001 Drug Receptor Interactions
PHARMACEUTICAL CHEMISTRY
TAXONOMY OF BIOLOGICAL MECHANISMS
Receptors
Agonists
Antagonists
Partial Agonists
Inverse Agonists
Enzymes
Inhibitors
Ion Channels
Openers
Blockers
Protein-Protein Interactions
Inhibitors
In Vivo Effectors
RECEPTORS
Membrane-bound proteins that bind endogenous ligands (usually extracellular) to induce physiological effects (usually intracellular).
Function: First step in a long intracellular signaling cascade.
Major Types: G-Protein Coupled Receptors (GPCRs), Seven-Transmembrane Spanning Receptors.
Notable Recognition: Brian Kobilka and Robert Lefkowitz, 2012 Nobel Prize in Chemistry.
Structure: GPCRs span the membrane seven times, with both intracellular and extracellular domains.
ENZYME INHIBITION – FOUR MODES OF ACTION
Competitive Inhibition: Inhibitor competes reversibly with the substrate for the active site.
Uncompetitive Inhibition: Inhibitor binds only to the enzyme-substrate complex (ES), leading to EIS intermediates. Extremely rare.
Non-Competitive Inhibition: Inhibitor binds non-covalently to allosteric sites; may impact kinetics resulting in partially inhibited enzymes.
Irreversible Inhibition: Inhibitor binds covalently to the active site machinery (e.g., Suicide inhibitors).
Examples: MAO inhibitors, β-lactamase inhibitors.
ION CHANNELS
Life exists within an electric potential window of less than one volt; for most cells, the membrane potential is about 60-70mV.
Ion channels regulate passive ion flow across membranes based on electrical and concentration gradients.
Characteristics:
Ion Selective
Composed of glycoprotein subunits in homo- or heteropolymer arrays.
Channels almost never have an open rest state.
Relevance: Involvement in cardiac, neuronal, and psychiatric disorders.
Main Ions: Na+, K+, Ca++, Cl-.
Example: hERG (iKr) channel blockade can cause prolonged QT intervals, leading to “Long QT syndrome” and sudden death.
LEAD COMPOUNDS FROM VARIETY OF SOURCES
Chance Discovery
Example: Penicillins
Natural Products
Example: Taxol
Clinical Observation
Example: Viagra
Natural Ligands
Existing Drugs
High Throughput Screening (HTS)
NATURAL LIGANDS
Adrenaline
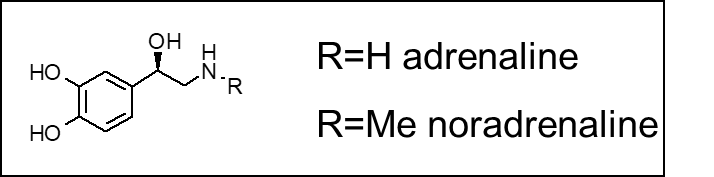
R=H: Adrenaline
R=Me: Noradrenaline
The neurotransmitters adrenaline and noradrenaline are a classic example of the rational design of a drug from a natural ligand.
known as the ‘fight-or-flight’ hormone which when released into the blood stream causes a number of physiological responses. For example increased heart rate, brochodilation, release of blood sugar, vasodilation and increased blood flow to the muscles. All of these changes prepare the body for fight or flight.
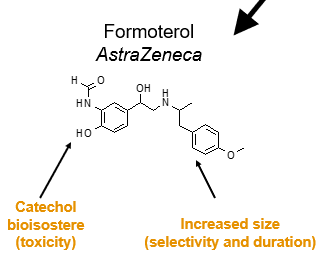
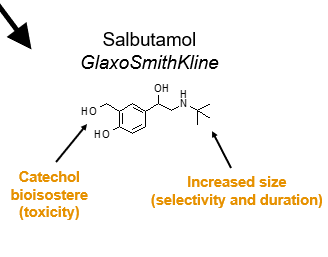
Rationally designed drugs based on adrenaline focus on two main problems:
The catechol unit is easily oxidized in the body and can form toxic by-products which cause side effects.
Adrenaline is non-selective and interacts with a multitude of adrenoreceptors. Bronchodilation (and relief of asthma symptoms) requires selectivity for the beta2-adrenoreceptor.
Long acting inhaled drugs of this type are obtained by increasing the lipophilicity (fattiness) of the side chain such that it remains in the lung tissue for a longer period of time.
Analogs:
Catechol bioisostere (toxicity) & increased size, (selectivity, and duration)
Examples include Formoterol (AstraZeneca) and Salbutamol (GlaxoSmithKline).
EXISTING DRUGS
Known as the “Me-Too” or “Me-Better” Approach.
Issues:
Short duration
Multiple side effects and drug incompatibilities
Examples:
Viagra (Pfizer)
Levitra (Bayer): Fewer side effects and incpatibility with other drugs
Cialis (Eli Lily): longer duration (36 hours, “the weekend pill”).
Caution advised regarding patent issues.
STRUCTURALLY NONSPECIFIC DRUGS
Characteristics:
Activity is not dependent on specific chemical structure [Physicochemical properties of the drug are not dependent on the specific chemical structure of the drug.]
similar biological activities can occur across different structures.
Do not target specific molecular mechanisms (e.g., enzymes or receptors).
Small structural changes lead to minimal changes in biological activity.
Administered in relatively large doses.
EXAMPLES OF STRUCTURALLY NONSPECIFIC DRUGS
General Anesthetics: Chloroform, Ether, Ethyl Chloride.
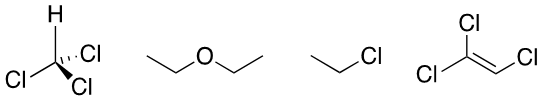
Volatile Insecticides (fumigants): Methyl Bromide, 1,3-Dichloropropene.
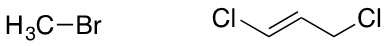
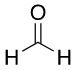
Bactericidal Agents: Formaldehyde, Chlorine.
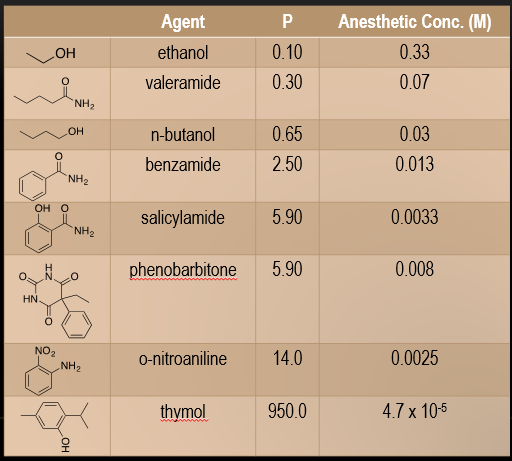
POTENCIES OF STRUCTURALLY NONSPECIFIC DRUGS
Potencies relate to physicochemical properties; correlation with partition coefficients is noted [The potencies of structurally nonspecific agents often correlate with physicochemical properties, such as their partition coefficients:]:
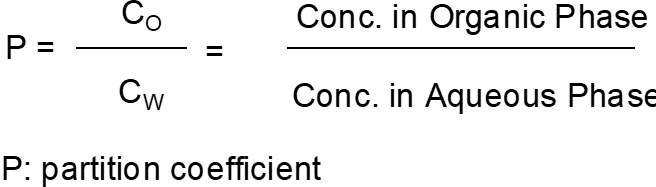
Higher P indicates higher lipophilicity.
Lower P indicates higher hydrophilicity.
Partition coefficient indicates equilibrium distribution between phases.
STRUCTURALLY NONSPECIFIC DRUGS AND LIPID SOLUBILITY
Substances soluble in lipids have depressant effects, strongest in lipid-rich cells (e.g., nerve cells) [Structurally nonspecific substances which are soluble in lipids have depressant properties.].
The depressant effect is quickest and strongest in cells which are particularly rich in lipids, such as nerve cells.
Potency of depressant substances increases with partition coefficient between lipid and water.
Meyer-Overton Principle: Anesthetic potency correlates with lipid solubility; more lipid-soluble anesthetics are more potent, affecting membrane properties (e.g., fluidity) [It is proven empirically that there exists a close correlation of anesthetic potency with lipid solubility. The more lipid soluble an anesthetic, the more potent is its anesthetic effect. ].
According to the lipid theory, general anesthetics induce anesthesia by perturbing the structure of cell membranes by dissolving in them. General anesthesia is thought to occur as soon as a particular number of anesthetic molecules have dissolved in the cell membrane, thus causing critical changes in the physicochemical properties like membrane fluidity, thickening of the membrane, axonal conduction velocity, etc. of the lipid bilayer.
SUMMARY OF STRUCTURALLY NONSPECIFIC DRUGS
Comprise a very small percentage of current drugs.
Modifications in structure yield no significant biological changes.
While structurally varied, they cause similar biological effects
Act as depressants.
They do not act on specific molecular targets (enzymes or receptors) and act in relatively large doses.
STRUCTURALLY SPECIFIC DRUGS
The biological activity results primarily from specific chemical structures
Usually effective at lower concentrations compared to nonspecific drugs.
Modifications in structure can lead to significant changes in pharmacological effects.
Targets: Receptors, enzymes, nucleic acids (DNA, RNA).
They have some structural characteristics in common, and the fundamental structure present in all of them is responsible for the analogous biological response they cause.
Pharmacophore: The critical portion of the structure of the drug which binds to the receptor.
Statin Drugs:
Nitrogen Mustards:
Diuretics:
Salient Features in Drug-Receptor Interactions
Drug-receptor binding is only transient in the case of majority of therapeutically useful drugs.
The combination of various bonds including ionic, hydrogen and Vander Waal’s attractive forces can contribute to the binding of a drug to the receptor.
Once the drug-receptor bonding has taken place:
1) a biological response may result if the drug molecule is an agonist or…
2) no biologic response if the drug molecule is an antagonist.
Conformational change in the receptor initiates the activation of the biologic response and aids in the dissociation of the drug-receptor complex.
AGONISTS AND ANTAGONISTS
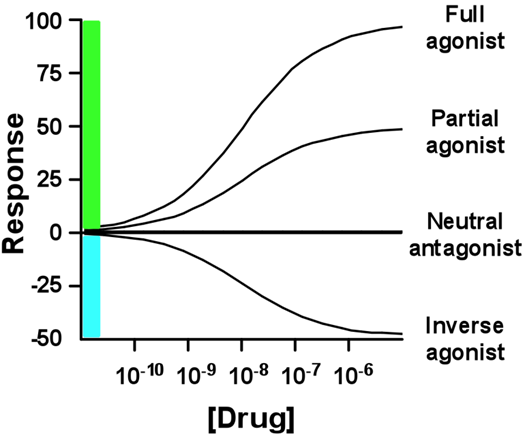
Agonists: compounds that have affinity for the receptor and are also capable of producing a biological response as a result of its interaction with the receptor.
Compounds that are attracted to a receptor macromolecule are said to have affinity for that receptor and may be classified as agonists or antagonists.
Agonists can be drugs or endogenous ligands for the receptor.
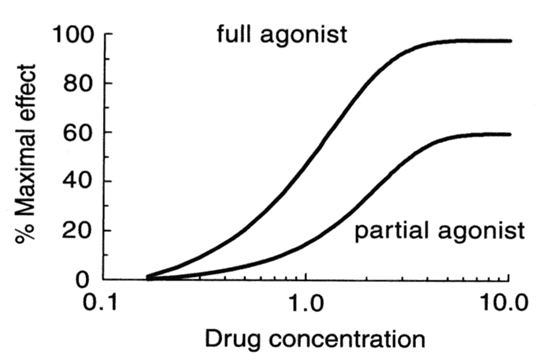
Full Agonist: Evokes 100% of the maximum possible effect.
Increasing concentrations of the agonist will produce an increase in the biological response
Drugs:
Isoprenaline, Treatment of Bradycardia (slow heart rate) b-adrenergic agonist [Adrenaline]
Morphine, potent opiate analgesic μ-opioid receptor agonist [endorphins]
Partial Agonist: Produces the same type of biological response, but cannot achieve 100% even at very high doses.
Drugs:
Busiprone [serotonin 5-HT1A receptor partial agonist, presynaptic dopamine agonist D2, D3 α1 receptor agonist]: generalized anxiety disorder (GAD)
Aripiprazole (Abilify) [Partial dopamine agonist]: anti-depressant, schizoprenia, bipolar disorder, major depressive disorder
Inverse Agonist: reduce baseline or trigger a negative response. Not the same as antagonists.
Drugs:
Benzodiazepine drugs, reduce anxiety GABA receptor agonists
Diazepam, (Valium) Alprazolam, (Xanax)
anxiogenic, memory enhancing effect benzodiazepine (GABA) receptor inverse agonist
β-carboline
A GnRH agonist is a synthetic peptide that acts like the gonadotropin-releasing hormone (GnRH) but has a much longer biological half life. As a result initially there is an increase in FSH and LH secretion. However after about ten days to one month, a profound hypogonadal effect is achieved through downregulation (desensitization). Generally this induced and reversible hypogonadism is the therapeutic goal and it leads to castrate levels of testosterone.
Buserelin, GnRH agonist for 2-3 weeks, then acts as inverse agonist: Treatment of hormone dependent cancers such as breast and prostate cancer
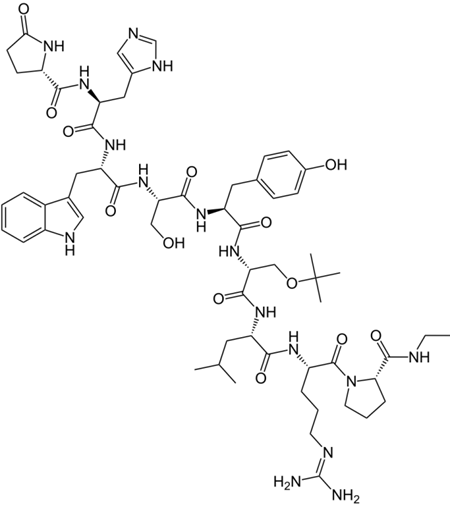
Super agonists: A superagonist is a type of agonist capable of producing a response greater than the endogenous agonist for the target receptor, and thus has an efficacy of more than 100%.
Goserelin, GnRH super agonist, then inverse agonist. During the treatment of prostate/breast cancers, initially this drug increases the symptoms as it is a super agonist. Thus, an anti-androgen such as bicalutamide is co-prescribed to relieve some of the symptoms. Later, once it starts acting as an inverse agonist, it leads to near castrate levels of testosterone and estrogen.

ANTAGONISTS
Antagonists: Drugs that are capable of interacting with the receptor but not activating it to produce a response. This class of drugs is said to have affinity but lacks intrinsic activity.
Block or reverse the effects of agonists. They have no effects on their own.
Competitive Antagonists: Compete with agonist for receptor binding. Agonist appears less potent, but can still achieve 100% effect (albeit at higher concentrations)
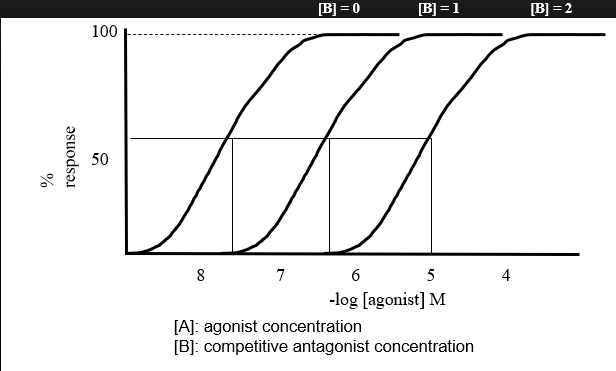
Reversible & can be overcome
Bind to the same site as the endogenous ligand or agonist.
Their presence produces a right-ward shift in both the binding and dose-response curves.
No change in Emax.
Similar dose-response curve shapes indicates the presence of a competitive agonist (competing for the same binding sites).
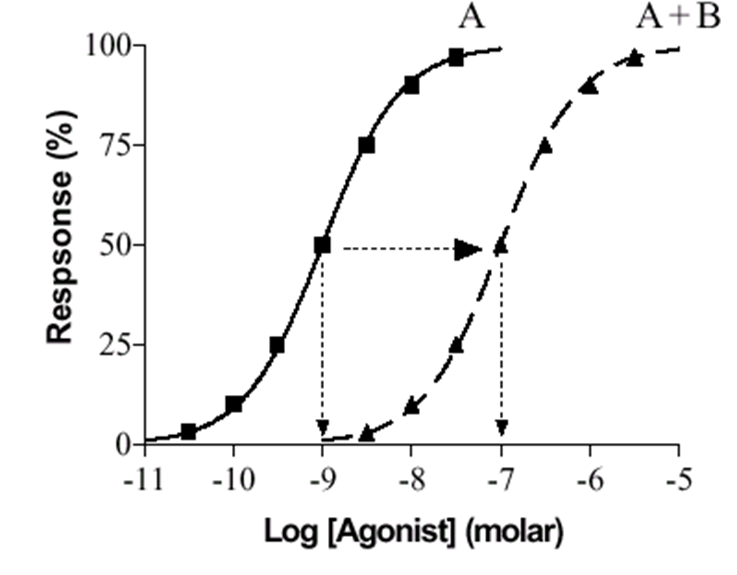
A = agonist alone
B = antagonist (one concentration)
A+B = agonist + antagonist
Drugs:
Naloxazone: irreversible m-opioid receptor antagonist
Naltrexone: m- and k-opioid receptor antagonist
Phenoxybenzamine: irreversible a-antagonist, treatment of hypertension
Fluoxetine (Prozac): reversible 5-HT2C antagonist, anti-depressant
Quetiapine (Seroquel): reversible D1, D2, D3, D4 antagonist [antipsychotic, Treatment of Schizoprenia]
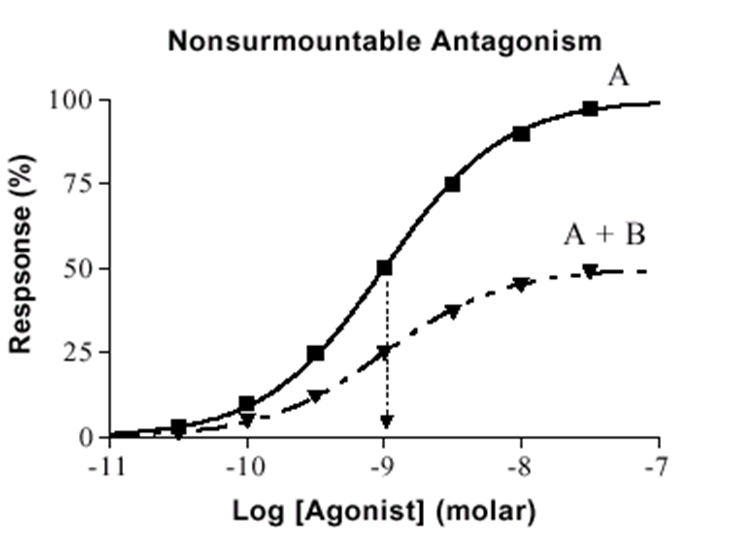
Non-competitive Antagonists: Bind to receptor at different site and either prevent agonist binding or the agonist effect maximal achievable response is reduced.
Does not prevent formation of the DR complex, but impairs the conformation change which triggers a response.
Bind to a site different than the agonist binding site at an allosteric site.
Cannot be overcome by adding more agonist
Emax is reduced but EC50 remains the same for the unaffected receptors.
Dose-response curves will have different shapes indicating different binding sites.
Drug:
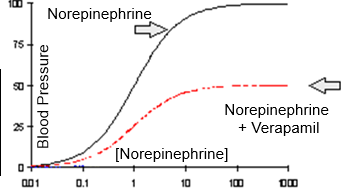
Cyclothiazide: Non-competitive antagonist of mGluR1 receptor [Anti-hypertensive and diuretic]
Verapamil: Verapamil decreases the maximal response of norepinephrine
Verapamil [antagonist]
Norepinephrine [Agonist]

AFFINITY AND EFFICACY
Affinity: The tendency of a drug to bind to a receptor, measured using equilibrium and inhibition constants [measure of propensity of a drug to bind receptor; the attractiveness of drug and receptor].
D + R <==> DR
Energy
DG = - RT ln K (K = Equilibrium Constant)
DG = RT ln Ki (Ki = Inhibition Constant = 1/K)
If Ki = 1 nM, DG = -53.4 kJ/mol v|s If Ki = 1 mM, DG = -35.6 kJ/mol
Roughly 5.9 kJ/mol change in DG causes a change of 10 in Ki.
DG = DH - TDS
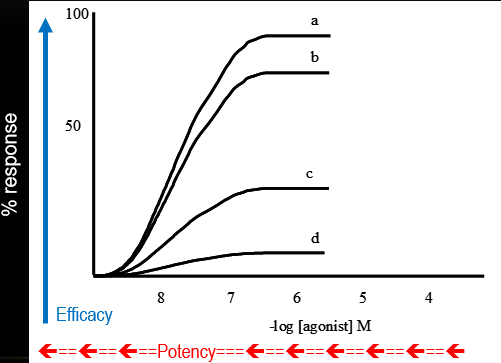
Efficacy: Ability of a drug to initiate a biological response upon binding (scale 0-1) [or Intrinsic Activity) – ability of a bound drug to change the receptor in a way that produces an effect or the ability of a drug molecule to produce a response at the molecular, cellular, tissue or system level]. Efficacy or Intrinsic activity (α): Efficacy/ Intrinsic activity: the ability of a drug molecule to produce a response.
DR <=> Effect
α = ED/Emax
ED = Effect (response) produced by a drug D.
Emax = maximal response
α = efficacy (α ~ 0-1)
For full agonist: α = 1
Partial agonist: α > 0, < 1
Antagonist: α = 0
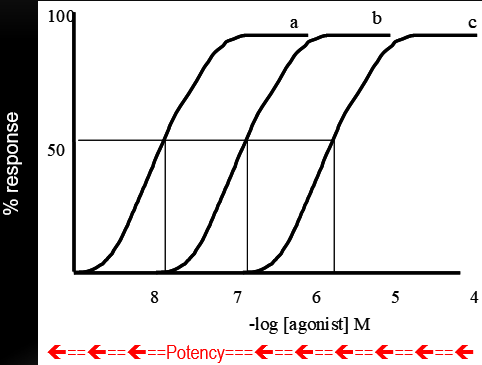
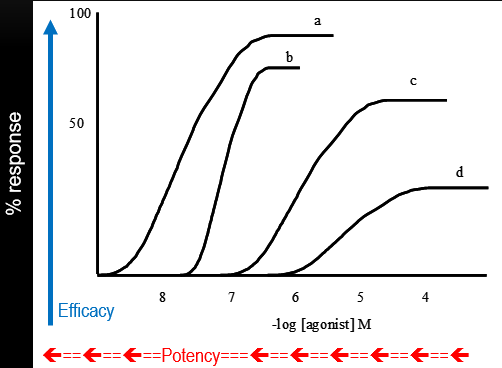
Potency: is how much drug is needed for a specific effect; lower EC50 indicates high potency. -a measure of drug activity expressed in terms of the amount required to produce an effect of given intensity
A highly potent drug = larger response at low concentrations. Ex: morphine, alprazolam, chlorpromazine
A low potent drug= smaller response at low concentration. Ex: ibuprofen, acetylsalicylic acid
It is proportional to affinity and efficacy.
The Emax is the maximum possible effect for the agonist. The concentration of drug at which E is 50% of Emax is termed the half maximal effective concentration and is abbreviated EC50 .
The term "potency" refers to this value. The lower the EC50, the lesser the concentration of a drug that is required to produce 50% of maximum effect.
lower EC50 = lower concentration to be effective (lower [D] for 50% effect)
DOSE-RESPONSE RELATIONSHIPS
Dose-effect phenomenon: Greater the number of agonist molecules present at the site of the receptors, the greater will be the biologic response, i.e., there is a direct relationship between the number of agonists and the biologic response. Linear scale plot yields a rectangular hyperbolic function. It is difficult to accurately extrapolate quantitative information from this type of mathematical function because of the constantly changing slope of the curve. Log scale plot yields a sigmoidal function, quantitative extrapolations are more accurate.
Direct correlation: More agonist molecules lead to a larger biological response.
Plots:
Linear Scale: Results in rectangular hyperbolic curves; difficult for quantitative extrapolation.
Log Scale: Results in sigmoidal curves; allows for improved quantitative analysis.
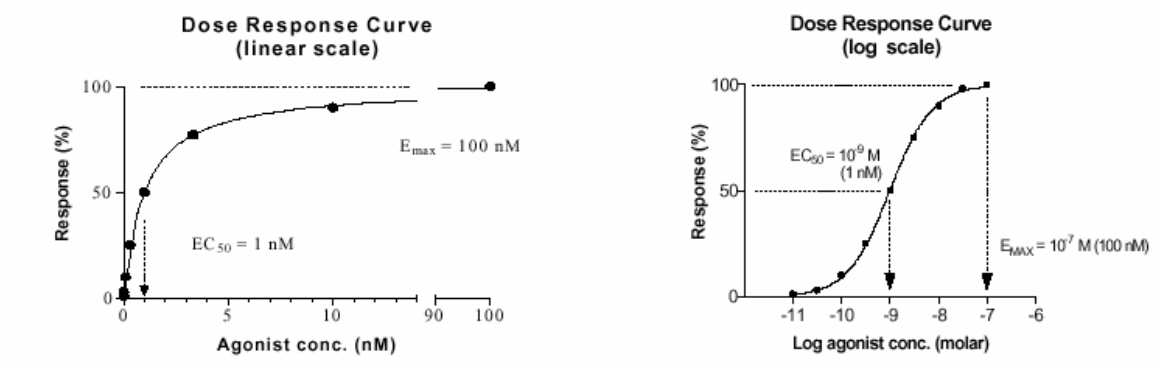
Dose Response Curve
Kd= 10^-7 M
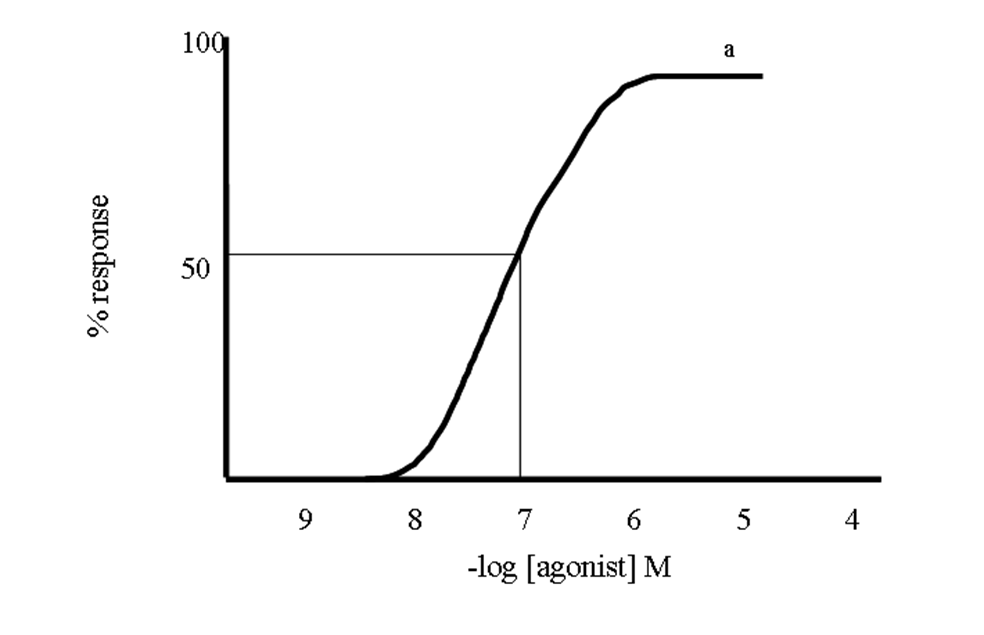
Dose Response Plot
A series of agonists with the same efficacy (α = 1), but different Kd values, indicating different potency
A series of agonists that differ in both potency and efficacy
A series of agonists that have the same affinity but different efficacy
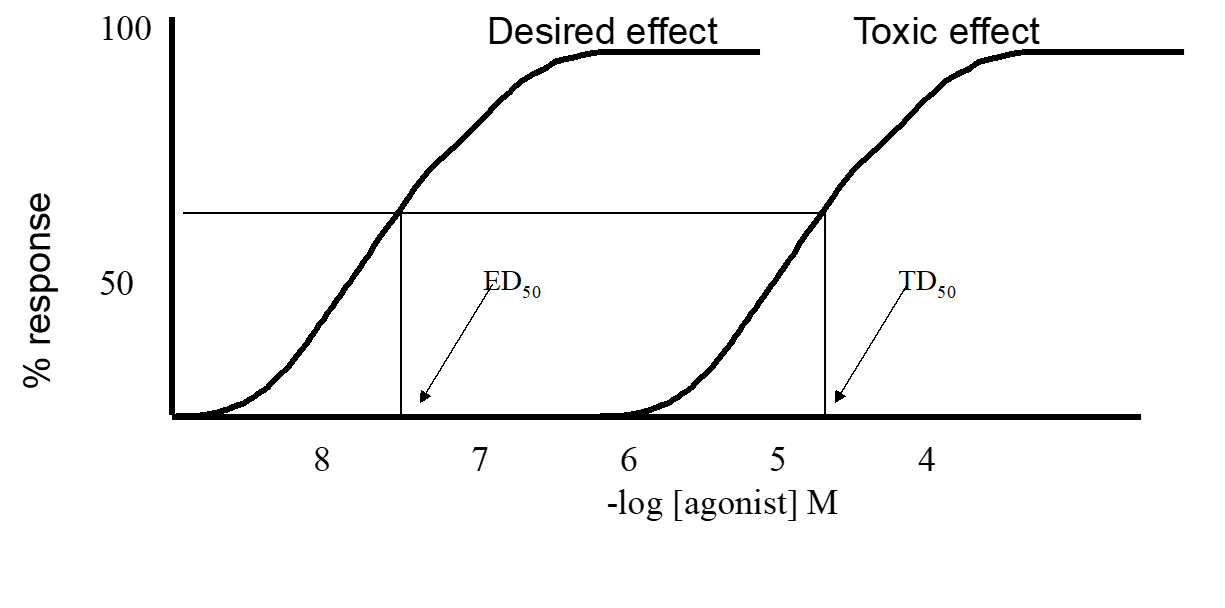
THERAPEUTIC INDEX
Therapeutic Index: the ratio of drugs undesirable effects with respect to its desirable effects [Ratio of the lethal dose to the therapeutic dose].
TI = TD50 / ED50 (in humans) & TI = LD50 / ED50 (in animal studies).
TD50 : The mean toxic dose of a drug required to cause toxicity in 50% of the test sample.
LD50 : The median lethal dose of a drug required to kill 50% of the test sample.
ED50 : The mean effective dose of a drug necessary to produce a therapeutic effect in 50% of the test sample.
The higher the therapeutic index, the greater the margin of safety of the drug.
Therapeutic index compares the drug dose levels which lead to toxic effects in 50% of cases studied, with respect to the dose levels leading to maximum therapeutic effects in 50% of cases studied.
The therapeutic index of a drug could be determined by plotting % of individuals responding vs. log [Drug].
Measure of the drug's safety; ratio of LD50 to ED50. A higher index indicates greater safety.
Calculated from the proportion of toxic and effective doses in test populations and can be plotted for visual analysis.