molecular genetics!.docx
MOLECULAR GENETICS
-Lesson 1 Vassallo
Regulation of gene expression in prokaryotes and eukaryotesThe regulation of gene expression is crucial for cellular function, allowing different cell types to perform specialized roles even though they share the same genome. Essentially, cells differ in their behavior and functions because they express distinct sets of genes, leading to the synthesis of different RNAs and proteins. This process is tightly controlled and responds to various environmental signals that influence gene transcription.
How Gene Expression is Regulated
Gene expression can be influenced at multiple stages, from the initial transcription of DNA to the eventual translation of RNA into proteins, as well as in subsequent protein folding, modification, and degradation. However, transcriptional control is often the most significant regulatory point. Focusing on transcription is efficient because it prevents the cell from wasting energy and resources on the synthesis of unnecessary RNA and proteins.
Transcriptional Control
Regulating transcription is efficient because it occurs before large amounts of mRNA or protein are synthesized. Since there are only one or two copies of most genes in a cell, regulating transcription involves fewer targets compared to controlling the many copies of mRNA or protein molecules.
Cells use transcriptional regulators, specialized proteins that respond to signals from the environment. These signals are received by receptors and then transmitted into the cell, where they activate or inhibit the expression of specific genes. Transcriptional regulators bind to DNA sequences to either activate or repress gene expression.
Mechanism of DNA Recognition
Transcriptional regulators recognize and bind to DNA at specific sequences. These sequences are located in the major groove of the DNA helix, which has enough space to accommodate protein interactions. The unique pattern of hydrogen bond donors and acceptors present in each DNA base pair allows these proteins to distinguish one sequence from another.
Structural motifs within transcriptional regulators, such as beta-sheets, helix-loop-helix motifs, and zinc fingers, enable these proteins to bind to DNA. The interaction is highly specific; for example, certain amino acid side chains can protrude into the major groove and form bonds with specific DNA sequences.
Activators and Repressors
- Activators: These proteins increase the transcription of a gene, enhancing the production of the corresponding mRNA and protein.
- Repressors: These decrease or block gene transcription, thereby reducing mRNA and protein synthesis.
Both activators and repressors work by recognizing and binding to specific DNA sequences known as consensus sequences. A consensus sequence is a common pattern made up of the most frequently occurring bases within a DNA region. Importantly, transcriptional regulators do not function alone; they typically work as dimers (pairs) or tetramers (groups of four). This multimeric formation increases specificity because a single protein may only recognize a short sequence of 6 to 8 base pairs, which might lead to accidental activation of the wrong genes. By acting together, multiple proteins can recognize longer sequences, usually around 12 to 15 base pairs, ensuring that the correct gene is targeted.
Thus, the regulation of gene expression involves a highly coordinated interplay between environmental signals, transcriptional regulators, and DNA, ensuring that each cell expresses only the genes necessary for its function at any given time.
Regulation of transcription in prokaryotes
In prokaryotic cells, a single type of RNA polymerase (RNA pol) is responsible for transcribing all genes. However, the RNA polymerase needs a sigma factor to specifically recognize and bind to the promoter region, where transcription begins. The promoter is a DNA sequence where RNA polymerase binds and unwinds the DNA to initiate transcription. The efficiency of this binding can vary, which influences the level of gene expression.
Regulation Through RNA Polymerase Recruitment
The interaction between RNA polymerase and the promoter can sometimes be weak, particularly if the promoter lacks key elements necessary for efficient binding. In such cases, the enzyme may only support a minimal or basal level of gene transcription. Regulation can occur through:
- Repressors: These proteins bind to the operator, a DNA sequence that overlaps with the promoter. When a repressor binds the operator, it blocks RNA polymerase from binding to the promoter, effectively stopping transcription.
- Activators: These proteins bind to DNA near the promoter to enhance RNA polymerase binding and increase transcription efficiency. An example of this is the lac operon in Escherichia coli.
Steps in Transcription Regulation
During the initiation phase, RNA polymerase undergoes a conformational change called isomerization to transition from a closed complex (DNA strands still paired) to an open complex (DNA strands separated). In prokaryotes, this isomerization happens spontaneously and does not require ATP, unlike in eukaryotes, where ATP is needed.
Distant Binding Sites and DNA Looping
Regulatory proteins can bind to DNA sequences that are far from the promoter. In such cases, DNA can bend, bringing these regulatory proteins closer to the RNA polymerase via protein-protein interactions. This looping mechanism ensures effective regulation of transcription.
Examples of Gene Regulation
A. Lac Operon in E. coli
The lac operon contains genes (lacZ, lacY, and lacA) involved in lactose metabolism. Its expression is controlled by two key regulators:
- Lac Repressor (LacI): Encoded by the lacI gene, it binds to the operator to prevent transcription in the absence of lactose.
- CAP (Catabolite Activator Protein): It binds to the CAP site and recruits RNA polymerase when activated by cyclic AMP (cAMP). This activation is necessary for efficient transcription.
Regulatory Scenarios:
- No Lactose Present: The Lac repressor (LacI) binds to the operator, blocking RNA polymerase and preventing transcription. Since lactose is absent, there is no need for the cell to produce enzymes for lactose metabolism.
- Lactose Present, No Glucose: Lactose binds to LacI, inactivating it and preventing it from binding to the operator. Additionally, the absence of glucose increases cAMP levels, activating CAP. CAP then binds the CAP site, recruiting RNA polymerase to initiate transcription and produce enzymes for lactose breakdown.
- Both Lactose and Glucose Present: The Lac repressor is inactivated by lactose, but glucose inhibits cAMP production. As a result, CAP remains inactive, and RNA polymerase binds weakly to the promoter, leading to minimal transcription.
B. Trp Operon in E. coli
The trp operon encodes enzymes for tryptophan synthesis. Its regulation involves a mechanism called attenuation, where transcription can terminate early based on the availability of tryptophan.
- Low Tryptophan Levels: The Trp repressor cannot bind to the operator, allowing RNA polymerase to transcribe the genes needed for tryptophan synthesis. If tryptophan levels are low, the ribosome pauses during translation, forming a 2-3 hairpin structure (antiterminator) in the mRNA. This allows transcription to continue.
- High Tryptophan Levels: When tryptophan is abundant, it binds to the Trp repressor, enabling it to bind the operator and block transcription. Additionally, the ribosome quickly translates the leader peptide, causing a 3-4 hairpin (terminator) to form, which halts transcription.
C. Trp Operon in Bacillus subtilis
In Bacillus subtilis, the trp operon is regulated differently. It is part of the aro supraoperon, which controls aromatic amino acid synthesis. Regulation involves the TRAP protein, a circular structure made of 11 subunits, each binding tryptophan.
Regulatory Mechanisms:
- High Tryptophan Levels: TRAP becomes active, binds to mRNA, and causes a terminator loop to form, stopping transcription.
- Low Tryptophan Levels: TRAP remains inactive, and an antiterminator loop forms, allowing transcription to continue.
- Low Charged tRNA-Tryptophan: An anti-TRAP protein binds to TRAP, preventing it from binding mRNA, and transcription continues. The anti-TRAP gene is located elsewhere in the genome.
This system ensures that tryptophan synthesis is tightly controlled and responsive to the cell’s metabolic needs.
Gene Expression Regulation in Eukaryotes
In eukaryotic cells, such as those of yeast and multicellular organisms, gene expression is regulated by multiple sequences that can be quite distant from the genes they control. These distances are managed by the formation of DNA loops, which allow distant regulatory sequences to come into proximity with the gene promoter. These regulatory sequences can be located either upstream or downstream of the promoter.
Regulatory Sequences and Enhancers
These distant regulatory sequences contain numerous binding sites for different regulatory proteins, and they are often grouped into clusters known as enhancers. Each cluster of binding sites can respond to various cellular signals, allowing fine-tuned regulation of gene expression in both time and space. This means that some regulatory sequences might only be activated or bound by proteins at specific times during the cell cycle or under certain conditions.
Mechanisms of Activators
- Enhancing Recruitment: Activator proteins in eukaryotes generally do not interact directly with RNA polymerase. Instead, they bind to transcription factors, helping to recruit these factors to the promoter and enhance the initiation of transcription.
- Chromatin Remodeling: Another significant mechanism of gene regulation in eukaryotes involves modifying the structure of chromatin. Chromatin consists of DNA wrapped around histone proteins, forming nucleosomes. The N-terminal domains of histones can be chemically modified (e.g., by phosphorylation or methylation) to either loosen or tighten the chromatin structure. Looser chromatin (euchromatin) is more accessible for transcription, whereas compacted chromatin (heterochromatin) inhibits transcription.
Enhancers and Insulators
Enhancers can activate the transcription of a gene even if they are located far away from it. However, there are regulatory sequences called insulators that can block this activation. When an insulator is positioned between an enhancer and a promoter, it prevents the enhancer from influencing that promoter, thus ensuring that enhancers only activate the correct genes.
Mechanisms of Repression
In eukaryotes, transcription can also be repressed by specific mechanisms. Unlike prokaryotes, where repressors typically bind directly to the promoter to block RNA polymerase, eukaryotic repressors use different strategies, such as:
- Competition: Repressors can compete with activators for binding sites, thereby preventing the activators from functioning.
- Interference: Repressors can interfere with the action of activators, making them ineffective.
- Interaction with Transcription Factors: Repressors can directly bind to general transcription factors, blocking the initiation of transcription.
- Chromatin Remodeling: Repressors can recruit complexes that modify chromatin, making it more compact and less accessible for transcription.
Silencing and Chromatin Modifications
Silencing refers to the permanent inactivation of genes based on their position within the chromosome, rather than as a response to external signals. This form of regulation occurs when chromatin transitions from a loose, transcriptionally active state (euchromatin) to a condensed, inactive state (heterochromatin).
- Histone and DNA Modifications: Repressors can recruit enzymes that modify histone tails, adding chemical groups (like methyl groups) to make chromatin more compact. These modifications are recognized by reader proteins, which in turn recruit additional proteins to propagate further modifications. This series of events creates a self-reinforcing loop of chromatin condensation, ensuring that the genes in this region remain transcriptionally inactive.
This coordinated system of gene regulation allows eukaryotic cells to express genes in a highly controlled and specific manner, responding to various internal and external cues while maintaining the proper function of each cell type.
-Lesson 2 – PCR
PCR, or Polymerase Chain Reaction, is a powerful and widely used technique for amplifying specific DNA fragments, allowing us to generate millions of copies of a particular DNA sequence. This process relies on the natural principles of DNA replication, where a DNA polymerase synthesizes a new DNA strand using a template strand. The enzyme extends the DNA by adding nucleotides to the 3'-OH end provided by a primer.
Steps of PCR
The PCR process consists of three main steps that are repeated in cycles, leading to the exponential amplification of the target DNA fragment:
- DNA Denaturation (94°C): The reaction mixture is heated to a high temperature to break the hydrogen bonds between the two strands of the DNA double helix, resulting in the formation of single-stranded DNA.
- Primer Annealing (50°C): The temperature is lowered to allow short, single-stranded primers to bind, or anneal, to their complementary sequences on the single-stranded DNA templates. This step is crucial because it defines the starting points for DNA synthesis by the DNA polymerase.
- Extension (72°C): DNA polymerase adds nucleotides to the 3'-OH end of each primer, synthesizing a new DNA strand complementary to the template strand. This step occurs at the optimal temperature for the enzyme to work efficiently.
These three steps are repeated for 20-40 cycles, depending on the amount of DNA needed, resulting in a massive amplification of the target DNA sequence.
Essential Reagents for PCR
To carry out PCR successfully, several key components are needed:
- Template DNA: The DNA sample containing the region that needs to be amplified.
- Taq DNA Polymerase: A heat-stable enzyme derived from the bacterium Thermus aquaticus. It can withstand the high temperatures used in the denaturation step.
- DNA Polymerase Buffer: This buffer maintains the pH and provides the right environment for the DNA polymerase to function. It contains essential salts and stabilizers to ensure optimal enzyme activity.
- MgCl₂ (Magnesium Chloride): Magnesium ions are critical cofactors for the DNA polymerase. They stabilize the negatively charged DNA backbone and help in the incorporation of nucleotides during DNA synthesis.
- Primers: Short, single-stranded DNA sequences that are complementary to the target DNA region. Primers guide the DNA polymerase to the specific site for DNA synthesis.
- dNTPs (Deoxynucleotide Triphosphates): These are the building blocks (nucleotides) that the DNA polymerase adds to synthesize the new DNA strand.
- Water (H₂O): Used to adjust the volume of the reaction mixture.
Designing Primers for PCR
Primers play a critical role in determining the specificity and efficiency of the PCR. When designing primers, the following factors must be considered:
- Length: Primers are typically 18-24 nucleotides long. This length ensures adequate specificity and efficient binding to the target DNA sequence.
- GC Content: The proportion of guanine (G) and cytosine (C) bases should be between 40-60%. GC pairs are more stable than AT pairs due to three hydrogen bonds, which provides a stronger and more stable binding to the template DNA.
- GC Clamps: Primers should ideally start and end with 1-2 GC pairs to enhance the stability of the primer binding at both ends.
- Melting Temperature (Tm): The melting temperatures of the two primers in a pair should be within 5°C of each other to ensure they anneal at the same temperature during the PCR. This ensures efficient amplification of the target DNA.
- Avoid Complementary Regions: Primers should not have sequences that are complementary to each other to prevent the formation of primer dimers, which can interfere with the PCR process.
By carefully considering these parameters, you can design primers that maximize the efficiency and specificity of the PCR, resulting in a successful amplification of the desired DNA fragment.
Electrophoresis
Electrophoresis is a method used to separate electrically charged molecules, such as nucleic acids and proteins, in a fluid or gel under the influence of an electric field. During this process, molecules with a negative charge migrate toward the positive electrode, while positively charged molecules move toward the negative electrode.
Types of Gels for Electrophoresis
Electrophoresis utilizes two main types of gels:
- Agarose Gel: Used for separating relatively long DNA fragments. Agarose is a polysaccharide derived from agar, a gelatinous substance obtained from the cell walls of red algae. Agarose gel is formed by melting at high temperatures and solidifying when cooled. The concentration typically ranges from 0.7% to 2%, influencing the gel’s pore size and DNA fragment resolution.
- Polyacrylamide Gel: Ideal for high-resolution separation of shorter DNA fragments and proteins.
Factors Affecting DNA Mobility in the Gel
- Size: Smaller DNA fragments move faster through the gel matrix than larger fragments.
- Conformation: The shape or structure of the DNA influences its migration speed. For instance, supercoiled DNA, being more compact, travels faster than relaxed circular DNA. Among DNA conformations, supercoiled and coiled forms exhibit the highest mobility, while circular forms move more slowly.
DNA Visualization
DNA fragments can be visualized in the gel by incorporating an intercalating dye, such as ethidium bromide, into the gel. Ethidium bromide binds to DNA and fluoresces under ultraviolet (UV) light. However, it is a mutagen, and safer alternatives are available. These dyes enable the clear observation of DNA bands after electrophoresis.
DNA Ladder
A DNA ladder is a reference mixture containing DNA fragments of known sizes. It is loaded alongside the samples to provide a comparison for estimating the sizes of the DNA fragments in the samples.
Southern Blot
The Southern blot is a molecular biology technique used to detect the presence of a specific DNA sequence within a DNA sample. It relies on the hybridization of a labeled DNA probe with the target sequence. Although considered an older method, it remains fundamental in some research applications. Here is an overview of the Southern blot procedure:
- DNA Fragment Separation: The DNA sample is digested with restriction enzymes to produce fragments, which are then separated using gel electrophoresis.
- Blotting: After electrophoresis, the DNA fragments are transferred from the gel onto a solid support, typically a positively charged membrane. This transfer ensures that the negatively charged DNA binds securely to the membrane.
- Membrane Transfer Setup:
- The setup involves a layered "sandwich" structure, consisting of:
- A tray containing the transfer buffer at the bottom.
- Filter paper soaked in the buffer to maintain a moist environment.
- The gel containing the separated DNA fragments.
- A positively charged membrane placed on top of the gel, to which the DNA will transfer.
- Additional layers of filter paper and paper towels placed above the membrane.
- A weight on top to facilitate capillary action, drawing the buffer upward and transferring the DNA from the gel to the membrane.
- The setup involves a layered "sandwich" structure, consisting of:
- Hybridization with the Probe: The membrane is then incubated with a labeled DNA probe designed to specifically bind to the target DNA sequence. The probe can be labeled with radioactive isotopes (like ^32P), biotin, or fluorescent dyes, which facilitate the visualization of hybridization.
- Detection: The bound probe reveals the presence and location of the target DNA fragment on the membrane, making it detectable through various imaging methods, depending on the probe used.
The transfer buffer often has an alkaline pH to denature double-stranded DNA, converting it into single strands that are more accessible for hybridization with the probe. This process allows for the specific identification of DNA sequences within a complex mixture.
Northern Blot
Northern Blot is a technique similar to Southern blotting, but instead of targeting DNA, it is used to detect and study RNA molecules. Here’s an overview of how it works and what it is used for:
- RNA Separation: The process begins with the separation of RNA molecules using gel electrophoresis. RNA fragments are sorted by size as they migrate through the gel.
- Blotting onto a Membrane: The separated RNA is then transferred (or blotted) onto a membrane, usually a positively charged one, where the RNA is immobilized.
- Hybridization with Probes: A specific probe, which is complementary to the target RNA sequence, is used to detect and visualize the RNA. These probes can be labeled with radioactive, fluorescent, or chromogenic markers.
Applications of Northern Blot
- Comparing Gene Expression: It is used to analyze how the expression levels of specific genes change under different conditions, across various tissues, or over time.
- Detecting Alternative Splicing: Northern blotting can help identify the presence of alternatively spliced mRNA isoforms by comparing the lengths of the RNA transcripts.
- Quantitative Analysis: This method is semi-quantitative, allowing for the comparison of band intensities to estimate RNA abundance. However, more advanced techniques have been developed for precise quantification.
DNA Microarray
DNA Microarray is a high-throughput method used to study the expression levels of thousands of genes simultaneously. It involves an inverted hybridization setup:
- Probe Immobilization: Specific probes are immobilized on a solid surface, typically a silicon or plastic slide, arranged in a grid-like pattern.
- cDNA Synthesis and Hybridization: The target sample, usually converted into cDNA from RNA, is labeled with fluorescent dyes and then allowed to hybridize with the probes on the microarray.
- Detection and Analysis: The level of hybridization, indicated by the fluorescence intensity at each probe spot, provides information about gene expression.
Applications of DNA Microarray
- Single Nucleotide Polymorphism (SNP) Detection: It can identify genetic variations at the single-nucleotide level.
- Gene Expression Comparison: Microarrays are used to compare gene expression profiles between different conditions, such as diseased versus healthy tissues.
The microarray chip is custom-designed to match the target sequences for the study.
qPCR (Quantitative PCR) or Real-Time PCR
qPCR (Quantitative PCR) or Real-Time PCR is a powerful technique for the precise quantification of DNA during the amplification process. Unlike conventional PCR, qPCR measures the DNA concentration after each cycle using fluorescence:
- Principle of Detection: As the target DNA is amplified, the fluorescence intensity increases. This is because of the use of either intercalating dyes or specific probes.
- Intercalating Dyes: These dyes bind to double-stranded DNA and emit fluorescence. However, they are non-specific and can also bind to non-specific PCR products or primer dimers, leading to background fluorescence.
- Probes for Specific Detection: More specific detection is achieved using probes like TaqMan Probes. These probes take advantage of DNA polymerase's 5’-3’ exonuclease activity and a mechanism called Förster Resonance Energy Transfer (FRET).
- TaqMan Probes (Pacman Probes): The probe has a fluorophore at the 5’ end and a quencher at the 3’ end. When intact, the quencher suppresses the fluorescence of the fluorophore.
- Mechanism: During PCR, if the probe binds to the target DNA between the primer sites, the polymerase degrades the probe while synthesizing new DNA. As the fluorophore is separated from the quencher, fluorescence is emitted and detected.
- Multiplexing: TaqMan probes enable monitoring multiple targets in a single reaction by using different fluorophore-quencher pairs. This is not possible with intercalating dyes, which only measure total DNA.
Applications of qPCR
- Absolute Quantification: The exact amount of target DNA is determined by comparing the fluorescence data to a standard curve.
- Relative Quantification: The amount of target DNA is measured relative to a reference gene, providing a ratio that indicates changes in expression levels between different samples.
This technique is widely used in research and clinical diagnostics to quantify gene expression, detect genetic variations, and study disease biomarkers.
Plasmids
Plasmids are extrachromosomal DNA elements that exist separately from the main chromosome of a cell. Although they are most commonly found in prokaryotes, plasmids can also occur in organisms like yeast and some plants. Typically, plasmids are circular DNA molecules, although some can be linear. Here are key features and details about plasmids:
- Non-Essential Genes: Plasmids generally do not contain genes crucial for the basic survival of the host cell. However, they often carry genes that provide a survival advantage in specific environments, such as genes for antibiotic resistance or virulence factors.
- Independent Replication: Plasmids replicate independently of the host cell’s chromosome. They use the host’s replication machinery but have their own origin of replication (ori), a specific DNA sequence where replication begins. This origin includes regulatory sequences that dictate plasmid replication and its control mechanisms.
- Horizontal Gene Transfer: Plasmids are transferable between cells, facilitating the horizontal transfer of genetic traits, like antibiotic resistance, across bacterial populations.
Essential Features of Plasmids:
- Origin of Replication (ori): This is the DNA site where the replication process initiates. The ori contains sequences that regulate plasmid replication and affect:
- Host Range: The variety of host organisms in which the plasmid can replicate.
- Copy Number: The average number of plasmid copies present in each cell.
- Incompatibility: The inability of two plasmids with similar replication control mechanisms to coexist in the same cell.
- Host Range: This defines the set of organisms in which the plasmid can be maintained. Plasmids can have either:
- Narrow Host Range: Restricted to a limited number of species.
- Broad Host Range: Capable of replication across diverse species, making them useful in various genetic engineering applications.
Host range depends on several factors, such as:
- The presence or absence of restriction-modification systems that degrade foreign DNA.
- Specific host factors necessary for plasmid replication.
- Availability of compatible origins of replication for function.
- Copy Number: This indicates how many copies of the plasmid are typically found in a single cell. Based on this, plasmids are categorized as:
- Low Copy Number: Few copies per cell.
- Medium Copy Number: A moderate number of copies.
- High Copy Number: Many copies per cell.
The replication control mechanism determines the copy number:
- Relaxed Control: Plasmid replication does not rely on the host’s replication initiation proteins; only elongation and termination need the host machinery.
- Stringent Control: Requires initiation proteins made by the host for plasmid replication to start.
Regulating the copy number is crucial because having too many plasmids can create a metabolic burden on the cell, affecting its normal functioning.
- Incompatibility Groups: Plasmids that cannot coexist stably in the same bacterial cell are part of the same incompatibility group. This incompatibility arises when plasmids share similar replication control mechanisms or partitioning systems. When plasmids from the same group are present in a cell, one may be lost over generations of cell division, a phenomenon called "curing." However, if plasmids belong to different incompatibility groups, they can coexist without this issue, ensuring equal distribution in daughter cells after division.
-Lesson 3 – Recombinant DNA technology
Recombinant DNA technology involves creating new DNA molecules in the laboratory by combining genetic material from different sources. This technology enables the construction and maintenance of recombinant DNA in living cells through a process called DNA cloning.
Structure of Recombinant DNA
Recombinant DNA is composed of two essential parts:
- Insert: This is the DNA fragment of interest, such as a product of PCR amplification.
- Vector: A DNA molecule, like a plasmid, viral vector, or artificial chromosome, which provides the means to replicate and maintain the recombinant DNA in a host cell.
The vector must include:
- Origin of Replication: A sequence required for the replication of recombinant DNA within the host cell.
- Selectable Marker: A gene, such as an antibiotic resistance gene, that allows for the selection of cells containing the recombinant DNA.
- Unique Restriction Sites: Specific sequences where the insert can be integrated without disrupting essential functions of the vector.
Natural plasmids already possess some necessary features, such as an origin of replication and a selectable marker. The choice of a vector depends on the intended application and the nature of the host cell. Factors to consider include:
- Purpose (e.g., constructing a DNA library, expressing a protein, or conducting functional studies)
- Desired copy number of the insert
- Type of selection required for clones
- Compatibility with the host organism’s ability to express the insert
Restriction-Modification System in Bacteria
Bacteria use restriction-modification systems as a defense mechanism against foreign DNA, such as from bacteriophages:
- Restriction Endonucleases: Enzymes that recognize and cut DNA at specific short sequences, creating double-stranded breaks in the sugar-phosphate backbone.
- Methylases: Enzymes that methylate bacterial DNA, protecting it from being cleaved by restriction endonucleases.
Restriction Enzymes are categorized into four types:
- Type I: Complex enzymes with three subunits (restriction, modification, and specificity) that methylate DNA symmetrically and cleave at random sites away from the recognition sequence.
- Type II: Enzymes that function separately for restriction and methylation. They cut DNA at predictable palindromic sequences of 4-8 base pairs and are widely used in molecular genetics. They need magnesium ions as cofactors.
- Type III: These have two subunits and need ATP to cleave DNA. They recognize non-palindromic sequences and cut at a defined distance from the recognition site.
- Type IV: Enzymes that cleave modified DNA, such as methylated or hydroxymethylated DNA, and operate at undefined sequences.
Restriction enzymes are named based on the bacterial species from which they are derived. Type II enzymes are preferred in labs because of their predictable cleavage sites and simpler mechanisms compared to Types I and III.
E. coli strains used in the lab contain three specific methylases:
- DAM: Methylates adenine in GATC sequences.
- DCM: Methylates cytosine in CCAGG sequences.
- EcoKI: Methylates adenine in specific sequences.
Some restriction enzymes cannot cut DNA methylated by DAM or DCM, while others are unaffected by methylation.
DNA End Types and Ligation
Restriction enzymes create two types of DNA ends:
- Blunt Ends: No overhangs; the ends are straight.
- Sticky Ends: Single-stranded overhangs that can easily hybridize with complementary sequences.
Restriction enzymes cut DNA at different locations relative to their symmetry axis:
- Before the symmetry axis: Creating 5' overhangs.
- After the symmetry axis: Creating 3' overhangs.
- At the symmetry axis: Producing blunt ends.
Enzymes can have specific behaviors:
- Isoschizomers: Recognize and cut the same sequence in the same way.
- Neoschizomers: Recognize the same sequence but cut differently.
- Isocaudomers: Recognize different sequences but produce the same sticky ends.
DNA Ligase is used to rejoin DNA fragments:
- T4 DNA Ligase is a common enzyme that uses ATP and can ligate both sticky and blunt ends.
- Ligation between sticky ends is generally more efficient.
To prevent self-ligation of vectors, phosphatases can be used to remove 5' phosphate groups. Cells' DNA repair mechanisms will then seal the nicks in recombinant DNA.
If incompatible ends need to be joined, DNA polymerase I (Klenow fragment) can modify them to create blunt ends. This enzyme retains both 3'-5' exonuclease and 5'-3' polymerase activities but lacks 5'-3' exonuclease activity.
DNA Library Construction
A DNA library is a collection of DNA fragments cloned into vectors and maintained in microorganisms. Applications of genomic libraries include:
- Genome sequencing
- Identifying genes associated with specific traits
- Studying regulatory sequences
- Discovering genes involved in biosynthesis
Reporter Genes are used to study regulatory sequences. These genes encode easily detectable proteins, such as GFP or luciferase, and help identify important promoter regions.
In summary, recombinant DNA technology uses restriction enzymes to cut DNA precisely and ligases to join fragments, facilitating cloning and gene studies.
-Lesson 4 - Restriction Cloning
Restriction cloning uses restriction enzymes to create compatible ends on both the vector and the insert DNA. These enzymes cut at specific sites, leaving either sticky ends or blunt ends. The vector and insert are then joined using DNA ligase, which forms covalent phosphodiester bonds, resulting in a stable, circular plasmid that can replicate in cells.
PCR-Based Cloning
PCR-based cloning is a flexible method that can incorporate any DNA fragment into a vector. Here's how it works:
- Amplification: The DNA fragment is amplified using primers. These primers are specially designed to contain:
- A 3' end that is complementary to the template DNA to ensure efficient synthesis.
- A 5' end that can include non-complementary sequences, such as restriction sites, allowing the DNA fragment to be inserted into a specific location on the vector.
This approach enables targeted and efficient insertion of DNA into a backbone vector.
TA Cloning
TA cloning eliminates the need for restriction enzymes and uses the properties of Taq DNA polymerase, which adds a single adenine (A) nucleotide to the 3' ends of newly synthesized DNA strands.
- The vector used in TA cloning is engineered to have a single thymine (T) overhang at its 3' ends, allowing the A-overhangs from the insert to pair and bind.
- DNA ligase then covalently links the vector and insert.
- Note: Proofreading DNA polymerases (those with exonuclease activity) cannot be used for this method because they remove the A-overhangs. Instead, commercial vectors with T-overhangs are typically used.
TOPO-TA Cloning
TOPO-TA cloning is an advanced version of TA cloning that uses topoisomerase instead of DNA ligase to join DNA fragments. Here's how it works:
- Topoisomerases are enzymes that break and rejoin DNA strands to relieve supercoiling. They cleave DNA through a transesterification reaction. In this process, a tyrosine residue in the enzyme forms a temporary covalent bond with the 3' end of the cleaved DNA, leaving a free 5'-OH.
- When a suitable 5'-OH from another DNA strand is nearby, it displaces the enzyme in a second transesterification reaction, restoring the DNA’s phosphodiester backbone and releasing topoisomerase.
- TOPO kits provide vectors with T-overhangs and pre-bound topoisomerase I (from the vaccinia virus) to simplify cloning. These kits require primers without 5' phosphates, as the free OH groups are essential for the ligation process.
Gibson Assembly
Gibson assembly is a versatile and efficient method for assembling multiple DNA fragments into a vector. It requires that all fragments have overlapping homologous regions, which can be added using PCR. The reaction occurs in a single tube with a master mix containing three enzymes:
- Exonuclease: Trims the 5' ends of the DNA fragments to create single-stranded 3' overhangs, which help the fragments anneal based on their homologous regions.
- DNA Polymerase: Fills in any gaps within the annealed regions to create a continuous DNA sequence.
- DNA Ligase: Seals any nicks, completing the assembly.
Key Points:
- Gibson assembly can join up to six fragments in one step, producing a seamless, scar-free DNA construct.
- The process is quick and efficient, with all reactions occurring in the same tube.
- To ensure success, DNA fragments should be relatively long (over 200 nucleotides) to prevent degradation by exonuclease or the formation of secondary structures. Additionally, specific long primers must be designed for each fragment, tailored to create the necessary overlapping regions.
Golden Gate Cloning
Golden Gate Cloning is a technique that leverages type IIs restriction enzymes. These enzymes recognize specific palindromic sequences but cut outside of these recognition sites, leaving sticky overhangs that can be customized to guide the assembly of DNA fragments in a specific order. The advantage of this method is that the recognition sites are eliminated after ligation, resulting in a scarless DNA construct.
The DNA fragments (both vector and insert) are designed such that the enzyme recognition sites are positioned outside the regions that will be cleaved. When the fragments are digested and ligated together, the original restriction sites are removed. The reaction mix, containing both the vector and insert DNA, is combined with the type IIs restriction enzyme and DNA ligase in one tube. If the original DNA fragments re-ligate without the desired insert, they will retain the recognition sites and be re-digested, ensuring only correctly assembled constructs persist.
Key Points:
- Unique 4-base overhangs are used to control the order in which DNA fragments are assembled.
- This method is scarless but requires precise design to ensure overhang compatibility.
- It works best when the restriction sites are not present elsewhere in the DNA to prevent undesired cuts.
- Efficiency may decrease if multiple fragments are assembled simultaneously, or if the fragments are either very small or very large. Even minor sequence variations can result in incorrect ligation products.
Cell Transformation
Cell transformation is a process where bacteria, such as E. coli, take up DNA from their surroundings. This can increase genetic diversity, help with nutrient acquisition, or repair DNA damage. Some bacteria are naturally competent, meaning they can take up DNA under stressful conditions like nutrient scarcity. However, most laboratory strains of E. coli used for cloning are not naturally competent, and artificial competence must be induced.
Inducing Competence:
- Competence is typically induced by altering the cell membrane's permeability through chemical or physical methods. This is achieved using a combination of chemical agents, adjusting growth conditions, and controlling temperature.
Methods for Transforming E. coli:
- Heat Shock: E. coli is treated with calcium ions (Ca²⁺) at cold temperatures, which shields the negative charges on both the cell membrane and the DNA, allowing DNA to enter the cell. The cells are then briefly exposed to heat, causing the membrane to become more permeable.
- Electroporation: An electric field is applied to create transient pores in the cell membrane. The electrical potential drives DNA into the cell. While more efficient than heat shock, electroporation requires specialized equipment and salt-free DNA samples.
Post-Ligation Transformation:
- After ligating the DNA, you should perform at least two types of transformation:
- A control with the vector only to ensure there is no background growth.
- A test with both the insert and vector to identify successful recombinant clones.
Screening for Recombinant Clones
To identify cells with the recombinant plasmid, selectable markers are used. These are usually genes that confer antibiotic resistance. If the cells successfully take up the plasmid (whether it carries the insert or not), they will grow in the presence of the antibiotic. However, growth alone does not confirm the presence of the insert, so insertional inactivation is used to differentiate recombinant clones from those carrying an empty vector.
Example Using pBR322 Vector:
- pBR322 is a classic cloning vector that contains two antibiotic resistance genes (for ampicillin and tetracycline). The plasmid also has unique restriction sites within these resistance genes.
- If a fragment is inserted into the ampicillin resistance gene, the gene becomes disrupted. Recombinant clones will lose resistance to ampicillin but will still be resistant to tetracycline.
Screening Steps:
- Grow the transformed cells on a medium with tetracycline to select for those carrying the plasmid.
- Use a replica plating technique to transfer the colonies onto a second plate containing ampicillin. This involves using a velvet disk to imprint the pattern of colonies onto a new plate.
- Colonies that do not grow on the ampicillin plate are the ones that have the insert, as the insertion disrupted the ampicillin resistance gene.
This method ensures that only the bacteria with the correctly inserted DNA fragment are identified, making the screening process efficient and reliable.
Blue-White Screening
Blue-white screening is a method used to easily identify recombinant bacterial colonies. It utilizes the insertional inactivation of the lacZ gene in specially engineered strains of E. coli and relies on the production of a color change when beta-galactosidase is expressed.
How It Works:
- lacZ Gene and Beta-Galactosidase:
- The lacZ gene encodes the enzyme beta-galactosidase, which breaks down lactose. In the lab, lactose is replaced with IPTG, a chemical analog that induces the expression of lacZ.
- When the enzyme beta-galactosidase acts on a dye-linked substrate called X-gal, it produces galactose and a blue-colored insoluble pigment.
- Mutant E. coli Strains:
- The E. coli strains used in blue-white screening are mutant strains that cannot produce a complete, functional beta-galactosidase enzyme. They synthesize only the non-functional part of the enzyme due to a missing N-terminal fragment.
- The pUC vectors used for cloning carry the missing alpha peptide in the multiple cloning site (MCS). When the peptide is expressed from the vector, it complements the mutant E. coli strain, restoring functional beta-galactosidase activity.
- Screening Process:
- After transformation, the bacterial cells are grown on a medium containing IPTG and X-gal. The colonies that have:
- An empty vector (no DNA insert) will produce a functional beta-galactosidase enzyme, turning the colonies blue.
- A vector with a DNA insert disrupting the lacZ gene will result in white colonies because beta-galactosidase remains inactive.
- After transformation, the bacterial cells are grown on a medium containing IPTG and X-gal. The colonies that have:
- Interpretation:
- Blue Colonies: These have active beta-galactosidase, indicating the absence of an insert (i.e., the vector is empty).
- White Colonies: These have inactive beta-galactosidase, indicating the presence of an insert. These are the colonies of interest.
Note: This method cannot be used with wild-type E. coli, as they already have a functional beta-galactosidase enzyme.
Insertional Inactivation Using the Toxin-Antitoxin System
Another strategy for negative selection of empty vectors involves the toxin-antitoxin (TA) system. This system utilizes two types of proteins: toxins, which inhibit cell growth, and antitoxins, which neutralize the toxins.
- ccdA/ccdB System:
- The ccdB gene is a toxin gene, while ccdA is the antitoxin. This system is part of the F factor in bacteria and is used to ensure that cells carrying the plasmid survive.
- The multiple cloning site (MCS) in the vector is engineered within the ccdB gene.
- How It Works:
- If a bacterial cell receives the empty vector, the ccdB toxin is expressed, and since the cell does not produce the antitoxin, the cell dies.
- However, if the vector contains a DNA insert, the ccdB gene is disrupted, preventing toxin production. As a result, these cells survive.
- Practical Use:
- This approach is effective only in F- strains of E. coli because F+ or F’ strains already have the antitoxin ccdA.
- The toxin-antitoxin system serves as a useful alternative to antibiotic selection, helping maintain plasmids in recombinant clones without the need for antibiotics.
By linking these concepts, we understand that blue-white screening helps easily identify colonies with inserts by a visible color change, while the toxin-antitoxin system provides a way to eliminate cells that do not have the desired recombinant DNA.
Screening of recombinant clones
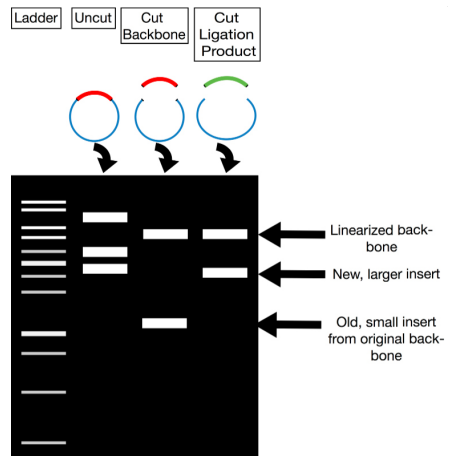 To ensure that a recombinant clone contains the correct plasmid with the DNA insert, we use restriction analysis and PCR-based strategies.
To ensure that a recombinant clone contains the correct plasmid with the DNA insert, we use restriction analysis and PCR-based strategies.
1. Restriction Analysis for Screening Once you have purified the plasmids from putative recombinant colonies, you can screen them by performing restriction digestion:
- Digest Plasmids with Restriction Enzymes:
- Use enzymes that cut at known sites on the plasmid and the insert. This will help differentiate between vectors with and without the insert.
- Gel Electrophoresis:
- Run the digested DNA on an agarose gel. The DNA fragments will separate based on their size.
- Analyze Band Patterns:
- Compare the observed band pattern with the expected size. If the insert is present, the band pattern will differ from an empty vector. Since you already know the sizes of both the vector and the insert, you can predict and verify the expected length of the recombinant plasmid.
Verifying Insert Orientation
After confirming the presence of the insert, it's crucial to verify its orientation within the vector. Incorrect orientation may affect downstream applications.
PCR-Based Strategy for Screening
Instead of extracting DNA from all colonies, which is costly and time-consuming, you can use colony PCR. Here, you:
- Lysis of the Colony:
- The colony is lysed, and the lysate serves as the source of template DNA for PCR. This method quickly checks for the insert and its orientation.
- Using Primers to Verify Presence and Orientation:
- Three main strategies are employed:
a. Insert-Specific Primers
- Design primers that bind specifically to the DNA insert.
- Perform PCR and electrophoresis: If a band appears, it indicates the presence of the insert. However, if there is no band, it might mean the insert is absent, or it could be due to a PCR error. This method does not provide information about the insert’s orientation.
b. Vector-Specific Primers
- Design primers that bind to the vector’s DNA sequence.
- Perform PCR to see the size of the amplified product. If the product size matches the expected size of the vector plus the insert, then the insert is present. This method confirms the insert's presence but not the orientation.
c. Combined Vector and Insert-Specific Primers
- Use a combination of primers binding to the vector (outside the insertion site) and primers binding to the insert.
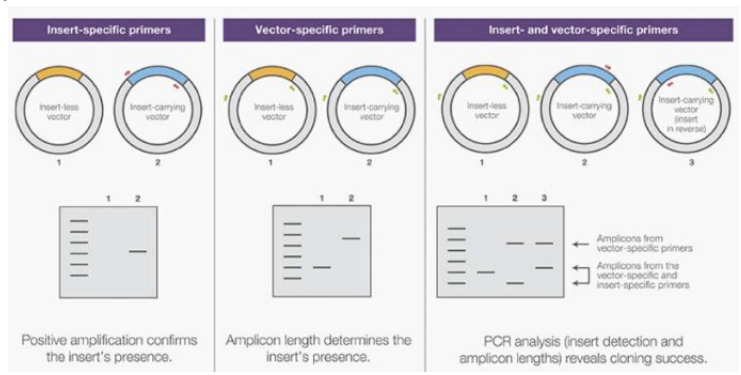 This approach helps determine the orientation:
This approach helps determine the orientation:- Based on which vector primer pairs with the insert primer, you can identify the orientation of the insert. The size of the PCR product will indicate the specific orientation, as it corresponds to a specific amplicon length depending on how the insert is positioned.
Using this PCR approach, you can efficiently verify both the presence and the orientation of the insert without extracting DNA from every colony.
-Lesson 5 - Expression Vectors
Expression vectors are specialized plasmids designed to not only isolate and purify specific DNA fragments but also to drive the expression of genes contained within the insert DNA. They include all necessary regulatory elements to ensure that a cloned gene can be transcribed and translated efficiently in the transformed host organism.
Primary Uses of Expression Vectors:
- Protein Production: To generate large amounts of a specific protein for purification and further analysis.
- Functional Analysis: To express foreign (heterologous) or mutated genes in order to study their effects and functions within the host cell.
Simply inserting a gene into a vector does not guarantee successful gene expression. Several factors need to be considered to ensure efficient transcription and translation.
Why E. coli Is a Common Host
Most proteins produced using recombinant DNA technology are expressed in E. coli due to several advantages:
- Rapid Growth: The doubling time of E. coli is around 20 minutes, allowing for quick scaling of cultures.
- High Cell Density: E. coli cultures can achieve a high cell density, maximizing protein yield.
- Cost-Effective: Media and growth conditions for E. coli are inexpensive and straightforward.
- Ease of DNA Transformation: Introducing foreign DNA into E. coli is fast and efficient.
While E. coli is the preferred host, other systems are sometimes necessary for specific proteins that require different conditions for proper folding or post-translational modifications.
Essential Elements in Expression Vectors
For effective gene expression, vectors must include several key components:
- Promoter:
- Located immediately upstream (before) the gene's insertion site. If the gene is inserted in the correct orientation near the promoter, it will be transcribed into mRNA and then translated into protein by the host cell. The proper placement of the coding sequence relative to the promoter is crucial for efficient expression.
- Transcription Terminator:
- Positioned downstream (after) the multiple cloning site. This sequence ensures proper termination of transcription, preventing unnecessary read-through of the mRNA transcript.
- Shine-Dalgarno Sequence:
- Located between the promoter and the cloning site. It ensures the proper alignment of mRNA with the ribosome for translation initiation in prokaryotes.
The vector design must ensure the correct reading frame, especially for fusion proteins. To address this, researchers often use a set of three vectors, each differing only in the reading frame, ensuring that at least one vector will properly express the desired protein.
Types of Promoters
Promoters can be categorized based on their activity levels and regulation:
- Strong Promoters:
- These have a high affinity for RNA polymerase, resulting in frequent and robust transcription of downstream genes. However, constant high expression can stress the host cell.
- Inducible Promoters:
- These promoters remain inactive until triggered by external stimuli, such as light, temperature, or specific chemicals (e.g., IPTG, tetracycline). Inducible promoters provide precise control over gene expression, which is useful for:
- Avoiding the buildup of toxic gene products.
- Reducing the metabolic burden on the host cell, as constant gene expression can drain resources and impede essential cellular functions.
- Ensuring that cells lacking the induced gene do not outgrow the modified cells in a culture.
- Restricting gene expression to specific growth phases or conditions.
- These promoters remain inactive until triggered by external stimuli, such as light, temperature, or specific chemicals (e.g., IPTG, tetracycline). Inducible promoters provide precise control over gene expression, which is useful for:
Common Inducible Promoters in E. coli:
- Lac Promoter:
- A weak promoter regulated by the lac repressor. In the absence of lactose, the lac repressor binds to the operator, blocking transcription. Adding lactose or the lactose analog IPTG releases the repressor, allowing transcription.
- Trp Promoter:
- A strong promoter regulated by the trp repressor. When tryptophan is abundant, it binds to the trp repressor, which then attaches to the operator, preventing transcription. Decreasing tryptophan levels or adding indoleacetic acid (IAA) can deactivate the repressor, allowing transcription to proceed.
Hybrid Promoters
Combining features of different promoters can enhance expression control. For example, a hybrid promoter can be created using the -35 region of the trp promoter and the -10 region of the lac promoter. This hybrid promoter is still repressed by the lac repressor and can be induced with IPTG.
AraBAD Promoter
- Regulation by AraC: The araBAD promoter controls the expression of the arabinose operon. When arabinose is absent, AraC protein binds in a way that prevents RNA polymerase from accessing the DNA. When arabinose is present, it binds to AraC, altering its configuration and allowing RNA polymerase to bind and initiate transcription.
Lambda Phage Promoter
The lambda phage promoter (PL) is temperature-sensitive and plays a role in the viral life cycle, which includes the lytic and lysogenic phases:
- Lytic Cycle: Driven by the PL promoter, which is typically repressed by the CI repressor protein.
- Induction Mechanism: At normal temperatures (around 30°C), the CI repressor is active, preventing transcription. When the temperature rises to 42°C, the repressor becomes inactive, allowing transcription from the PL promoter to occur.
T7 Promoter System
The T7 promoter is known for being exceptionally strong and efficient, as it is specifically recognized and transcribed only by the T7 RNA polymerase. This polymerase has unique properties, including high specificity for the T7 promoter, impressive efficiency in transcription, and a low error rate, making it particularly reliable for producing RNA. The gene encoding the T7 RNA polymerase is integrated into the chromosome of the host bacterial strain. However, the expression of this gene is controlled by the lac promoter. This means that transcription of the T7 RNA polymerase gene can be turned on by adding IPTG to the growth medium, which acts as an inducer.
Due to the strength of the T7 promoter, there is often a small amount of background or "leakage" expression of the T7 RNA polymerase, even in the absence of IPTG. To manage this, the T7 lysozyme, encoded by another gene, is used to inhibit any unwanted basal expression. This lysozyme effectively reduces the activity of the T7 RNA polymerase when expression is not needed.
Fusion Proteins
Fusion proteins are engineered by combining multiple open reading frames (ORFs) into a single, continuous sequence, producing a chimeric protein. There are several reasons for creating fusion proteins:
- Adding Functional Domains: To give the protein new capabilities or interactions that weren’t part of the original structure.
- Labeling for Visualization: By attaching fluorescent proteins, researchers can easily track or visualize the fusion protein inside cells.
- Improving Stability or Targeting: Some fusion proteins are engineered to be more stable or to be directed to specific parts of the cell.
- Facilitating Purification: These proteins often include tags that simplify the purification process, typically using affinity chromatography.
Affinity Chromatography Example:
- In Immobilized Metal Affinity Chromatography (IMAC), metal ions are chelated to a chromatographic medium. Certain amino acids in the fusion protein, like histidine residues, form complexes with these immobilized metals, allowing for easy separation. Alternatively, antibodies can be used to selectively bind the tagged fusion protein.
However, sometimes the presence of a purification tag is undesirable, especially if it interferes with the protein’s function or makes it unsuitable for clinical applications. In such cases, the tag can be removed. This is done by incorporating a cleavage site that can be targeted by specific proteases. It’s essential that the protein itself does not have any internal cleavage sites that would complicate this process.
SDS-PAGE: Protein Separation Technique
SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis) is a widely used method to separate proteins based on their size. The process involves a few critical components:
- SDS (Sodium Dodecyl Sulfate): An anionic detergent that binds to proteins, causing them to denature and linearize. SDS also provides a uniform negative charge to all proteins, ensuring that their migration through the gel is solely determined by size.
- Polyacrylamide Gel: A polymer that acts as a molecular sieve, separating proteins based on their molecular weight. The separation power depends on the concentration of polyacrylamide and the level of cross-linking within the gel.
Factors Influencing Protein Mobility (In Native Form)
When proteins are not denatured, their migration in an electric field depends on:
- Protein Structure: Globular proteins move more quickly through the gel than fibrous proteins.
- Net Electric Charge: Proteins with different charges migrate in opposite directions.
- Molecular Weight: Smaller proteins travel faster than larger ones.
Denaturation by SDS:
SDS interacts with both hydrophobic and hydrophilic parts of proteins, making them linear. It also ensures that all proteins carry a negative charge, enabling size-based separation. The complete denaturation of proteins requires:
- SDS: To break down the structure.
- Boiling: To disrupt secondary structures.
- Reducing Agents (like DTT or β-mercaptoethanol): To break disulfide bonds.
Buffer Systems in SDS-PAGE
There are two buffer systems used in SDS-PAGE: continuous and discontinuous.
- Continuous Buffer System:
- Uses a single buffer with a constant pH for the gel, samples, and reservoirs. This method is more common for nucleic acid analysis rather than for proteins.
- Discontinuous Buffer System:
- Divides the gel into two sections with different buffer compositions:
- Stacking Gel: Contains large pores that concentrate proteins into tight bands.
- Resolving Gel: Has smaller pores for separating proteins based on size, giving higher resolution.
- Divides the gel into two sections with different buffer compositions:
How Proteins Migrate:
In both systems, proteins enter the gel at different times. In the discontinuous system, proteins initially move rapidly through the stacking gel and then slow down when they reach the resolving gel. This improves separation.
Role of Ions in the Discontinuous System:
- The stacking gel and sample contain Tris-Cl (pH 6.8), while the resolving gel contains Tris-Cl (pH 8.8) and the running buffer contains Tris-glycine.
- Chloride ions (Cl-) move faster than the proteins, forming a leading ion front. At pH 6.8, glycine is neutral and trails behind the proteins. When the proteins enter the resolving gel, the higher pH ionizes glycine, which then moves past the proteins. Proteins are separated solely based on size as they pass through the resolving gel.
Visualizing Proteins:
The separated proteins are stained with Coomassie Brilliant Blue, making them visible as distinct bands on the gel.
Western Blot Technique
The Western blot is a widely used method to detect a specific protein in a sample that has already been separated using SDS-PAGE. Here’s how the process works:
- Transfer: First, the proteins from the gel are transferred onto a nitrocellulose membrane using an electro-blotting technique. This transfer allows the proteins to be more accessible for detection.
- Staining and Blocking: To confirm successful transfer, the membrane is reversibly stained with Ponceau S stain, which shows the presence of the proteins. The membrane is then blocked using bovine serum albumin (BSA) or dry milk to cover all potential binding sites that might nonspecifically attract antibodies. Blocking prevents the antibodies from binding to areas other than the target protein.
- Antibody Incubation: The membrane is incubated with a primary antibody that specifically binds to the protein of interest. Following this, a secondary antibody (linked to a reporter enzyme) is added to bind the primary antibody. The alternative, though more costly, is to use a primary antibody already conjugated with a reporter enzyme.
- Detection: The reporter enzyme on the secondary antibody enables the visualization of the protein of interest, often through chemiluminescence or colorimetric methods.
Yeast Two-Hybrid Assay
Many cellular activities depend on protein-protein interactions, and the yeast two-hybrid assay is a method used to identify these interactions in vivo. The assay takes advantage of the Gal4 transcription factor, which has two distinct domains: one for DNA binding and the other for activating transcription.
- Gal4 Transcription Factor: This factor activates transcription of galactose-responsive genes in yeast (S. cerevisiae) by binding to an upstream activating sequence when galactose is present.
Assay Mechanism:
- To determine if Protein A interacts with Protein B, you create two fusion proteins:
- Protein A is fused with the DNA-binding domain of Gal4.
- Protein B is fused with the transcription activation domain of Gal4.
- If Protein A and B interact, the two domains of Gal4 are brought close together, leading to the activation of a reporter gene. This gene’s expression indicates the interaction between the two proteins.
Bacterial Two-Hybrid Assay
This method, known as the bacterial adenylate cyclase-based two-hybrid system, is used to identify protein interactions in bacteria. It is based on reconstituting a cAMP signaling pathway in an E. coli strain that lacks adenylate cyclase (cya- strain).
- Two proteins thought to interact are fused to separate fragments of the adenylate cyclase enzyme. When these fusion proteins interact, they form a functional enzyme, restoring cAMP synthesis.
- The presence of cAMP triggers the expression of a reporter gene like lacZ, providing a readout of the interaction.
Applications:
- Identifying unknown proteins that interact with a known protein (using a DNA library for the prey proteins).
- Testing if a drug disrupts the interaction between two proteins.
- Pinpointing specific amino acids crucial for the interaction using mutant proteins.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA is a technique used to study DNA-protein interactions, specifically determining if a protein can bind a given DNA sequence.
- The protein is mixed with a 32P-labeled DNA fragment that contains the suspected binding site.
- The mixture is run on a non-denaturing polyacrylamide gel. If the protein binds the DNA, the complex will move more slowly through the gel compared to the free DNA. The results are visualized using autoradiography.
To test the specificity of the interaction:
- A cold probe (unlabeled DNA with the same sequence) can compete with the labeled DNA. If the protein binds the cold probe, it confirms the interaction is sequence-specific.
- An unrelated DNA fragment can be used to ensure the protein does not bind nonspecifically.
- A supershift assay includes an antibody that recognizes the protein, forming a larger complex that migrates even slower on the gel.
Sanger Sequencing
Sanger sequencing is a classic method for determining the DNA sequence, utilizing dideoxynucleotides (ddNTPs) that terminate DNA strand synthesis.
- The method uses four separate reactions, each containing the template DNA, a processive DNA polymerase (lacking exonuclease activity), a radiolabeled primer, and the four standard nucleotides along with one of the four ddNTPs.
- Each ddNTP interrupts DNA synthesis when incorporated, generating DNA fragments of varying lengths. The concentration of ddNTPs is optimized to ensure that every position in the sequence is sampled.
- After DNA synthesis, the fragments are separated on a polyacrylamide gel. The bands are visualized by autoradiography, and the sequence is read from the gel, starting from the shortest fragment at the bottom.
Modern Sanger Sequencing:
- Today, primers are often labeled with fluorescent dyes instead of radioactive markers. Each ddNTP is tagged with a dye that emits a unique color, allowing all four reactions to be combined into one.
- Fragments are separated using capillary electrophoresis, and the fluorescent signals are read by a detector. The results are displayed as an electropherogram, with peaks representing each nucleotide.
Example of exam
Suppose that we want to produce a specific protein.
- First of all we want the sequence encoding the protein and we need to know its nature (prokaryotic or eukaryotic).
In this case it is a bacterial protein. If instead the protein need to undergo posttranslational modifications, like glycosylation, we use strains able to do it. For ex bacillus Subtilitis able to do it. To express a mammalian protein we can use insect (but they have a different pathway for glycosylation so if this is a crucial step, choose another source)
- Once we have the DNA sequence, we have to insert it inside a cloning vector, containing promoter and terminator so the expression cassette necessary for the expression of the protein.
Before inserting the DNA of interest, we ensure that there are not restriction sites inside it.
We need also a selectable marker like antibiotic resistance gene to select those cells who receive the recombinant plasmids from empty cells.
Purify the protein.
We can create a fusion protein, so we can add a tag that allow the purification like an yeast tag.
- So insert in the expression vector the yeast tag, the antibiotic resistance gene.
The promoter must work in the bacillus subtilitis.
We could ass a secretion signal also in order to purify the protein from the medium and not from the biomass.
There is the preparation of the vector
The vector is inside the cell line so we extract it from the culture, and purify it.
- We cut the vector by using restriction enzymes and use them to prevent self ligation and to know the orientation of the insert. We need the dephosphorylation of the vector to avoid the self ligation.
- The next step is the PCR, so we design primer to introduce the restriction site at the end of the vector. After we sequence it, to be sure that it is correct.
So we have a linear vector, we have to insert restriction sites by PCR and then digest with the insert. Then insert the ligase to seal the nick.
The insert will be always in excess to avoid the self ligation of the vector.
- Then after the formation of the recombinant vector, transform cells by heat shock or electroporation.
Screening the cells
so first check if the vector is present, then screen the cells containing the recombinant vector and the empty vector.
To be sure that the insert is present we make the PCR.
The prier could pair externally to the insert, so in this case we expect to have PCR product with different length so in the case of the recombinant plasmids we have a longer insert.
After the PCR
we select a couple of clones which has the recombinant plasmid. To verify the sequence we do a sequence to be sure that the insert has the right orientation, also because a point mutation cannot be visible by the run of the gel.
We can also use the blue-white method.
- At the end check if the protein is the target protein, by using the western blot, so specific antibodies that bind the protein of interest.
If the protein has enzymatic activity can do a test to see it.
- If all is clear then we can do a scale up to produce in large amount.
-Lesson 1 – Pucciarelli - Molecular Genetics
Molecular genetics is a branch of science that investigates the structure, function, and interactions of genes at the molecular level. It can be categorized into two main approaches:
- Classical Genetics:
- This traditional method involves screening for mutations that cause specific phenotypic traits. It focuses on observing outward characteristics to infer genetic mechanisms.
- Reverse Genetics:
- In this approach, scientists deliberately induce mutations in a known gene to study the effects on the organism's phenotype. It is especially useful when the genetic sequence is already identified, but the precise function is still unknown. This method can be applied to inactivate a gene and observe the consequences on the organism, providing insights into gene function.
Model Organisms and Their Importance
Model organisms are species extensively used in research to uncover fundamental cellular and developmental processes, such as replication and translation, often in simpler contexts like bacteria. These organisms provide insights into basic biological mechanisms, which can be extrapolated to more complex systems.
- The underlying principle is that many biological processes are conserved across species, meaning they have remained relatively unchanged throughout evolution. Because of this conservation, studying these processes in simpler organisms can be much more manageable and still yield valuable information applicable to more complex beings, including humans.
- For instance, genetic research is far more straightforward in organisms that reproduce quickly, like bacteria, compared to humans, where such studies are complex and time-consuming.
Defining a Model Organism:
- A model organism is a specific species that:
- Is well-researched and frequently studied in laboratory settings.
- Helps advance understanding of cellular processes, development, and disease.
- Allows scientists to apply the knowledge gained to other species, broadening the scope of research.
Examples of Commonly Used Model Organisms:
- E. coli (Escherichia coli):
- A widely used bacterium in molecular biology research, known for its simplicity and ease of manipulation.
- Saccharomyces cerevisiae (Yeast):
- Yeast serves as a model for eukaryotic organisms. They are highly advantageous because they are haploid (having a single set of chromosomes) and grow very rapidly. Researchers use yeast to test the impact of various mutations efficiently.
- Arabidopsis thaliana (Thale Cress):
- This small flowering plant is a model for plant biology. It can be genetically modified with relative ease, often using a bacterium as a tool to insert desired genes.
- Drosophila melanogaster (Fruit Fly):
- Fruit flies reproduce quickly, making them ideal for studying genes involved in development and discovering how these genes orchestrate the formation of various structures over time.
- Frog (Xenopus laevis):
- Frogs are valuable for studying embryonic development. However, they are sensitive to stress, and hormonal changes can significantly influence experimental outcomes.
- Zebrafish (Danio rerio):
- Zebrafish embryos are transparent, facilitating developmental studies. They produce numerous eggs that develop rapidly and can act as biosensors, with the ability to fluoresce in response to pollutants, making them useful for environmental research.
- Mice (Mus musculus):
- Mice are mammals often used to study processes closely related to humans. They are a preferred model for research on genetics, disease, and the effects of various treatments.
Why Bacteria Are Ideal for Genetic Research:
- Bacteria are advantageous for research because they are relatively simple and easy to manipulate in the lab. Key features include:
- Rapid Reproduction: Bacteria have a short generation time, allowing for the observation of genetic changes over many generations in a brief period.
- Haploidy: They typically possess a single circular chromosome, making genetic studies more straightforward.
- Plasmids: Many bacteria contain plasmids—small, circular DNA molecules independent of the chromosomal DNA. Plasmids can be copied many times, which is useful for genetic experiments.
- Asexual Reproduction: Bacteria reproduce by binary fission, producing genetically identical offspring, or clones. However, spontaneous mutations can sometimes arise, leading to genetic diversity.
- Growth on Solid Media: Bacteria can be cultured on nutrient-rich plates, allowing colonies to grow under various conditions, such as the presence of antibiotics or specific nutrients.
- Storage and Genetic Exchange: Bacteria can be preserved by freezing and are capable of genetic exchange through processes like:
- Transformation: Uptake of foreign DNA from the environment.
- Conjugation: Direct transfer of DNA between bacterial cells via physical contact.
- CRISPR-Cas System: A natural defense mechanism in bacteria used to fight viral infections. It has been adapted as a powerful tool for genetic engineering, allowing precise genome modification.
Bacteriophage
Bacteriophages are viruses that specifically infect and replicate within bacterial cells.
- Structurally, they are simple organisms, consisting of genetic material (nucleic acid), which can be either DNA or RNA and may be double-stranded or single-stranded. This genetic core is enclosed in a protective protein coat called a capsid.
Types of Bacteriophages:
- Lytic (Virulent) Phages:
- These phages only replicate by infecting bacteria, multiplying inside the host, and ultimately causing cell lysis (destruction) to release new phage particles. This cycle is known as the lytic cycle.
- Lysogenic (Temperate) Phages:
- Temperate phages have the ability to choose between two life cycles: the lytic cycle or a dormant state.
- In the lysogenic cycle, the phage’s DNA integrates into the host bacterial chromosome and is replicated passively along with the host’s DNA, without causing immediate damage. The integrated viral DNA, called a prophage, can be inherited by daughter cells. Under certain conditions, it can reactivate and enter the lytic cycle.
Escherichia coli (E. coli) Characteristics:
- E. coli is a Gram-negative, non-spore-forming, rod-shaped, motile bacterium.
- It is facultatively anaerobic, meaning it can thrive in both aerobic and anaerobic environments.
Non-Pathogenic Strain: E. coli K12
- This strain is widely used as a model organism in research and biotechnological applications.
- The K12 strain closely resembles the original, minimally manipulated E. coli K12, making it ideal for laboratory use.
- It is easy to cultivate and study, with a rapid generation time of about 20 minutes.
- E. coli K12 has a single, circular chromosome predominantly composed of monocistronic genes (each gene transcribed individually), though a small fraction is polycistronic (multiple genes transcribed together).
E. coli Movement and Structures:
- Flagella:
- Flagella are long, filamentous protein structures that protrude from the cell’s surface and enable movement.
- They are driven by a proton motive force generated across the bacterial membrane, facilitating movement in response to environmental stimuli (tactic behavior).
- Fimbriae and Pili:
- These are thin, hair-like structures on the bacterial surface.
- Fimbriae aid in attaching the bacterium to various surfaces, substrates, and tissues.
- Pili, particularly the sex pilus, stabilize bacterial mating during conjugation, a process where genetic material is exchanged between bacteria.
Cell Wall and Membrane Details:
- The cell wall is a crucial structure that shields the bacterial protoplast from mechanical stress and osmotic lysis (bursting due to osmotic pressure).
- It has unique components not found in other organisms, making it a prime target for antibiotics.
- The cell wall also facilitates adherence, provides receptors for drugs and viruses, and can trigger disease symptoms in host organisms.
- Murein (Peptidoglycan):
- Murein is a specialized peptidoglycan, a polymer made of sugar chains cross-linked by amino acids, and is an essential part of bacterial cell walls.
- A key component is N-acetylmuramic acid, which defines murein’s unique structure.
Gram Staining and Cell Wall Composition:
- Bacteria are classified into two main groups based on cell wall composition:
- Gram-Negative Bacteria:
- Have a thin layer of peptidoglycan sandwiched between the plasma membrane and an outer membrane. They stain pink.
- Gram-Positive Bacteria:
- Feature a thick layer of peptidoglycan outside the plasma membrane. They stain blue.
- Gram-Negative Bacteria:
Plasma Membrane Functions:
- The plasma membrane serves multiple critical roles:
- Acts as a permeability barrier controlling the flow of substances.
- Generates energy through respiratory and photosynthetic electron transport systems, establishing a proton motive force and housing ATP-synthesizing ATPase.
- Synthesizes membrane lipids and murein.
- Facilitates the assembly and secretion of proteins beyond the cytoplasm.
- Coordinates DNA replication and cell division.
- Hosts specialized enzymes for various biochemical pathways.
Periplasm (in Gram-Negative Bacteria)
- The periplasmic space lies between the inner and outer membranes.
- It houses a variety of enzymes and proteins, which perform functions such as:
- Binding proteins for nutrient uptake.
- Biosynthetic enzymes for cell wall and membrane components.
- Degradative enzymes for breaking down harmful substances.
- Detoxifying enzymes for protection against toxins.
Cytoplasm and Genetic Material:
- The bacterial cytoplasm appears granular due to the presence of ribosomes, which are essential for protein synthesis.
- Nucleoid:
- E. coli’s genetic material consists of a single, large, circular DNA molecule, which is tightly coiled, supercoiled, and attached to proteins.
- DNA replication and segregation are coordinated by proteins and structures within the cytoplasm.
Plasmids:
- Plasmids are small, circular DNA molecules that range in size from 1 to over 1000 kilobases.
- They often carry genes for traits like antibiotic resistance and can be transferred between bacteria through horizontal gene transfer.
- In biotechnology, plasmids are engineered as vectors to replicate DNA sequences in host cells.
Gene Transfer Mechanisms in Bacteria:
- Conjugation:
- Direct transfer of plasmid DNA from a donor to a recipient bacterium through cell-to-cell contact.
- Transduction:
- A bacteriophage accidentally transfers DNA from one bacterial cell to another.
- Transformation:
- Bacteria take up DNA directly from their environment, typically released by other cells.
- Genetic Recombination:
- After DNA transfer, genetic recombination can occur, creating new genotypes and increasing genetic diversity.
Reverse Genetics:
- In reverse genetics, scientists start with a known gene sequence and aim to determine its function.
- To study this, they disrupt or modify a specific gene and then observe the resulting phenotype to understand the gene’s role.
- For example, scientists can inactivate a gene to observe how it affects the organism or replace a normal gene with a version containing a specific mutation.
- Gene Replacement:
- Achieved through homologous recombination, where the introduced DNA replaces the original sequence in the chromosome.
- Gene Knockout:
- Involves removing a gene entirely or inserting an antibiotic resistance marker into it to prevent its function.
-Lesson 2: The Genetic Code
The genetic code is the set of rules that translates a sequence of nucleotides into a sequence of amino acids, ultimately forming proteins. It is crucial because it maintains the instructions for protein synthesis, and any significant change to this code could have serious implications.
- Gene Expression Overview:
- The process begins with DNA being transcribed by RNA polymerase, producing mRNA.
- This mRNA is then translated into a polypeptide (a chain of amino acids).
- DNA contains four types of nucleotides, and the genetic code uses these to specify 20 amino acids. Each amino acid is represented by a set of three nucleotides (a triplet).
- The start codon specifies where translation begins, while stop codons indicate where it ends.
- Triplet Code Discovery:
- Crick and Brenner demonstrated that the genetic code is read in sets of three nucleotides. Using a poly-U mRNA sequence, they showed that it translated into a chain of phenylalanine, proving that each set of three bases specifies an amino acid.
- While each triplet codon generally corresponds to a single amino acid, some amino acids are specified by more than one codon. This redundancy is known as codon degeneracy.
- Reading Frames and Specificity:
- Only one of the three potential reading frames produces the correct sequence of amino acids.
- The ribosome identifies the start codon (usually AUG) to determine the correct reading frame and proceeds to translate the mRNA into a protein.
- An interesting exception to the standard codons is selenocysteine, the 21st amino acid, which is encoded by what is normally a stop codon (UGA). Specific signals tell the ribosome when to insert selenocysteine instead of stopping translation.
Properties of the Genetic Code
- The genetic code is smooth, meaning it minimizes the effects of point mutations.
- It is degenerate, which means that multiple codons can specify the same amino acid. This provides a buffer against mutations because a change in one base might still result in the same amino acid being incorporated.
- The code is compact: even if a mutation occurs, the resulting amino acid may have similar properties to the original, minimizing the impact on the protein's function.
- There are only three stop codons (UAA, UAG, UGA), which signal the end of translation.
- Some amino acids, like tryptophan (Trp) and methionine (Met), are exceptions to the rule of degeneracy, being encoded by only one codon.
Codon and Anticodon Interaction
- Codons that specify the same or similar amino acids often have similar nucleotide sequences. For example, the nature of the second base in a codon influences whether the amino acid is polar (when it’s a purine) or nonpolar (when it’s a pyrimidine).
- Wobble Theory: Proposed by Crick, it suggests flexibility at the third base of the codon, allowing non-standard base pairing. This flexibility means a single tRNA can pair with multiple codons, broadening the range of codon-anticodon interactions.
Types of Substitutions
- Synonymous Substitution: A change in the DNA sequence that does not alter the amino acid sequence of the protein.
- Nonsynonymous Substitution: A nucleotide change that does result in a different amino acid. However, if the replacement amino acid is similar in properties to the original, the protein function may not be severely affected.
- Radical Substitution: A change that results in an amino acid with very different properties, likely altering the protein's function.
Types of Mutations
- Point Mutations:
- Base Substitution: One base is replaced with another, which can be:
- Transition: Substitution of a purine for another purine or a pyrimidine for another pyrimidine.
- Transversion: Substitution of a purine for a pyrimidine or vice versa, less frequent due to structural changes in DNA.
- Missense Mutation: Alters the amino acid sequence.
- Nonsense Mutation: Converts a codon into a stop codon, halting translation.
- Frameshift Mutation: Caused by the insertion or deletion of a base, disrupting the entire reading frame.
- Base Substitution: One base is replaced with another, which can be:
Mechanisms to Increase Protein Diversity
- Programmed Frameshift: The ribosome changes reading frames during translation, allowing a single mRNA to code for multiple proteins. This strategy is used by some viruses and cellular organisms to maximize protein diversity.
Genetic Variability and Mutations
- Triplet Repeat Expansion: Mutations where a sequence of three nucleotides is repeated excessively, often linked to human genetic disorders. The presence of such repeats in an abnormal allele can cause disease.
- While excessive genetic changes can be harmful, a moderate level of genetic variation is crucial for the evolution of a species.
Mutation and Evolution
- A mutation is any change in the nucleotide sequence resulting from errors in DNA replication or repair. Some substitutions are tolerated and may even be beneficial over evolutionary time.
- If a mutation is passed on through generations, it becomes a substitution used to track genetic diversity, often as SNPs (single nucleotide polymorphisms).
- The genetic code bias refers to differences in the frequency of certain codons between organisms, which can affect the efficiency of gene expression.
Expressing Human Proteins in Bacteria
- To express a human protein in bacteria, one must adapt the genetic code to the bacterial system, as codon preferences differ. Yeast can be used for human protein expression since they can perform post-translational modifications.
Universality of the Genetic Code
- The genetic code is largely universal among organisms, having remained unchanged for billions of years. However, some exceptions exist:
- In certain ciliates, one of the stop codons (UAG) encodes glutamine (Gln).
- In eukaryotic mitochondria, slight variations in the genetic code are present.
- The message in mRNA must be read correctly, beginning with the start codon and ending at the stop codon to maintain the correct open reading frame.
-Lesson 3-4 - Transcription
Transcription is the process where RNA is synthesized from a DNA template. While it resembles DNA replication, there are key differences:
- Substrates: Transcription uses ribonucleotides (RNA building blocks), while DNA replication uses deoxyribonucleotides.
- Primer Requirement: Unlike DNA polymerase, RNA polymerase does not require a primer to initiate synthesis. A primer normally helps create a strong bond with the first nucleotide, but without it, the initial bond is weaker.
- Usage of DNA: The entire DNA is replicated once per cell cycle, but during transcription, only a single DNA strand is used as a template.
- Accuracy: Transcription is less precise compared to DNA replication, meaning RNA polymerase has a higher error rate.
RNA Polymerase Structure and Function
- In prokaryotes, there is a single type of RNA polymerase, different from those in more ancient organisms like archaea.
- Eukaryotes have three distinct RNA polymerases (RNA pol I, II, III), each specialized for transcribing different types of genes.
- RNA polymerase is composed of multiple subunits and is initially inactive until it forms a complex.
- The structure and folding of RNA polymerase are crucial for its function. A conformational change is necessary to activate the enzyme.
- Although the overall structure of RNA polymerases is similar, the amino acid sequences differ across species.
Steps of Transcription in Prokaryotes
- Initiation:
- Transcription begins when RNA polymerase binds to the promoter region upstream of the gene.
- The DNA strands separate, forming a transcription bubble. This allows the enzyme to start copying the DNA template.
- The enzyme initially releases a few nucleotides before properly engaging in the elongation phase. This happens because RNA polymerase starts without a primer.
- Elongation:
- RNA polymerase synthesizes RNA in the 5’ to 3’ direction, unwinding the DNA ahead of it and rewinding it behind.
- The enzyme also separates the growing RNA strand from the DNA template. However, its proofreading ability is not as accurate as DNA polymerase.
- Termination:
- When a termination signal is reached, transcription stops, and the RNA transcript is released.
- RNA polymerase then detaches from the DNA.
Role of Sigma Factor in Prokaryotes
- Sigma factor is a crucial cofactor that guides RNA polymerase to the promoter.
- It facilitates the binding and melting of DNA, enabling transcription to start.
- When the sigma factor associates with RNA polymerase, they form the holoenzyme. This complex is responsible for recognizing and unwinding the promoter DNA.
Promoter Recognition
- Promoters in bacteria have specific sequences that determine how efficiently RNA polymerase binds and initiates transcription. The closer the promoter sequence is to the consensus sequence, the higher the transcription efficiency.
- Sigma factor has multiple domains with specific roles:
- Sigma 4 interacts with the upstream promoter region, stabilizing the complex.
- Sigma 2 facilitates the melting of DNA, forming the open complex necessary for transcription to proceed.
Isomerization
- Isomerization refers to the structural changes in DNA and RNA polymerase that occur without needing ATP. This transition is essential for transcription to start effectively.
Transcription in eukaryotes
Core Promoter Elements in Transcription
- The core promoter is the DNA region containing several essential elements needed to start transcription. These include:
- BRE (B Recognition Element)
- TATA box
- DPE (Downstream Promoter Element)
- These elements can be located upstream or downstream relative to the +1 site, which is the transcription start point. However, not every promoter contains all of these elements; each promoter might have a unique combination that aids in initiating transcription.
Regulatory Sequences
- Transcription is also influenced by regulatory sequences like:
- Enhancers: Increase transcription efficiency by binding transcriptional activators.
- Silencers: Decrease transcription by binding repressors.
- Insulators: Block enhancers from influencing the wrong promoters, ensuring proper regulation.
- These regulatory elements work by interacting with specific proteins to modulate the level of transcription from the core promoter.
Role of General Transcription Factors in Eukaryotes
- In eukaryotic cells, the function of the sigma factor (used in prokaryotic transcription) is replaced by general transcription factors.
- These factors bind to the promoter region and help recruit RNA polymerase II (Pol II) to initiate transcription.
- One of their crucial roles is DNA melting, which is necessary to open the double helix and expose the DNA template for transcription. This process requires ATP hydrolysis for energy.
- The RNA polymerase II enzyme has a tail called the C-terminal domain (CTD). When the CTD undergoes phosphorylation:
- It helps the polymerase "escape" from the promoter and continue transcribing the DNA.
- It also recruits other factors involved in RNA processing, like splicing and polyadenylation.
Role of Additional Proteins
- Transcription in eukaryotes requires more than just RNA polymerase and transcription factors. Other essential proteins include:
- The mediator complex: This large protein complex interacts with the phosphorylated CTD of Pol II and facilitates communication between transcriptional activators and the polymerase.
- Regulators and chromatin modifiers: These proteins help stabilize the association of Pol II with the promoter and modify the structure of chromatin to make DNA more accessible.
- By forming a DNA loop, these proteins bring distant regulatory elements closer to the promoter, ensuring efficient transcription initiation.
Translation: Connecting Transcription to Protein Synthesis
- Once mRNA is transcribed, the next step is translation, where the genetic code in the mRNA is used to synthesize an amino acid sequence (a protein).
- In prokaryotes, translation occurs simultaneously with transcription. A single mRNA strand can have multiple ribosomes attached, forming a polysome.
- In eukaryotes, mRNA must first undergo several processing steps:
- 5' capping: A protective cap is added to the mRNA.
- Splicing: Introns are removed, and exons are joined.
- Polyadenylation: A tail of adenine nucleotides is added to the 3' end.
- After processing, the mature mRNA is transported out of the nucleus for translation.
The Ribosome and Its Structure
- Translation occurs on the ribosome, a complex made of rRNA and ribosomal proteins, consisting of a small subunit and a large subunit.
- In bacteria, these subunits are 30S (small) and 50S (large).
- In eukaryotes, they are 40S (small) and 60S (large).
- The ribosome has three important sites for tRNA interaction:
- A site (Aminoacyl site): The entry point for charged tRNA, which carries an amino acid.
- P site (Peptidyl site): Where the tRNA holds the growing polypeptide chain, and the peptide bond forms between amino acids.
- E site (Exit site): Where uncharged tRNA exits the ribosome.
Reading the Correct Open Reading Frame
- The small ribosomal subunit has a channel that guides the mRNA, forming a kink to ensure the mRNA is read correctly.
- The ribosome uses the correct open reading frame (ORF), defined by the start codon, to ensure the proper sequence of amino acids is synthesized.
tRNA Charging and Function
- tRNA must be "charged" before it can deliver amino acids to the ribosome.
- The enzyme aminoacyl-tRNA synthetase catalyzes this charging process:
- It first activates the amino acid through adenylation.
- Then, it attaches the amino acid to the correct tRNA.
- The enzyme is highly specific, ensuring each tRNA carries the correct amino acid.
- The enzyme aminoacyl-tRNA synthetase catalyzes this charging process:
Ribosome Recruitment and Translation Initiation
- In prokaryotes, ribosomes are recruited to the mRNA by the Shine-Dalgarno sequence. This sequence aligns the start codon (AUG) in the P site to set the correct reading frame.
- Prokaryotic mRNA is often polycistronic, meaning it can encode multiple proteins.
Special Initiation tRNA
- Both eukaryotic and prokaryotic translation start with a special tRNA that carries formyl methionine (fMet).
- This tRNA is unique because:
- It is the only tRNA that enters the ribosome's P site directly.
- The formyl group ensures that peptide synthesis occurs only in one direction, from the N-terminal to the C-terminal.
- This tRNA is unique because:
The Shine-Dalgarno Sequence: Ribosome Binding and Translation Initiation in Prokaryotes
The Shine-Dalgarno sequence is a highly conserved consensus sequence found in the mRNA of prokaryotes. It plays a crucial role in the initiation of translation by serving as the ribosome binding site. This sequence is essential because it ensures that the ribosome correctly positions itself on the mRNA to start protein synthesis.
- Recognition by 16S rRNA:
- The Shine-Dalgarno sequence is recognized specifically by the 16S rRNA component of the 30S ribosomal subunit. This recognition occurs because hydrogen bonds form between the nucleotides of the Shine-Dalgarno sequence in the mRNA and complementary bases in the 16S rRNA.
- These hydrogen bonds anchor the ribosome to the mRNA and ensure that the ribosome binds accurately.
- Setting the Open Reading Frame:
- The interaction between the Shine-Dalgarno sequence and the 16S rRNA helps to set the correct open reading frame. This means that the mRNA's start codon (usually AUG) is aligned precisely in the ribosome's P site, where translation begins. By placing the start codon in the P site, the ribosome is ready to start synthesizing the correct sequence of amino acids.
Factors Influencing Translation Initiation
Several factors can affect how efficiently translation is initiated in prokaryotes:
- Start Codon Efficiency:
- The most efficient start codon is AUG, as it is the standard initiation codon. However, alternative start codons like AUU, GUG, and UUG can also initiate translation, but they do so less efficiently. The ribosome recognizes these alternative codons, but they are used less frequently.
- Distance Between the Shine-Dalgarno Sequence and the Start Codon:
- The length of the sequence between the Shine-Dalgarno site and the start codon is crucial. If the distance is too great, the ribosome may not position the start codon efficiently in the P site, which decreases the overall efficiency of translation. A shorter and optimal distance enhances the ribosome's ability to initiate translation properly.
- Secondary Structures in the mRNA:
- The presence of secondary structures in the mRNA, such as hairpins or loops, can complicate the initiation process. These structures may form around the Shine-Dalgarno sequence and the start codon, preventing the ribosome from binding properly. The folding energy refers to the energy required to unfold or break these secondary structures. If a significant amount of energy is needed to unfold them, it can hinder the ribosome's binding and slow down or inhibit translation initiation.
Translational Reinitiation
- Polycistronic mRNA: In some organisms, a single mRNA molecule can contain multiple coding regions, known as polycistronic mRNA. This means the mRNA has several open reading frames (ORFs), each coding for a different protein.
- Reinitiation Process: Translational reinitiation occurs when the ribosome finishes translating one open reading frame, reaches a stop codon, and releases the newly synthesized protein. Instead of completely dissociating from the mRNA, the ribosome can remain associated with it and continue scanning along the mRNA strand.
- The ribosome scans downstream until it finds a new start codon, which is often located close to or even partially overlapping the termination codon of the previous ORF. Once a new start codon is identified, the ribosome can restart the translation process, synthesizing a new protein.
- Purpose and Efficiency: This strategy allows cells to maximize protein synthesis efficiency by enabling ribosomes to translate multiple proteins from the same mRNA molecule without having to repeatedly assemble from scratch. It also provides a way for the cell to regulate gene expression efficiently.
- Influencing Factors: The effectiveness of translational reinitiation can depend on several factors:
- The sequence around the stop codon, which may affect how easily the ribosome can scan for a new start codon.
- The presence of specific RNA secondary structures that could either facilitate or hinder the ribosome's scanning and reinitiation.
- The availability of initiator factors, which are necessary to help the ribosome start translating the new ORF.
Recoding
- Definition: Recoding is a molecular mechanism that temporarily changes the way the genetic code is read during translation. This leads to the incorporation of a different amino acid than what would typically be specified by the genetic code.
- Mechanisms and Function: Various mechanisms can trigger this alteration, and the result is the production of alternative protein products with modified or expanded functions. By using recoding, cells can generate a diverse array of proteins from the same mRNA sequence.
- Protein Diversity: In many organisms, especially those that lack alternative splicing (a process that creates different proteins from the same gene), recoding provides a way to increase protein diversity. This is crucial for adapting to different cellular needs and functions.
Mechanisms Involved in Translational and Transcriptional Control
- Programmed Frameshifting:
- This process involves the ribosome shifting to a different reading frame within an mRNA sequence. As a result, the ribosome translates a protein with a different sequence of amino acids or a protein with a modified length.
- Purpose: Used by viruses and certain organisms to synthesize multiple proteins from overlapping reading frames on the same mRNA.
- Control: Frameshifting is regulated by specific signals within the mRNA.
- Example 1: Release Factor 2 (RF2) is an autoregulatory protein.
- If RF2 concentration is low, it cannot bind to the stop codon, causing the ribosome to shift frames and bypass the stop codon, which results in continued translation and the production of more RF2.
- If RF2 concentration is high, it binds to the stop codon, stopping translation normally.
- Example 2: DNA X gene in E. coli:
- This gene encodes two subunits of DNA polymerase III: Tau and Gamma.
- Normally, the Tau subunit is synthesized in the standard reading frame.
- A frameshift of -1 nucleotide leads to the synthesis of the Gamma subunit. This shift occurs due to a secondary structure in the mRNA and happens about 50% of the time.
- Translational Bypassing (Hopping):
- Here, the ribosome pauses translation at a specific site, then resumes translation further downstream, skipping a block of intermediate nucleotides.
- This jump can happen without changing the reading frame, but it may sometimes involve a frame change.
- The formation of secondary structures in the mRNA facilitates this ribosomal "hopping."
- Redefinition of the Codon:
- In this mechanism, a codon temporarily changes its meaning.
- Example: The stop codon UGA can be redefined to encode the amino acid selenocysteine if the SECIS element (a specific RNA structure) is present. The SECIS element forms a hairpin, attracting the tRNA for selenocysteine and allowing its incorporation into the protein.
- Translational Control – Autorepression:
- In some cases, a protein can regulate its own synthesis by interacting with the mRNA.
- Mechanism: Secondary structures in the mRNA can block translation at the start of a gene. If the protein concentration is high, it binds near the Shine-Dalgarno (SD) sequence, preventing the ribosome's small subunit from binding and thus inhibiting translation.
 Translational Control – Translational Coupling:
Translational Control – Translational Coupling:
- This occurs in polycistronic mRNA (mRNA that encodes multiple proteins, such as gene1 and gene2).
- The mRNA may adopt a conformation that blocks the SD sequence of the second gene. Only after gene1 is translated does the secondary structure melt, allowing gene2 to be translated.
- Translational Inhibition:
- When two proteins are needed, mRNA remains available for translation.
- If these proteins are not required, and there is insufficient rRNA, the ribosomal proteins bind to the mRNA with lower affinity, inhibiting translation since rRNA is necessary for ribosome assembly.
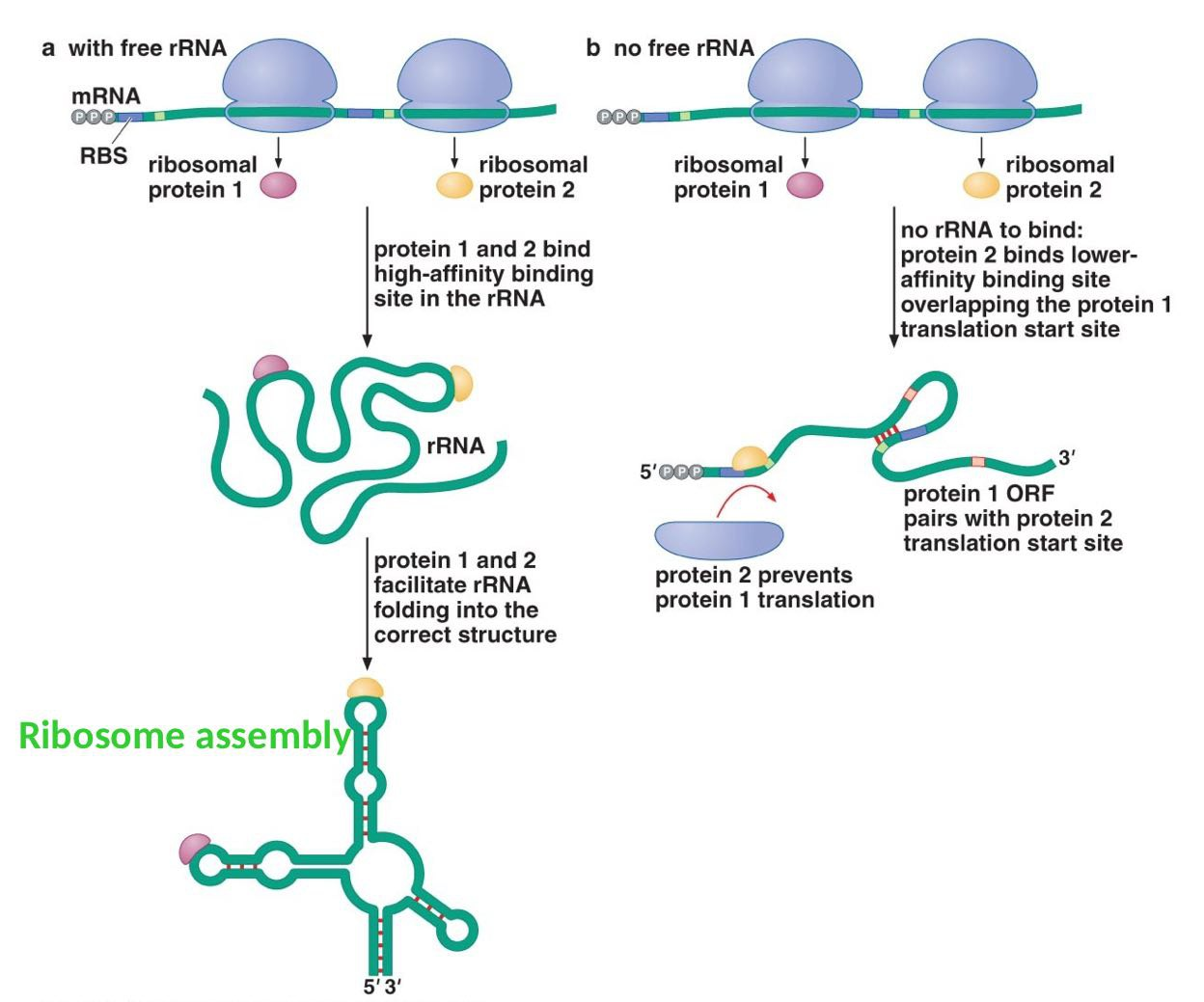
- Transcription Regulation:
- Gene expression is often regulated at the mRNA production stage or through the activity of RNAase enzymes, which degrade RNA.
- Commonly, operon regulation occurs at the promoter region, where RNA polymerase binds to various promoter components to control gene transcription.
- Inhibition of Initiation:
- The mRNA can form complex structures that block the ribosome's binding site for charged tRNA, preventing the initiation of translation.
- Stalled Ribosome:
- When an mRNA lacks a stop codon, the ribosome stalls. A transfer messenger RNA (tm-tRNA) is recruited to rescue the stalled ribosome.
- The tm-tRNA carries an amino acid (alanine) and a short mRNA sequence. It binds in the ribosome's A site, and the attached mRNA provides a stop codon, allowing the defective protein to be tagged for degradation.
- Inhibition of Translation – Antisense RNA:
- Antisense RNA pairs with the target mRNA to form a double-stranded molecule. The ribosome cannot bind this double-stranded RNA, so translation does not occur.
- Entrapment Mechanism:
- When there is excess S50 protein, it binds to the 5' untranslated region of the mRNA, stabilizing a pseudoknot structure that blocks the SD sequence. This prevents ribosome binding.
- When S50 protein levels decrease, the pseudoknot structure destabilizes, and the +1 start codon can be placed in the P site, allowing translation to proceed.
-Lesson 5 - RNA-Mediated Regulation of Gene Expression in Bacteria
RNA-based gene regulators are crucial components in bacterial cells because they provide precise control over gene expression. This precision ensures that genes are activated or deactivated at the right times and at the appropriate levels, depending on the cell's needs. By regulating gene expression in such a fine-tuned manner, RNA-based mechanisms can influence almost every aspect of how genes are expressed.
The Power of RNA in Gene Regulation
- Versatility of RNA: RNA is highly adaptable, capable of forming different structures and participating in a wide range of cellular processes. This versatility makes RNA an excellent tool for regulating gene expression.
- Designability of RNA: Scientists can design RNA molecules in such a way that they fold into specific shapes. These structures can then serve as regulatory elements, controlling the translation of target genes. This designability allows for precise manipulation of gene activity.
- Characterizability of RNA: With advancements like next-generation sequencing, researchers can now extensively study and characterize the various forms and functions of RNA. This means we can better understand and engineer RNA for specific regulatory purposes.
How RNA-Mediated Regulation Works
RNA controls gene expression through the formation of secondary and tertiary structures. These complex shapes are not just random; they have specific roles in regulating which genes are turned on or off. By folding into these structures, RNA can interact with other molecules in the cell, influencing processes like the initiation of translation, stability of the mRNA, or even transcription itself. This structural flexibility gives RNA-based regulators a powerful ability to fine-tune gene expression efficiently.
Riboswitches: Mechanism of Gene Regulation
Riboswitches are a mechanism used by cells to control the metabolism of essential molecules like cofactors or vitamins. They function as regulatory elements in the mRNA that directly bind to specific metabolites. Importantly, this binding occurs without the need for any protein factor. The interaction between the metabolite and the riboswitch causes a change in the RNA's structure, which can then influence various stages of gene expression.
How Riboswitches Work
- Direct Binding and Structural Changes:
- When a metabolite binds to a riboswitch, the RNA undergoes a conformational change (change in shape). This structural change can affect processes such as:
- Transcription elongation: How long and efficiently the RNA transcript is made.
- Translation initiation: The start of protein synthesis from the mRNA.
- Other processes that ultimately lead to protein production.
- When a metabolite binds to a riboswitch, the RNA undergoes a conformational change (change in shape). This structural change can affect processes such as:
- Structure and Function:
- Riboswitches form secondary structures in the mRNA, which allows them to interact specifically with metabolites. These structures include:
- A sensor (ligand-binding domain): This part of the riboswitch has a conserved sequence that binds directly to the metabolite. It acts as a sensor for the metabolite’s presence and concentration.
- A connection to the downstream coding sequence: When the metabolite binds to the sensor, it triggers a structural change in the riboswitch, which in turn affects how the coding sequence is processed.
- If the metabolite concentration is above a certain threshold, binding to the riboswitch sensor causes a conformational change in the RNA's expression platform. This change modulates downstream events, such as halting transcription or blocking translation.
- Riboswitches form secondary structures in the mRNA, which allows them to interact specifically with metabolites. These structures include:
Types of Riboswitch Structures
Riboswitches can take on two main structural forms:
- Multihelical Junctions: These are internal loops that connect multiple helices, creating a complex secondary structure.
- Pseudoknots: These are more complex, tertiary structures where the RNA folds in a knot-like arrangement.
Despite their structural differences, both types of riboswitches share similarities at the molecular level:
- They use a pyrophosphate sensor to detect metabolite presence.
- The interactions between the riboswitch and the metabolite are often stabilized by Mg²⁺ ions.
Presence Across Organisms
Riboswitches are commonly found in prokaryotes, but they are not exclusive to them. They have also been identified in some eukaryotic organisms, indicating their broader role in gene regulation across different forms of life.
SELEX Method: Generating High-Affinity Aptamers
The SELEX (Systematic Evolution of Ligands by EXponential enrichment) method is a technique used to generate aptamers. Aptamers are single-stranded DNA (ssDNA) or RNA oligonucleotides that bind specifically and with high affinity to a target, which could be a small molecule, ligand, or protein.
- Creating the Oligonucleotide Library:
- An oligonucleotide library is synthesized, consisting of a vast collection of sequences that are randomly generated but of fixed length. These sequences are flanked by conserved 5' and 3' regions that help in later amplification steps.
- Binding to the Target:
- The random oligonucleotides are mixed with the target molecule. This mixture is subjected to affinity chromatography to separate bound oligonucleotides from unbound ones.
- Oligonucleotides that do not bind the target are washed away, while the oligo-target complexes are retained in the column.
- Recovery and Amplification:
- The bound oligonucleotides are then recovered from the column. To enhance the selection, the sequences are amplified using PCR with universal primers, which are specific to the conserved 5' and 3' regions.
- This process is repeated multiple times to enrich for sequences with the highest affinity to the target, resulting in aptamers that can be used for gene regulation or other applications.
Small Non-Coding RNAs in Gene Regulation
Small non-coding RNAs are molecules that play a regulatory role in controlling gene expression. They can influence:
- Transcription
- RNA stability
- Translation
These small RNAs bind to their target mRNA through base pairing. Depending on their complementarity:
- If they are fully complementary (like antisense RNA), they can completely block the mRNA activity.
- If they are partially complementary (like small regulatory RNAs or sRNAs), they can inhibit translation or affect mRNA stability
Thermo-Sensor RNA: Temperature-Responsive Gene Regulation
Thermo-sensor RNAs are regulatory RNA molecules that respond to changes in intracellular temperature. They do this by altering their conformation as the temperature changes, which affects gene expression.
- Temperature and RNA Structure:
- As the temperature increases, structured nucleic acid molecules can melt or unfold, disrupting the hydrogen bonds that maintain secondary structures.
- Certain temperatures favor the formation or melting of these structures, which in turn regulates whether a gene is expressed or repressed.
Examples of Thermo-Sensor RNA Regulation
- Lambda Phage CIII Gene:
- At 45°C, the Shine-Dalgarno (SD) sequence and promoter of the CIII gene are blocked by a secondary structure, preventing ribosome binding.
- When the temperature decreases, the RNA adopts a different conformation, freeing the SD sequence and allowing the ribosome to bind and initiate translation.
- Heat-Shock Sigma Factor in E. coli:
- At 30°C, the SD sequence of the heat-shock sigma factor gene is blocked by a secondary structure.
- When the temperature rises, this structure unfolds, exposing the SD sequence and enabling the ribosome to bind, initiating translation.
- Virulence Gene in Pathogenic Bacteria (Listeria):
- The virulence gene is activated by high temperatures. At lower temperatures, the RNA folds in a way that blocks the sequence, preventing gene activation. When the temperature increases, the RNA unfolds, exposing the gene and allowing expression.
- Cold-Shock Response in E. coli:
- The S1 protein regulates translation during cold shock. At 37°C, antisense RNA blocks the S1 binding site, preventing translation by forming a hairpin structure that occludes the SD sequence and start codon.
- As the temperature drops, the antisense RNA shifts position, but a hairpin still blocks the SD and start codon. The S1 protein binds and melts the hairpin, freeing the SD sequence and allowing translation to proceed.
-Lesson 6 - Bacteriophages
How They Infect Bacteria
Bacteriophages are viruses that specifically infect bacteria. They cannot replicate on their own, so they rely on infecting a host bacterium to reproduce. The genes of bacteriophages are organized in a way similar to operons in bacterial genomes, which means the genes are grouped and regulated together for efficient expression.
Infection and Genetic Material Injection
- The bacteriophage attaches to the surface of a bacterial cell and injects its genetic material into the bacterium's cytoplasm.
- Upon entering the bacterial cell, the viral DNA is initially linear. However, it quickly circularizes, meaning the two ends of the DNA attach to each other covalently, forming a circle. This circular DNA is easier to manage and integrate into the host’s genome.
Lysogeny and the Prophage
- When the viral DNA integrates into the bacterial chromosome, the bacterium becomes a lysogen, and the integrated viral DNA is called a prophage.
- In the lysogenic cycle, the viral DNA is copied alongside the bacterial DNA each time the bacterium divides. The virus does not produce new phage particles in this stage, and the bacterial cell continues to live and divide normally.
Lytic and Lysogenic Cycles
- Lytic Cycle:
- This is the virulent phase of the phage life cycle. During the lytic cycle, the bacteriophage takes over the bacterial cell's machinery to replicate its own DNA and produce new phage particles.
- Eventually, the cell is destroyed (lysed), releasing the new phages to infect other bacterial cells.
- Lysogenic Cycle:
- In this cycle, the phage DNA integrates into the host's chromosome and is replicated with the bacterial DNA each time the cell divides.
- The virus remains dormant, and no new phages are produced until certain environmental factors trigger the prophage to exit the lysogenic cycle and enter the lytic cycle.
- Switching from Lysogenic to Lytic: Under specific conditions, such as stress or damage to the bacterial DNA, the phage DNA is excised from the bacterial genome. This excised DNA is then transcribed and translated, leading to the production of new viral particles and the start of the lytic cycle.
Genetic Control in Bacteriophages
The bacteriophage chromosome contains several promoters that control the production of proteins essential for both cycles:
- Promoters:
- PR (Right Promoter): This promoter is responsible for producing the CRO protein and the N protein. The N protein acts as an antiterminator, allowing the transcription of downstream genes necessary for the lytic cycle.
- PL (Left Promoter): This promoter produces the CII and CIII proteins. These proteins are important in regulating the switch between the lytic and lysogenic cycles.
- PRM (Repressor Maintenance Promoter): This promoter controls the production of the CI protein, which is a repressor. The CI protein helps maintain the lysogenic state by repressing genes needed for the lytic cycle.
Summary of the Process
- When the phage DNA is injected into the host cell, the expression of viral genes begins. Depending on the environmental conditions and the balance of regulatory proteins (like CI, CII, and CRO), the virus will either enter the lytic cycle or establish a lysogenic state.
- If conditions favor the lytic cycle, proteins like CRO dominate, leading to the replication of the virus and eventual lysis of the bacterial cell.
- If the lysogenic cycle is favored, the CI repressor maintains the dormancy of the phage DNA within the host genome.
-Lesson 7 - Transposons:
Mobile Genetic Elements
Transposons are segments of DNA within a chromosome that have the ability to move from one location to another in the genome. These mobile genetic elements can cause significant genetic changes, contributing to the evolution of genomes in various ways. They can:
- Insert themselves into genes or regulatory sequences, which may disrupt normal gene function.
- Modify gene expression by altering where or how genes are turned on or off.
- Lead to chromosomal mutations, such as deletions, inversions, or even creating opportunities for crossing over.
Transposons are especially common in organisms with few genes because they help increase genetic diversity. However, even in humans, transposons make up more than 50% of the genome. These elements are usually inserted into specific loci that contain promoters, which can activate them when needed.
Classes and Mechanisms of Transposon Movement
There are two main classes of transposable elements, each with different mechanisms of movement:
- DNA Transposons:
- These transposons encode proteins that move the DNA directly to a new position within the genome or replicate the DNA, integrating the replicated copy elsewhere. They move using either:
- The cut-and-paste mechanism: The original transposon is cut out and inserted into a new site.
- The copy-and-paste mechanism: A copy of the transposon is made and inserted into a different location, leaving the original transposon in place.
- DNA transposons are found in both prokaryotes and eukaryotes.
- These transposons encode proteins that move the DNA directly to a new position within the genome or replicate the DNA, integrating the replicated copy elsewhere. They move using either:
- Retrotransposons:
- These elements encode reverse transcriptase, an enzyme that creates DNA copies from RNA transcripts. These DNA copies are then integrated into the genome.
- Retrotransposons only exist in eukaryotes and can also include polyA retrotransposons
Transposable Elements in Prokaryote
- Insertion Sequence (IS) Elements:
- IS elements are the simplest type of transposable elements. They carry the gene for transposase, the enzyme needed for mobilization and insertion.
- Integration of IS elements can disrupt genes or regulatory regions, leading to changes in gene expression or even chromosomal rearrangements like deletions or inversions.
- They can move using either the cut-and-paste or copy-and-paste mechanism.
- The transposase enzyme recognizes inverted repeats at the ends of the IS element and recruits DNA polymerase, which makes a copy of the sequence.
- The copy is inserted into a new site, and DNA polymerase and ligase fill the gaps. The transposase acts similarly to a restriction enzyme.
- The integration occurs through a one-step transesterification reaction, made possible because each inverted repeat ends with a free 3' OH group.
- Transposons (Tn):
- Transposons are more complex than IS elements and often carry additional genes, such as antibiotic resistance genes.
- Transposons can be:
- Composite Transposons: These have genes flanked by IS elements. The IS elements provide transposase and the necessary recognition signals for movement.
- Non-Composite Transposons: These carry genes but do not have IS elements at their ends. Instead, they end with inverted repeats and often contain cassettes for expressing resistance genes.
- Transposons can move using both the cut-and-paste and copy-and-paste mechanisms. For the cut-and-paste movement:
- The non-transferred DNA strands need to be cleaved. This can happen by:
- Using an enzyme other than transposase.
- Using transposase itself, which can form a DNA hairpin structure. The hairpin is then cleaved at the 3' OH, breaking the hairpin and causing a double-strand break in the DNA. The break is repaired by ligase.
- The non-transferred DNA strands need to be cleaved. This can happen by:
- Replicative Transposition:
- In this mechanism, the transposon is duplicated, creating a copy at a new site in the genome while the original remains. This mechanism is a type of copy-and-paste movement.
- First, transposase proteins assemble at the ends of the transposon, forming a complex.
- The transposase cleaves the DNA at the transposon ends, liberating the 3' OH ends, which then join to the target DNA through a transfer reaction.
- The main difference between this and the cut-and-paste mechanism is the formation of a double-stranded DNA intermediate, leading to the creation of a transposon copy.
- In this mechanism, the transposon is duplicated, creating a copy at a new site in the genome while the original remains. This mechanism is a type of copy-and-paste movement.
Transposable Elements in Eukaryotes
Barbara McClintock was a pioneering scientist who discovered the existence of mobile genetic elements in eukaryotic organisms, specifically studying them in corn (maize). These mobile genetic elements are segments of DNA that can move around within the genome, and they play a significant role in genetic variation and regulation.
Types of Transposable Elements in Eukaryotes
- Autonomous Elements:
- These are transposable elements that can move on their own. They are termed "autonomous" because they contain the transposition gene, which codes for the enzyme transposase. This enzyme is necessary for the element to cut itself out of one location in the DNA and paste itself into another location.
- Non-Autonomous Elements:
- These elements cannot move by themselves because they lack the transposition gene and, therefore, cannot produce transposase. They are termed "non-autonomous" and need the presence of another transposable element that can supply the transposase enzyme.
McClintock’s Discovery in Corn
Barbara McClintock identified mobile genetic elements in corn that she called dissociation elements (Ds). The Ds elements are examples of non-autonomous transposons, meaning they require the help of another element called an activator (Ac) to move. The activator element is autonomous and provides the transposase enzyme necessary for the movement of the Ds elements.
Effect of Transposable Elements on Corn Pigmentation
- When the dissociation elements (Ds) insert themselves into a gene responsible for pigment production in corn seeds, they can disrupt the function of that gene. As a result, the normal production of purple pigment is blocked, and the seeds remain yellow.
- This change in color illustrates how transposable elements can influence gene expression by inserting themselves into important genetic regions.
Ty Elements in Yeast
Ty elements are transposable elements found in yeast that are similar in some ways to bacterial transposons but also share key features with retroviruses. Like retroviruses, Ty elements integrate into specific regions of the yeast chromosome through a complex mechanism.
- This integration involves the formation of specific protein-protein complexes that bind to complementary DNA (cDNA).
- The process begins when the Ty element creates an RNA copy of itself. This RNA is then converted back into DNA using an enzyme called reverse transcriptase, which is characteristic of retroviruses. The newly synthesized DNA is then integrated into the yeast genome.
P Elements in Drosophila
P-elements are transposable elements found in Drosophila (fruit flies), and they are responsible for a genetic phenomenon called Hybrid Dysgenesis.
- The P strain of Drosophila contains numerous P-elements. These P-elements can move within the genome and cause mutations by inserting themselves into genes. However, the P strain also produces a P repressor protein, which prevents the P-elements from moving, thereby stabilizing the genome.
- In contrast, the M strain lacks both the P-elements and the P repressor.
Hybrid Dysgenesis Explained
- When a male P strain (which carries P-elements) mates with a female M strain (which lacks the P repressor), the offspring, or hybrid progeny, exhibit genetic instability. This happens because the P-elements can move freely in the absence of the P repressor, causing mutations, such as the loss of wings in the progeny.
- On the other hand, if a P strain mates with another P strain, the P repressor from both parents is present, so the P-elements are kept in check, and no genetic instability occurs.
Genetic Impact of P Elements
P-elements are of great genetic interest because of their significant effects on the Drosophila genome. They can:
- Cause mutations by inserting into genes.
- Lead to gene repression or even overexpression if they insert near regulatory regions.
- Cause chromosomal breakage, which can result in severe genetic consequences.
-Lesson 8
Mutagenesis
Mutagenesis refers to the process by which mutations arise in the DNA sequence. Mutations can occur spontaneously due to natural errors in DNA replication or repair. However, the rate of mutation can be significantly increased by various environmental factors, such as exposure to radiation or certain chemicals. Despite the increase in mutation rate, the mutations that occur are random and cannot be precisely reproduced.
- A mutation is a change in the nucleic acid sequence of DNA. If this mutation is stable and can be replicated during DNA replication, it may be passed down to the next generation, making it heritable.
- DNA damage, on the other hand, is different from a mutation. Damage refers to structural alterations in DNA (like breaks or modified bases) that cannot be directly replicated. If not repaired, DNA damage can prevent proper DNA replication
Role of Natural Selection
- In the context of evolution, natural selection plays a key role in determining which mutations persist in a population. Mutations that provide a survival advantage are more likely to be passed on and maintained in future generations.
Types of Substitutions
- Synonymous Substitution:
- This occurs when a change in a single nucleotide within a codon (the three-base sequence in mRNA that specifies an amino acid) does not alter the amino acid it encodes. Because the protein remains unchanged, synonymous substitutions are often considered neutral.
- Nonsynonymous Substitution:
- This type of substitution leads to a change in the amino acid sequence of a protein. Depending on the properties of the new amino acid, this change can have varying effects:
- Conservative Substitution: The new amino acid has similar chemical properties to the original one. As a result, the change may not significantly affect the protein's function.
- Radical Substitution: The new amino acid has very different chemical properties from the original one, which can have a major impact on the protein's structure and function.
- This type of substitution leads to a change in the amino acid sequence of a protein. Depending on the properties of the new amino acid, this change can have varying effects:
Substitutions and Evolution
- Some substitutions are changes that have been tolerated and maintained through evolution. These are referred to as single nucleotide polymorphisms (SNPs). SNPs are common genetic variations that can be used to study genetic diversity and may sometimes play roles in disease susceptibility or other traits.
Different Types of Mutations
Mutations can occur in various forms, affecting the DNA sequence in specific ways:
- Point Mutations:
- These involve a change in a single nucleotide and can be further categorized into:
- Transition: A mutation where a base is replaced with another base of the same type. This means a pyrimidine (such as cytosine or thymine) is swapped with another pyrimidine, or a purine (adenine or guanine) is swapped with another purine.
- Transversion: A mutation where a base is replaced with one of the opposite type. In this case, a pyrimidine is replaced by a purine, or vice versa.
- These involve a change in a single nucleotide and can be further categorized into:
- Specific Point Mutations:
- Silent Mutation: Although a nucleotide is changed, the amino acid sequence remains unchanged. This is because multiple codons can code for the same amino acid.
- Nonsense Mutation: This mutation causes the change of a normal codon into a stop codon, prematurely terminating protein synthesis.
- Missense Mutation: Here, a change in the nucleotide results in a different amino acid being incorporated into the protein. The substitution can be either a transition or a transversion, potentially affecting the protein’s function.
- Spontaneous Frameshift Mutation: This occurs when a single nucleotide is either added or deleted, altering the reading frame of the gene. As a result, the downstream amino acid sequence is completely changed, often rendering the protein nonfunctional.
Protein Engineering and Genetic Manipulation
Protein engineering uses genetic techniques to modify the DNA sequence of a cloned gene, which in turn alters the properties of the encoded protein. The goal is to create proteins with enhanced or novel functions. Scientists use site-directed mutagenesis and various genetic engineering techniques to bring about these changes.
Requirements for Site-Directed Mutagenesis
To successfully perform site-directed mutagenesis, certain information is crucial:
- Knowledge of the DNA sequence and the preferable structure of the protein, particularly the active site where the protein interacts with substrates.
- A detailed understanding of the protein’s mechanism, including how its structure influences its function.
- Identification of cofactors that are necessary for the protein's activity, to determine whether they need modification.
Potential Protein Alterations
Site-directed mutagenesis can be used to make targeted changes to a protein, which may include:
- Altering the Michaelis Constant (Km): This affects how tightly the substrate binds to the enzyme, either increasing or decreasing the affinity compared to the original protein.
- Modifying Thermal Tolerance or pH Stability: This enables the protein to withstand higher temperatures or different pH levels without losing its function. Such modifications can prevent denaturation (loss of structure) or inactivation under harsh conditions.
- Changing Enzyme Requirements: By altering the protein, scientists can sometimes eliminate the need for a specific cofactor, making the enzyme functional under different circumstances.
- Increasing Resistance to Proteases: The protein can be engineered to become more resistant to cellular proteases, which are enzymes that break down proteins. This prolongs the protein's stability and lifespan.
- Altering Allosteric Regulation: Adjusting how the protein is regulated allosterically (how its function is controlled by the binding of molecules at sites other than the active site) can change the protein's activity in response to cellular signals.
Rational Design in Protein Engineering
These intentional and specific changes to a protein are referred to as rational design. Scientists carefully design the protein to alter only the desired characteristics while maintaining or enhancing its overall function. This strategic approach allows for precise manipulation of the protein’s properties.
Mutagenesis
Mutagenesis refers to the process of altering the nucleic acid sequence within DNA. This change can be minor, like a point mutation, or involve larger modifications. Importantly, mutagenesis is not the same as DNA damage; rather, it is a deliberate change in the sequence that can naturally occur over time or be intentionally induced in a lab setting.
Types of Mutagenesis
- In Vivo Mutagenesis:
- This type of mutagenesis occurs inside living organisms or cultured cells, like in model organisms. For example, techniques like CRISPR-Cas9 can be used in bacteria, which do not have a nucleus, to make genetic changes. In vivo mutagenesis can also involve exposing cells to UV rays or chemicals.
- This approach is useful when scientists have limited information about the protein structure or function. The focus here is on observing phenotypic changes (observable traits), although this method has limitations, such as generating multiple, uncontrolled mutations and creating hotspots where mutations occur more frequently. The mutations are typically random.
- In Vitro Mutagenesis:
- This type of mutagenesis occurs outside of living cells, in a controlled lab environment. Scientists have complete control over which DNA sequences are altered because they already have detailed knowledge of the protein structure and gene sequence.
- In vitro mutagenesis allows for precise changes and can be screened for both phenotype (traits) and genotype (DNA sequence). This method can even be used to engineer hybrid proteins with new properties.
- Working directly on the DNA sequence is preferable because it's simpler to modify DNA before the protein is made, rather than trying to change the protein afterward.
Techniques in In Vitro Mutagenesis
- Random Mutagenesis:
- This technique introduces mutations randomly across a short segment of a cloned gene, resulting in a variety of mutated proteins. Researchers then screen these mutations to find the ones with the desired properties.
- Site-Directed Mutagenesis:
- Here, mutations are introduced at specific sites within the DNA sequence. This requires detailed knowledge of the gene’s DNA sequence and the ability to synthesize specific oligonucleotides (short DNA fragments).
Applications of Site-Directed Mutagenesis
This method is used to:
- Study the relationship between protein structure and function by altering enzyme activity.
- Investigate gene expression by modifying regulatory regions.
- Adjust vectors used in DNA cloning, such as inserting or deleting restriction sites.
Methods for Site-Directed Mutagenesis
- Using PCR and Plasmids:
- Scientists design primers with the desired mutation. These primers help create a mutant DNA sequence using PCR (Polymerase Chain Reaction).
- A key step involves distinguishing between the original DNA and the mutated DNA. To do this, restriction enzymes like DNP1 are used. DNP1 can cut methylated and hemimethylated strands, which are typical of the original DNA, leaving only the non-methylated mutated DNA intact.
- PFU polymerase is used because it has high processivity (stays on the DNA longer) and a low error rate, ensuring accuracy.
Old Method Using M13 Phage
- The older method involved using M13 bacteriophage vectors. A gene of interest was inserted into a double-stranded M13 vector. The single-stranded version was isolated and mixed with a synthetic oligonucleotide containing the desired mutation.
- The synthesis was catalyzed by the Klenow fragment of DNA polymerase I, and T4 DNA ligase was used to join DNA strands. The efficiency of this method was low compared to modern PCR techniques.
Modern Approach Using Oligonucleotides and Plasmids
- The target DNA is inserted into a plasmid vector with specific resistance genes (e.g., tetracycline-resistant and a non-functional ampicillin-resistant gene).
- The plasmid is denatured and combined with oligonucleotides designed to introduce specific mutations.
- DNA synthesis is again catalyzed by the Klenow fragment, but without certain exonuclease activities to maintain the mutation.
- This approach ensures that mutations are efficiently incorporated into the DNA.
PCR-Based Mutagenesis
- Using PCR, researchers can introduce desired mutations or add new components to the gene. The mutations are built into the primers used in the PCR reaction, ensuring a high percentage of products have the desired change.
- PFU polymerase is favored for its precision. To screen for successful mutations, DNP1 enzyme digests the methylated wild-type DNA, leaving the non-methylated mutant DNA intact.
Challenges and Random Mutagenesis
- Predicting the exact nucleotide changes needed to alter protein function can be difficult. Random mutagenesis methods are used to generate a variety of amino acid changes at one site to test different effects.
Error-Prone PCR
Error-prone PCR is a method used to introduce mutations randomly into a DNA sequence. It uses heat-stable DNA polymerases that lack proofreading ability, like Taq polymerase, which increases the likelihood of errors during DNA synthesis. These errors, or mutations, are introduced by altering PCR conditions. Here’s how this works:
- High Mutation Rate: Adjusting the concentration of DNA template and nucleotides, as well as the temperature of annealing, can create more mistakes. For example, using a low annealing temperature, adding excess Mg²⁺ ions, and cycling the PCR reaction more times (40-80 cycles) amplify errors. Adding Mn²⁺ ions can further destabilize correct base pairing, increasing the mutation rate.
- Outcome: After generating a pool of mutated DNA sequences, scientists screen these for the desired mutation that affects protein function or other characteristics.
Degenerate Primers
Degenerate primers are designed to introduce variability in specific parts of a DNA sequence. These primers contain a mix of nucleotides at certain positions, allowing for multiple sequences to be amplified simultaneously. Here’s how this works:
- Insert a target gene into a plasmid between two unique restriction sites.
- Use PCR with two sets of primers:
- Degenerate primers overlap near the gene's center.
- Non-degenerate primers bind to upstream and downstream regions.
- Perform PCR reactions separately, then combine and denature the products.
- Renaturation of the mixed fragments allows for overlapping sequences to join, forming new DNA molecules.
- Use DNA polymerase to complete the double-stranded DNA.
- Restriction enzymes digest the product, and the modified DNA is cloned into a plasmid and transformed into E. coli to express the new protein.
Nucleotide Analogs
A nucleotide analog is a chemical compound similar in structure to a DNA nucleotide but with differences that induce mutations. For example, 5-bromouracil is an analog of thymine. Although this method was once common for creating mutations, it is less used today due to newer, more efficient techniques.
DNA Shuffling
DNA shuffling is a technique used to create hybrid or chimeric genes from a family of related genes, increasing protein diversity. There are two main methods:
- Using Restriction Enzymes: Genes are cut into fragments at similar places using restriction enzymes. The fragments are then randomly recombined and ligated to form new hybrid genes. These hybrids are screened for unique properties.
- Using DNase I: Genes are fragmented using DNase I, then recombined during PCR using primers from different members of the gene family. The fragments with high sequence similarity will anneal and cross-prime, creating a diverse library of recombined genes.
- Advantages: A large variety of mutations and protein variants can be generated without needing detailed information about specific amino acids.
- Disadvantages: Extensive screening is needed to identify the variants with the desired traits.
Reverse Genetics
Reverse genetics focuses on understanding the function of a gene by starting with a known DNA sequence and then studying how altering this sequence affects the organism. Here’s the process:
- Researchers begin with a cloned DNA segment whose sequence is known but whose function is uncertain.
- They introduce mutations into the gene and observe the resulting phenotypic changes in the organism or in vitro.
- This method helps determine the gene's role in biological processes
Techniques in Reverse Genetics
- Gene Replacement: The normal gene is replaced with a mutated version via homologous recombination, allowing scientists to see how the mutation affects the organism.
- Gene Knockout: A gene is completely removed by inserting an antibiotic resistance cassette or deleting part or all of the gene. This results in the total loss of gene function.
- Gene Knockdown: Instead of removing a gene, RNA interference (RNAi) is used to block the translation of the gene's mRNA. This reduces the gene's expression, and the resulting effects on the organism are studied.