymes should not bind their substrates too tightly because:
very tight binding could prevent reaction; substrates bound in very tight binding pockets will be incompatible with reaction
consistent with this is the free energy diagram. this shows the strong binding could result in an E-S intermediate that would have an even harder time reaching the transition state that the uncatalyzed reaction (compared doted red line and blue line in the figure). instead, in the figure the E-S intermediates have free energy a little higher than the free energy a little higher than the free E + S (solid red line)
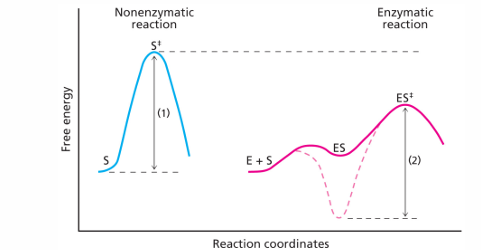
very tight binding pockets could prevent product release
distortion of the substrate and/or active site to promote reduction of activation energy
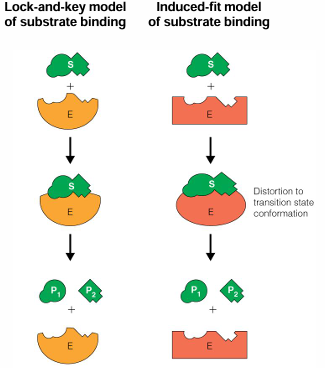
significance of induced-fit binding mechanism
enzymes are flexible and the ability to collapse on a suitable substrate offers several advantages
the substrate changes the enzyme structure, and this is connected to the enzyme activity e.g., the enzyme is not active unless the correct substrate is present
the enzyme structure is fairly open and accessible to the substrate, until it binds; this allows access but then prevents water and other undesired molecules from the active site
after reaction, enzymes can reversibly change its structure and release product
binding reaction of glucose in yeast hexokinase
organization of enzyme active sites
active sites for enzyme catalysis generally will be in interior locations
many reactions will need to exclude water, or carefully control it, at the reaction site
want to have a large are of contact between substrate specificity
many AA residues lining an active site will be non-polar
this will help exclude water from the active site
non-polar environment will help raise electrostatic effects in active site
however, they will be polar and charges AA at the active site that will be involved in binding the substrates and in the reaction mechanism
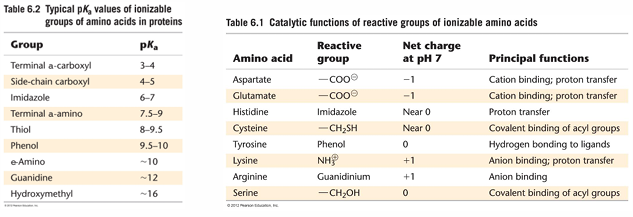
AA in enzyme active sites
an extremely important feature of the first list: the AA can have very different pKa values in enzymes compared to their values seen in free AA; many of the pKa values have shifted towards pH 7
this is due to nearby side chains that affect their behavior
pKa values in specific enzymes can be shifted even more. for example, cys in papain ha a pKa value of 4.2. this type of shift is usually due to the presence of a close-by group that is attracting the H+ ion from the cys SH group
chemical aspects of enzyme- catalyzed reaction mechanisms
nucleophiles and electrophiles
many enzyme reactions involve reaction between a nucleophile and a electrophile
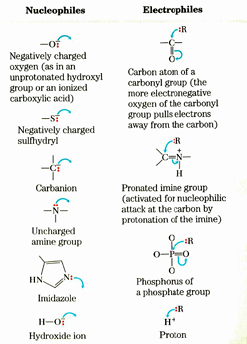
many reactions in metabolism invilve carbon-carb bond formation or breaking. the majority if these invlove the C atom in a carbonyl grou as an electrophile because it has a partial + charge
acid-base reactions
depends on AA side chain groups that act as bases or acids at pH 7
bases - His,Asp, Glu
acids - His, Lys (but not usually Arg because pKa of Arg is too high)
base catalysis happens:
directly: extraction of proton from the substrate to activate the reaction
indirectly through water: extraction of proton from water gives OH-
acid catalysis happens
usually through direct protonation (addition of H+) to the substrate to activate the reaction
covalent catalysis
in these reactions, an intermediate is formed by the covalent joining of the substrate and enzyme; this intermediate goes on to further reaction with another reactant, or the starting enzyme structure is regenerated through reaction with water
effect of pH on reaction rates
the pH of the solvent can significantly impact the activity of an enzyme. as the pH increases or decreases, the ionization states of the AA side groups change. if critical side groups are in the active site, changing the ionization will alter the strength of the substrate binding or the catalytic activity
the dependence of enzyme activity on pH can be evaluated by a titration experiment and gives insight into what types of side groups are involved in the reaction
for the following behavior, involvement of Asp would be suspected, and you could conclude that Asp has be in the acid state as part of the reaction mechanism

a maximum in the pH dependence curve can occur if there are wo ionizable groups, or if there are two steps in the reaction involving His in alternately protonated and unprotonated states
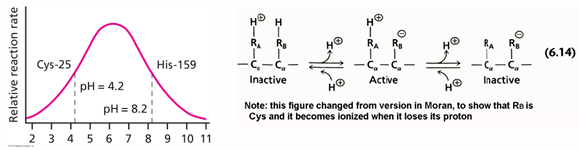
for example, of the first case the activity vs pH curve for the enzyme papain is shown on the left
the explanation for the profile is that there is a cys that becomes deprotonated at around 4.2 and the deprotonated form is needed for activity
on the other hand, there us a His pKa of 8.2 that needs to be in the protonated form for activity; the plot shown here indicates that His is involved in the reaction becomes deprotonated at pH 8.2
titration state stabilization
the transition state is enzymatic reaction is the unstable, high-energy arrangement of atoms in which bonds are being broken or formed
there is very tight binding between the reactant and the enzyme in the transition state and this helps lower the transition state energy; in fact, the quote by Linus Pauling at is a good one: “enzymes are molecules that are complementary in the structure of the activated complexes of the reactions that they catalyze
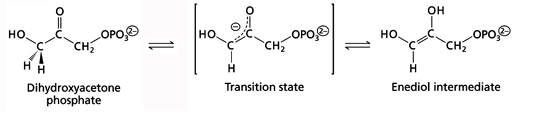
a powerful strategy to find enzyme inhibitors is to find a chemical compound that resemble is a transition state arrangement. this is called a transition state analog
the reason the transition state analog will be a good inhibitor is that such a compound typically binds much more strongly than the substrate or product
the resemble of a transition state analog can be seen for the reaction carried out by triose phosphate isomerase. in this case the compound 2-phosphoglycolate is an effective inhibitor of the enzyme
binding of the transition state analog 2-phosphoglycolate to triose phosphate isomerase
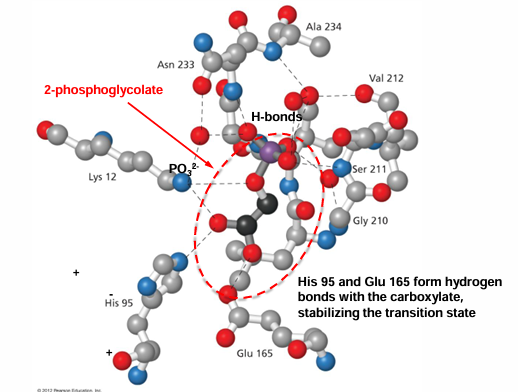
mechanisms of protease - activation
pancreatic proteases are regulated as zymogens
pancreatic proteases are secreted in the small intestine to digest food
to prevent them from digesting the pancreas, they are synthesized in an inactive form, as zymogens
zymogen: catalytically inactive precursor of enzymes, typically with an inactive leader sequence
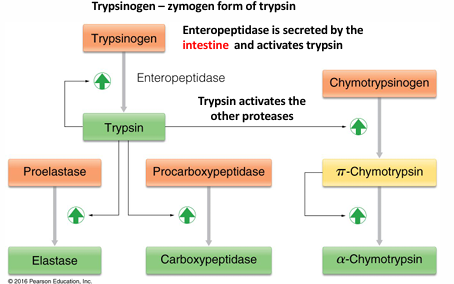
cascade process once it is activated
structure change in chymotrypsin
in chymotrypsinogen, the interior cleavage an the dissociation of the two dipeptides results in a structural change
there is a movement of the new N-terminal end at amino acid 16 towards the interior of the protein
this brings several non-polar AA side chains together and these form, for binding the substrate, a pocket which was not present before
this reorganization of the strands the results in the binding site pocket largely accounts for the activation of the protease
in chymotrypsinogen there are four protease cleavage (carried out by trypsin and also self-cleavage by chymotrypsin) within the protein. two dipeptides are released
arrangement of the “catalytic triad” in the serine protease
there is a critical serine AA (serine 195) that is at the center of the serine protease mechanism, but it cannot act alone. instead, there are 2 other AA ( Asp 102 and His 57) that help activate the Ser and together these are the “catalytic triad”
the geometrical arrangement (taken from a crystal structure); the way in which Asp 102 and His 57 act to induce oxyanion n Ser 195 is shown in the second figure. (Note that the serine protease is in the intestine, in which the pH is around 7: we do not need to think about
arrangement of the catalytic triad in proteases
there is a critical serine AA (ser 195) that is at the center of the serine protease mechanism, but it cannot act alone. instead, there are two other AA (Asp 102 and His 57) that help activate ser an thogether these are the catalytic triad
the geometrical arrangement (taken from a crystal structureO is shown in the first figure; the way Asp 102 and His 57 act to induce oxyanion in Ser 195 is shown in the second figure (note that the serine protease are in the intestine, in which the pH is around 7: we do not need to think about Asp or His in the protonated acid forms, which would be th case in the stomach)
note: that the Oh of serie is giving up its H+ atom, even though the reactio occurs at a pH well below the pKa = 16, previously listed for S in an active site; this is induced by the proximity of His imidazole group
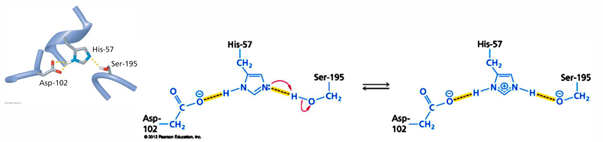
Reaction mechanism of chymotrypsin
altogether there are seven in the hydrolysis of a peptide bond by chymotrypsin, but worth inspection because it is possible to see how the enzyme is able to carry out reactions that could not otherwise possibly occur chemically
there are two unstable intermediates, both involve tetrahedral substitution in the carbonyl C that is the site for cleavage; both of the intermediates are stabilized by the oxyanion hole in the enzyme; in both cases this arrangement leads immediately to bond cleavages
the critical intermediates are
“short-lived intermediate” acylation, after step 2,
“Covalent intermediate”, after step 3
“short-lived intermediate” deacylation, after step 5
the reaction has 2 phases. in the acylation phase (steps 1-3), formation of a covalent acyl-enzyme intermediate is couple to cleavage of the peptide bond. in deacylation phase (steps 4-7), deacylation regenerates the free enzyme; this is essentially the reverse of the acylation phase, with water mirroring, in reverse the role of the anime component of the substrate
step 1: when substrate binds, the side chain of the residue adjacent to the peptide bond to be cleaved nestles in a hydrophobic pocket on the enzyme, positioning the peptide bond for attack
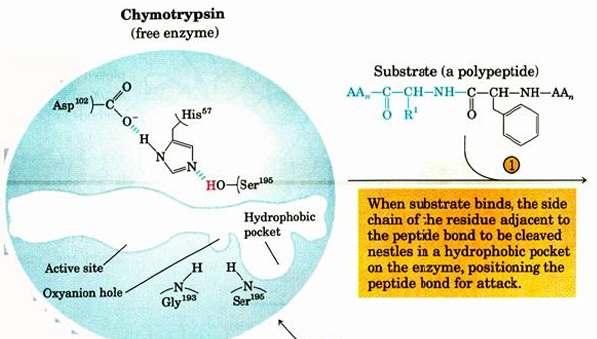
step 2: interaction of Ser 195 and His 57 generates a strongly nucleophilic alkoxide ion on Ser 195; the ion attacks the peptide carbonyl group, forming a tetrahedral acyl-enzyme. this is accompanied by the formation of a short-lived negative charge on the carbonyl oxygen of the substrate. which is stabilized by the H-bonding in the oxyanion hole
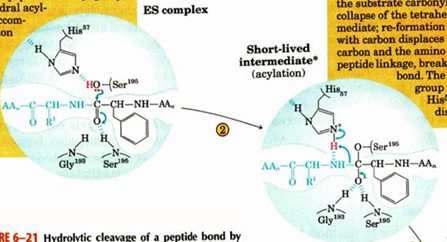
step 3: instability of the negative charge on the substrate carbonyl oxygen leas to the collapse of the tetrahedral intermediate; reformation of a double bond with carbon displaces the bond between carbon and the AA of the peptide linkages, breaking the peptide bond. the amino leaving group is protonated by His 57, facilitating its displacement
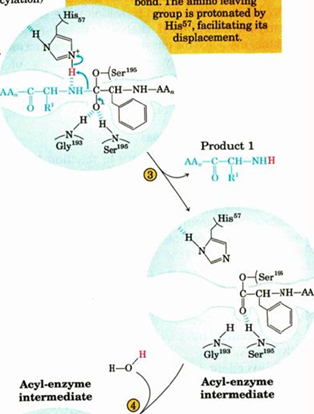
step 5: an incoming water molecule is deprotonated by general base catalysis generating a strongly nucleophilic hydroxide ion. attack if the hydroxide ion on the ester linkage of the acyl-enzyme generates a second tetrahedral intermediate, with oxygen in the oxyanion hole again taking on a negative charge
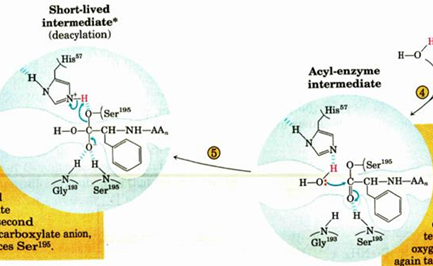
step 6: collapse of the tetrahedral intermediate forms the second product, a carboxylate anion, and displaces Ser 195
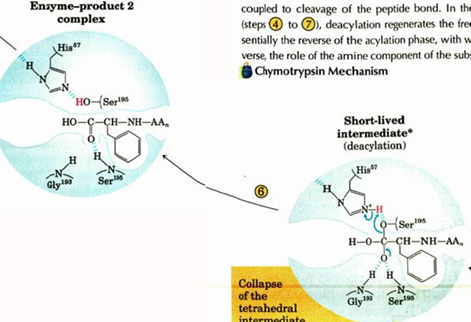
step 7: diffusion of the second product from the active site regenerates free enzyme
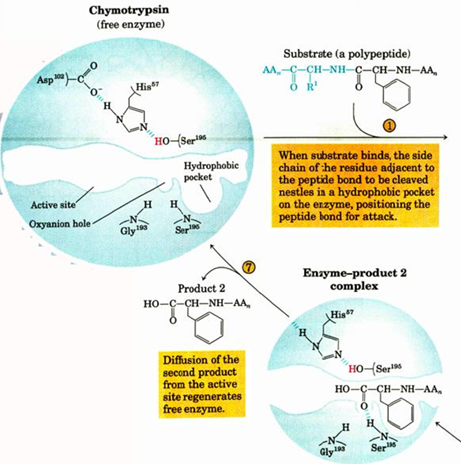
the proteases chymotrypsin, trypsin, elastase - serine proteases with different specificities
these enzymes are all very similar:
1) in their 3-d structure - the primary sequence is somewhat different, but structures can be superposed, this example of conservation of structure
2) the protease mechanism is the same in all three - they depend on critical serine to form a covalent intermediate in the reaction - hence the name of the family
however, each of these has a different specificity
chymotrypsin: cleaves the peptide bond on the C-terminal side of the AA that have aromatic and large hydrophobic side chains
trypsin: cleaves the peptide on the C-terminal side of Arg and Lys (positively charged)
elastase: cleaves the peptide bond at the C-terminal side of AA with small side chains (mostly Gly, Ala)
the specificity of these is accounted for by the size and charge of the substrate binding pocket in the enzyme
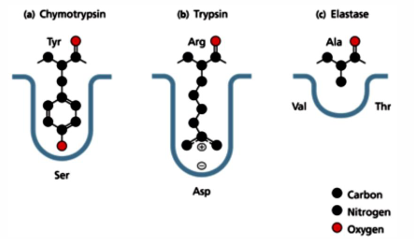
mechanism-based inhibitor of protease: peptide with a C-terminal boronic acid
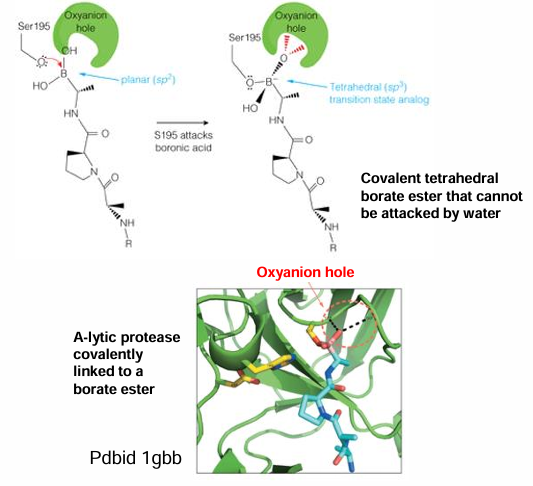
important aspects f the mechanism of chymotrypsin
chymotrypsin utilizes many features typical of enzymes to catalyze peptide bond hydrolysis:
1) specificity for substrate: binding pocket will fit Phe, Tyr, Leu, Ile
2) binding of substrate aligns the peptide bond for reaction
3) water is carefully controlled: H20 is excluded initially but used in second step of the reaction
4) chymotrypsin activates O of the -OH of Ser 195 for nucleophilic attack; the catalytic triad is responsible for this; the pKa of His 57 is shifted from7 to greater than 12, and without any other place to get an H+, the His 57 strongly attracts the H from the -OH of Ser 195; there is no pKa value given for deprotonation of Ser 195
5) a covalent intermediate is made between substrate and enzyme and stabilized by enzyme
6) there is a “valley” in free energy diagram corresponding to the covalent intermediate
7) a second step is needed to reverse covalent intermediate; the pKa value of His 57 is greater 12 is involved in activation of water for this step; after the hydrolysis to separate product from enzyme, enzyme will be in the original structure
8) this reaction cannot go backwards - the products will have higher Km values; this means that chymotrypsin cannot act a ligase to put two peptides together
9) overall, the rate of enhancement over the spontaneous hydrolysis by water at pH 7 is about 10^7
enzyme inhibition TA version
half of max velocity
how the molecules interact with each other in a medium
tehe look at the learning objectives
enzyme to substrate is reversible but one the inhibitor binds its non reversable
remember the arrows
enzyme inhibition mechanism
competitive inhibition ( binding to specific active site)
blocks the active site
only binds to the E
how will it affect the Km and Vmax
the Vmax doesn’t change bc the rate of the reaction does not change and increases the Km
it takes more substrate to reach the Km
just takes long to reach the Vmax
uncompet ( forms the ES complex)
it takes less S to bind half of the enzyme (Km decreases)
the equilibrium for binding E + S is shifted towards the ……..
the Km and Vmax both decrease
only seen in multisubstrates
noncompet binds to E and messes with the ES complex
bind to E and ES
binds equally to the E and the ES
do the Km does not change
the Vmax decreases because the overall ability of the enzyme decreases
aka mixed inhibition (tech not the same thing but this class put it together)
binds at different affinities
Vmax values look like the Vo2 max curve
not reversable
Km is concentration of substrate at 50% (on the graph it gives where it stops being a linear plot)
Vmax is when all the enzyme is bound
Kcat is the catalytic coefficient
Vmax = Kcat [Etot]
double-reciprocal plot Lineweaver Burk plot
y intercept = 1/ vmax
x intercept is -1/km
ratio of the Vmax and the km value
linear plot of the Vmax graph
steeper slope for the compet inhibition
higher slope
intercept at the same y but will have a diff x intercept
more inhibitor more steepness
uncompet
only binds with ES complex
gives parallel to the og line graph
decreasing but the same factor and the slop is the same
both the x and y intercept change bc the values are decreasing
noncompet
the Km is the same
the Vmax is decreased
the line is steeper
does not give related
same x value diff y value
compet = raises the Km Vmax remains the same
uncompet: lowers Vmax and Km, ratio remains the same
noncompet
mechanism-based inhibitor aka suicide inhibitors, enzyme-activated irreversible inhibitors
chemically unreactive without enzyme
AChE
remove ACh from synapse
stops ACh signal
no going back once broken down
regulatory schemes in enzyme inhibition
many pathways are used to either decrease or increase a specific enzyme in a pathway
typically a feedback inhibitor
can be pos or neg
allosteric activation: the accumulation of one substrate induces the activation of another enzyme in the sequence and to increase the function
enzyme inhibition
there are three ways in which inhibitor compounds can inhibit an enzyme reaction
competitive inhibition: the inhibitor and substrate compete for the same binding site on the enzyme. there are two versions classic and nonclassic
in both versions, binding of inhibitor precludes binding of substrate
uncompetitive inhibitor: inhibitor binds to the ES complex and prevents the reaction
noncompetitive inhibition: inhibitor binds to either E or ES complex, does not interfere with S binding but prevents the reaction
for both cases the inhibitor lowers concentration of available E, resulting in an increase in the apparent Km
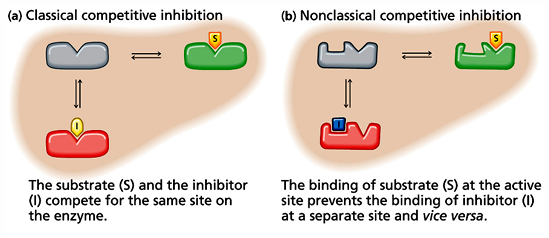
in this case the inhibitor decreased the Vmax but the [ES] is also lowered, resulting a decrease in the apparent Km as well
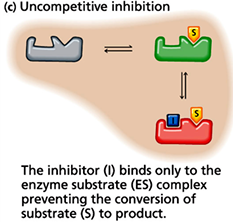
in this case, the inhibitor binds to both E and ES; Km is not changed but Vmax is decreased
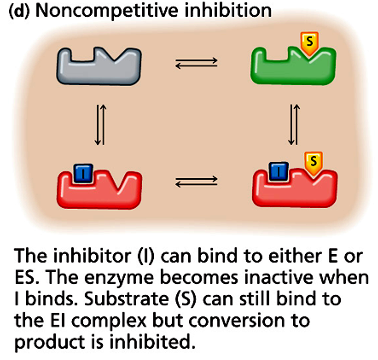
effects of inhibition mechanisms on enzyme reaction kinetics
competitive (I binds to E only): raises Km, Vmax remain unchanged
uncompetitive (I binds to ES only): lowers Vmax and Km but the Vmax/Km remains unchanged
noncompetitive (I binds to E or ES): lowers Vmax, Km remains unchanged
competitive inhibition
the effect of I binding to E is to shift the E + ←→ ES equilibrium to the left, this is the same as an increase in the Km
however high [S] overcomes the effect of I, so Vmax is not changed
the effect of I is seen in the vo vs [S] plot and clearly in the Lineweaver Burk plot
there will be a constant y-intercept for the control and inhibitor reactions
as [I] increases, x-intercept will move to smaller and smaller values
the slope of the plot will increase as [I] increases
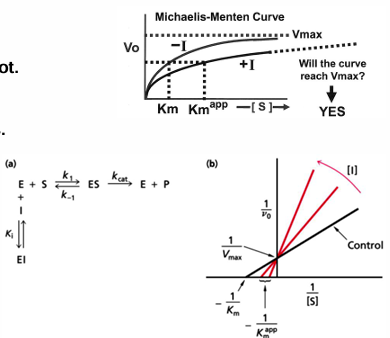
uncompetitive inhibition
I binds to the ES complex and inhibits reaction
this causes Kcat and Vmax to decrease
inhibitor binding shifts the E + S → ES equilibrium to the right, and this is the same as a decrease in the Km value
extent of reduction in Kcat and Km are same, so the slope of the Lineweaver- Burk plot does not change
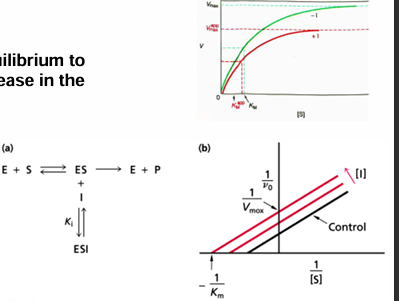
non-competitive inhibition
I binds to either E or ES
lowers Kcat and Vmax
however, binding of S is not inhibited, so the Km does not change
in the Lineweaver Burk plot, x-intercept is constant, and the y-intercept increases as [I] increases
slope of the plot increases with [I]
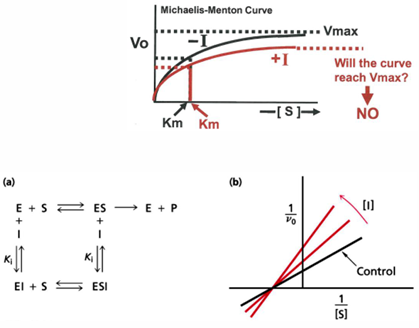
determining Ki
1) determine Km for the substrate (measure v at S values around Km)
2) measure Km (app) values at 3 or more inhibitor concentrations
3) plot Km (app) vs. inhibitor concentration
since Km(app)=Km (1+[I]/Ki=Km/Ki [I]+Km the slope =Km/Ki, and the intercept = Km
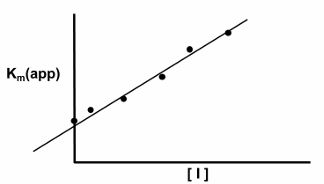
bottom line for the behavior of inhibitors on enzymes is that contructing a Lineweaver-Burk plot will give a good clue about what type of inhibition is occuring
two complications for real example:
1) there are many examples of mixed inhibition mechanisms, and these will give behaviors in the Lineweaver-Burk plots that are hybrids of the ones shown here, e.g. increasing [I] will result in the changing values of Km, Vmax and slope
2) for reactions with two substrates, plots become more complicated, so a series of plots with the second substrate at different concentrations are needed or “secondary” analysis is needed
3) for allosteric enzymes (we’ll see them in a little bit) the Lineweaver-Burk plots can be very different ways than seen in the simple examples
mechanism-based inhibitors
also called
suicide inhibitors
enzyme-activated irreversible inhibitors
Kcat inhibitors: since they use the catalyic mechanism
trojan horse inhibitors
effective mechanism-based inhibitors
chemically unreactive in the absence of E
activated specifically by the target E
once activated, it reacts with the E faster than it dissociates
function of acetylcholine esterase (AChE)
neurotransmitter released between nerve synapses. acetylcholine esterase breaks down the acetylcholine to stop the signal
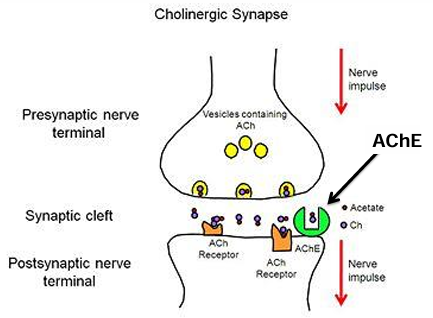
Diisopropyl fluorophosphate (DFP) is an irreversible inhibitor of AChE
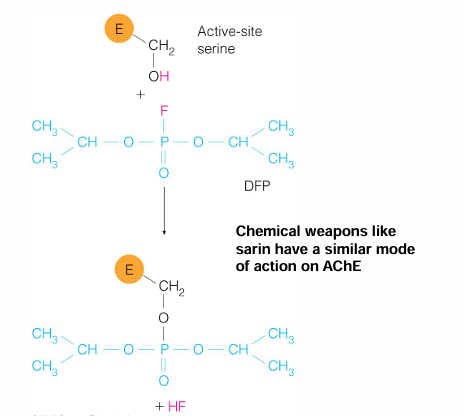
regulatory schemes in enzyme inhibition
the use of effectors to regulate enzymes play essential roles in regulating metabolic pathways
several regulatory schemes are used to either increase or decrease a specific enzyme in a pathway
A, B, etc. are the metabolites and the arrows and blockers show how the concentrations of A, B, C, D etc. alter upstream and downstream reactions
simple end-product inhibition
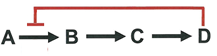
the final product in a metabolic pathway inhibits the first enzyme in the pathway. the inhibition is frequently just called feedback inhibition
this regulatory scheme makes sense when considering how to conserve substrates. If there is sufficient product D, there is no need to synthesize B and C. inhibiting the enzyme for the A to B reaction makes sense in conserving both biological materials and also cellular energy
more complex inhibition patterns
sequential end-product inhibition
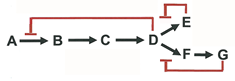
concerted end-product inhibition
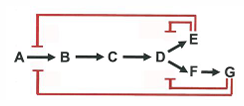
differential inhibition of multiple enzymes
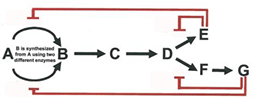
regulatory schemes in enzyme activiation
often it is important to activate an enzyme to increase the synthesis of a specific pathway product due to increased need for this specific metabolite. the ability to turn enzymes on, as well as off, allows the cell to respond rapidly to charging growth and environmental conditions
substrate activation

in this scheme accumulation of the substrate A induces activation of the enzyme to increase synthesis of B and activate the following pathway. this is typically allosteric activation involving a multi-subunit enzyme exhibiting cooperative substrate binding and alteration of enzyme structure to increase substrate binding interaction with the enzyme
more complex activation/inhibition patterns
stimulation of one branch of a pathway by the end-product if another branch
compensatory reversal of end-product inhibition
enzyme cofactors TA version
cofactors are used as lil helpers to reach max potential
enzyme - cofactor = apoenzyme (sad and alone - a for alone)
enzyme +cofactor = holoenzyme (haloooooo)
essential enzymes vs the coenzymes
the metal ions are tightly bound for essential ions
activator ions are weakly bound for essential ions (dissociate easily) (activator go by)(despensable)
cosubstrates are weakly bound for coenzymes
prosthetic groups are strongly bound for coE
coenzyme pre to vitamins
cofactors are minerals
make sure you know how to draw DNA, ATP, RNA and sugars
atp = three phosphates, ribose sugar plus adenine
alpha, beta, gamma phosphate, adenine is the base, and the ribose sugar
FMN
lol Vb complex
tehe 5
flavin
look out for the Ns (2)
3 oxidation states
NAD(P)
NAD ring
redox reaction
oxidized structure and reduced structure
lactate oxidization and NAD is reduced
SAM
donates methyl groups
recognize structure, what it does
CoA
Vd5
condensation of acetate and CoA
ACP is the carrier protein
PLP
vB 6
deamination
lipid soluble v
Vdake
what it does and recognize structure
protein CoE
group transfer or redox
contain metal ions or prosthetic groups
cytochromes
enzyme cofactors
coenzyme and essential ions
the catalytic activity of many enzymes depends on bound cofactors
enzyme-cofactor = apoenzyme
enzyme + cofactor = holoenzyme
a summary of cofactors divides into different categories:
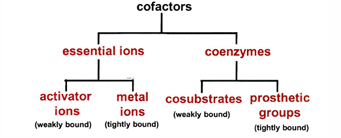
the molecular weight and chemical complexity of these cofactors varies greatly:
from single metal ions (charged atoms)
large and chemically complex organic molecules, or even small proteins
cofactor characteristics
essential ions:
organisms cannot synthesize minerals so they must be obtained from diet
both classes required by all organisms as enzyme cofactors
activator ions, readily exchanged from enzyme (Kd >/= 10^-5 M)
metal ion as metalloenzymes or part of prosthetic groups (Kd « 10^-5 M)
coenzymes:
may be synthesized de novo or form dietary precursors known as vitamins
functions as group transfer reagents (transfer H=, e-, VH3, acetyl, etc.)
cosubstrates
exhibit weak and reversible binding to the enzyme
participate as necessary substrate in the reactions
located at the catalytic center
these have to be recycled after each reaction by displacement/replacement
prosthetic groups:
these exhibit tight binding and no dissociation
tightly bound, or even covalently attaches, to enzyme
regenerated, as needed, while still bound to the enzyme
evolution of cofactor dependence
bacteria, fungi, and plants have retained the capability to synthesize coenzymes
animals have lost the ability to synthesize some of these coenzymes
therefore, animals must obtain some of these coenzymes from the diet, most often from plants
vitamins usually needed s coenzymes or the starting compounds for making coenzymes
therefore, a vitamin deficiency means some key enzymatic reactions will be lacking
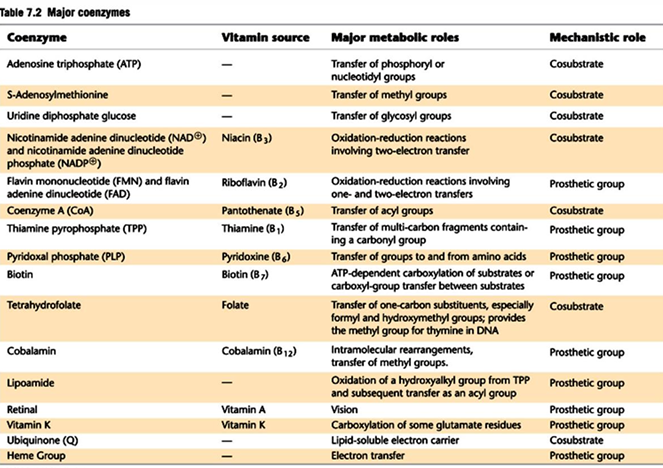
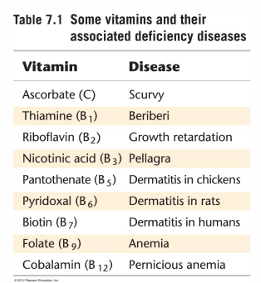
essential ion cofactors
approximately 25% of all enzymes use metal ions either as metal-activated (metals bind enzyme or substrate and change structure or properties of the complex) or a metalloenzymes (metal plays direct role in catalysis)
metal-activated enzymes
the most common metals associated with enzymes are K+, Mg2+, and Ca 2+
these are weakly bound ions exhibiting reversible binding; their presence in an enzyme often is necessary for substrate binding
an example for the necessary involvement of a metal occurs for the kinase enzymes. ATP is a substrate for kinases, but Mg 2+ must bind to the ATP (there is a strong electrostatic attraction); this reduces the negative charge around phosphates and makes them more susceptible to nucleophilic attack
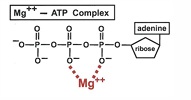
a second example of a metal that is necessary for function through a structural effect is the role that Ca 2+, plays in controlling neurotransmitter Ca 2+ channels
in this case Ca 2+ binds to the protein calmodulin (CaM); the parts of CaM are N-lobe and C-lobe colored blue); the Ca 2+ activated CaM interacts in a specific way with the control domain (red helix) of the channel
one more example is the role Zn 2+ in DNA and RNA-binding proteins called zinc finger proteins; the zinc finger protein is a domain motif found in many replication, transcription and RNA processing enzymes
the zinc ion is chelated by cystine and histidine AA in these domains and produces a domain with a specific special arrangement
the zinc finger domain binds DNA and RNA through residues in an alpha helix, which is some distance from the zinc ion, so even through zinc is needed for enzyme activity, the zinc is not part of the contact with DNA or RNA
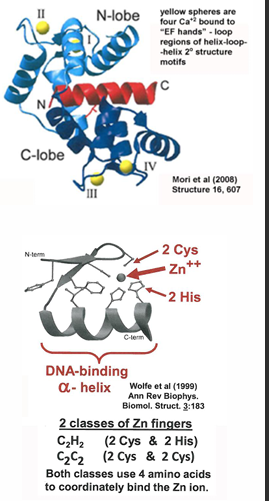
metalloenzyme
metals can also be tightly bound, and remain tightly bound, to the enzyme; in this case they can function in the catalytic reaction, frequently by being involved in substrate binding (remember how Fe functions in hemoglobin in O2 binding)
most common metals are Fe 2+. Zn 2+, Cu 2+, Co 2+, Ni 2+
Fe is the most common metalloenzyme ion; it often functions as an electron transfer agent in oxidation-redox reactions
Fe will be present as a simple ion in the cytochromes, as it was in heme
however Fe can be present with sulfur. in these cases, complexes of specific composition and geometery are present (2Fe-2S or 4Fe-4S), and the complex is bound as a prosthetic group by the enzyme. these are called iron-sulfur (or just Fe-S) cluster
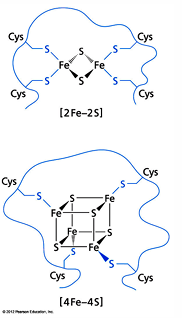
summary major coenzymes and their functions
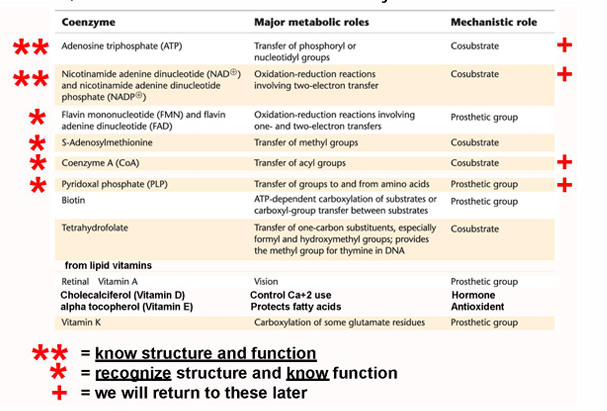
ATP and parts of ATP as cosubstrate (as one class coenzyme)
ATP is a cosubstrate in many reactions
ATP is the most abundant metabolite coenzyme; it can donate a variety of its parts in different reactions
phosphoryl (1 PO4)
pyrophosphoryl (2 PO4’s)
adenosyl (AMP)
adenine (A) groups
note that for ATP the atoms in the ribose will be numbered in primed numbers
atoms in adenine will be unprimed numbers
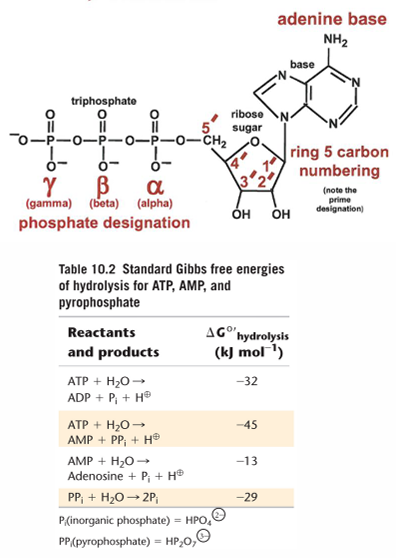
nucleic acids
m
DNA
DNA binding proteins and chromatin
RNA
classes of RNA
ribosomes are a combo of a RNA that is a protein
pre-mRNA splicing
snRNA U2 aka small nuclear; core of spliceosome
RNase
the intron goes by by folding into a 3-d structure to make it easier to splice away
no protein involved