Tumor Suppressor Gene
Oncogenes, like those of the original transforming retroviruses (e.g. RSV and v-src), seem to exhibit a dominant effect. (Normal cellular growth would therefore seem to be recessive.)
But is that also true of most human cancer-causing genes Yes, if a normal cell is chemically fused to a cancer cell that had been generated with a tumor virus. No, if the cancer cell was derived from most types of human tumors
Cancers are the result of multiple genetic lesions, and not all of those lesions are in oncogenes.
Some may be the result of genes that suppress the cancer phenotype, but which have somehow been rendered inactive (e.g., PTEN) These tumor suppressor genes normally function to keep cell proliferation in check, but if they are mutated or lost, cells proliferate uncontrollably.
Since cancers depend on a combination of genetic lesions, the cell fusion experiment ends up restoring many of the lost tumor suppressor genes because they are contributed by the normal cell.
Cancers as a whole then actually seem to be recessive
Retinoblastoma is an uncommon, sporadic cancer of the eye that manifests itself in early life in 1 out of every 20,000 children. It generally presents in one eye and is treatable
familial cases of retinoblastoma (where a parent also has the disease) are much more aggressive, appear in both eyes, and are associated with a 500-fold greater risk of developing other types of cancers (particularly osteosarcomas).
An epidemiological study showed that familial cases of retinoblastoma (bilateral-in both eyes) were consistent with a single genetic event. However, sporadic cases (unilateral-in one eye) were consistent with two genetic events. This is what the geneticist Alfred Knudson called ‘the 2-hit hypothesis’.
Consider there is a gene, Rb, that is linked to these cancers. Sporadic cases mean that there are no inherited mutations in Rb, and mutations must be acquired in both alleles for the cancer to develop. As this would be rare, only one eye would be affected (unilateral). Familial cases, however, mean that one Rb mutation has already been inherited, only one mutation must be acquired, and hence the more likely it is that both eyes might harbor tumors (bilateral).
It is true that the likelihood of mutations hitting both copies of the same gene in a single cell seems highly unlikely. However, it is quite possible (albeit rare) that one mutation occurs on one chromosome followed by the ‘transfer’ of the mutation to the other chromosome by homologous recombination during mitosis.
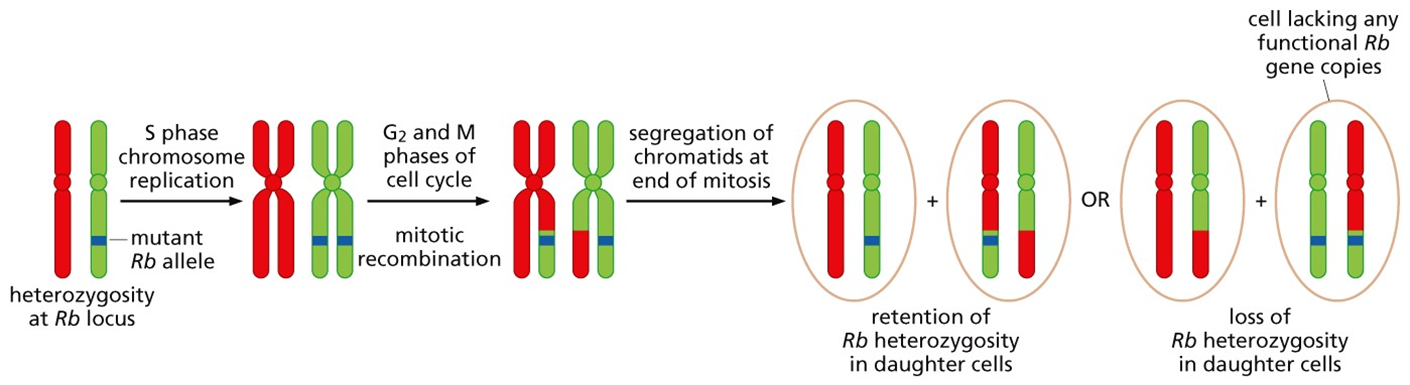
Other possibilities include hemizygosity, where a portion of a chromosome simply gets discarded and permanently lost, or nondisjunction (below), where chromosomes fail to separate correctly during mitosis.
All of these processes ultimately result in what is called loss of heterozygosity (LOH), the creation of two genetic mutations where once there was only one.
DNA is wrapped around proteins called histones.
These positively charged histones can make negatively charged DNA inaccessible to transcription factors.
Acetylation neutralizes the positive charge on histones, opens chromatin structure, and thereby enables transcription factors to bind DNA in order to increase gene expression. Histone deacetylation has the opposite effect.
DNA methylation is the addition of a methyl group to the cytosine of CpG nucleotide pairs. It is catalyzed by the enzyme DNA methyl transferase (e.g., DNMT3B, and others). CpG methylation often precedes the deacetylation of nearby histone proteins by histone deacetylases. This tightens the ‘grip’ that histones have on DNA. If this occurs in the promoter regions, these epigenetic modifications will diminish access by the transcription factors to the DNA and decrease overall gene expression. If this occurs in tumor suppressor genes, then it could lead to carcinogenesis.
Seventy percent of all genes have CpG islands in their promoters (~14,000 genes). Cancer genomes tend to have hyper-methylated CpG islands.
the LOH in tumor suppressor genes may be related to epigenetic factors (methylation, deacetylation) as much as genetic ones (homologous recombinations, deletions).
Some tumor suppressor genes fall into one of two classes: ‘gatekeepers’ and ‘caretakers’.
Gatekeepers control the fate of cells by determining whether they move through the cell cycle or not.
Caretakers control the fate of genomes by repairing the errors that compromise their integrity.
RB is a ‘gatekeeping’ tumor suppressor gene that keeps the cell cycle in check until conditions are favorable for growth.
TP53 is a ‘gatekeeping’ tumor suppressor gene that keeps the cell cycle in check until the genome can be freed of the mutations that would be catastrophic to pass along to the next generation of cells.
The cell cycle, the process by which new cells are made from old cells, involves (1) the duplication of a cell’s genetic material (synthesis), (2) their segregation to opposing poles (mitosis), and (3) the final division of one cell into daughter cells (cytokinesis).
The cell cycle is divided into 4 phases: G1, S, G2, M. Duplication of the genetic material (DNA replication) occurs during the S phase.
Segregation of chromosomes and cytokinesis occurs during M phase
G1 and G2 represent gaps, a time for cell growth and quality control decisions about cell cycle progression
G0 is a reversible exit point from the cell cycle when conditions are inappropriate for growth and cells quiesce.
In order to complete the cell cycle, the cell must pass through checkpoints where decisions are made about everything from environmental growth conditions to the integrity of a cell’s DNA. These checkpoints are some of the most regulated aspects of all cell biology.
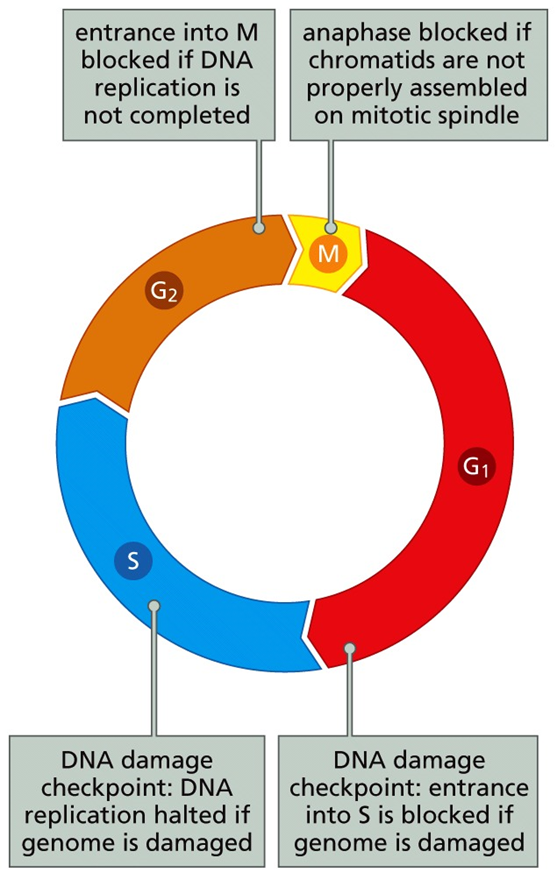
In G1 there is a key cell fate checkpoint where the cell inventory its environment for mitogen (e.g., FGF) and growth suppressor (e.g., TGF-β) concentrations.
How a cell ‘reads’ its environment is indelibly linked to the genes it is expressing. A cell can not ‘sense’ a mitogen if it lacks the receptor for it.
The same is true for growth suppressors. This decision process hits its pinnacle at the restriction (R) point of G1.
If passed, then the cell cycle will be completed even if growth conditions become unfavorable afterward.
As the rest of the cycle is largely pre-programmed, the R point is the most regulated cell cycle event.
The cell cycle control system regulates the cell cycle through the cyclical activation of signaling molecules. The major signaling molecules are cyclins and cyclin-dependent protein kinases (Cdks).
Cyclins bind to and activate Cdks, thus enabling Cdks to phosphorylate and activate other molecules whose functions enable progression through cell cycle checkpoints and during other aspects of the cell cycle.
Cdk levels do not generally change. Cyclin levels, however, rise and fall in accordance to the different phases of the cell cycle. Thus, Cdk activity cycles as well, depending on whether a cyclin is bound to it.
Some cyclins bind to more than one Cdk.
Some Cdks bind to more than one cyclin.
There are unique structural conformations when Cdk2 binds to cyclin E during late G1 phase and to cyclin A during S phase. This enables the same enzyme to phosphorylate different substrates and regulate different processes during different steps of the cell cycle.
Cyclins have short-half lives via ubiquitylation and proteasome degradation. Cyclin-Cdk activity inhibits expression of the previous cyclin in the cycle. Cyclin-Cdk activity promotes expression of the next cyclin in the cycle.
Cyclin D1 levels are uniquely regulated relative to the other cyclins. Many growth factor signaling pathways converge on cyclin D1 regulation in order to communicate a cell’s current environmental conditions
Note that cyclins D1, D2 and D3 have unique promoters and are regulated by different signals
D cyclins, however, are unstable (half-life of 30min). A reduction in growth factors will lead to their rapid decline. Fluctuations in cyclin D levels reflect current growth conditions that inform cell fate decisions during G1
Cyclin D-Cdk4/6 drives cell cycle progression through G1 and past R, if growth conditions are good. Then a cell-autonomous program carries the cycle through to completion, independent of growth signals.
TGFβ, an inhibitor of G1, and DNA damage block cell cycle progression via control of CKIs.
Mitogens act in an opposing fashion by inhibiting CKIs. For example, downstream of RTKs, phosphorylation of p21Cip1 and p27Kip1 by Akt leads to the export of CKIs to the cytoplasm so that nuclear cyclin-Cdk complexes are no longer inhibited and the cell cycle continues.
Note that Akt signaling status and p27Kip1 localization are prognostic in cancer.
Low-grade breast cancers have little activated Akt, nuclear-localized p27Kip1, inhibition of the cell cycle, and a fairly good prognosis over 6 years. High-grade breast cancers have activated Akt, cytoplasmic p27Kip1, no inhibition of the cell cycle, and a much poorer prognosis.
Cyclin D-Cdk4/6 is also a ligand of p21Cip1 and p27Kip1. But this binding does not inhibit its kinase activity. Rather, it serves to absorb the CKIs from Cyclin E-Cdk2 so that the cell cycle can move past the R point.
p21Cip1 and p27Kip1 are present in early G1. If mitogens are also present, cyclin D-Cdk4/6 will accumulate. This pulls the CKIs from the few cyclin E-Cdk2 complexes present. This causes a rapid accumulation of cyclin E-Cdk2 that promotes the phosphorylation events that help push the cell cycle past the R point and into S phase.
So what about pRb? pRb is a 105kDa nuclear protein whose phosphorylation status is the governor of the R point.
Hypophosphorylation is by cyclin D-Cdk4/6. pRb is active and restricts movement through the R point.
Hyperphosphorylation is by cyclin E-Cdk2. pRb is now inactive. The cell bypasses the R point of the cell cycle.
Dephosphorylation is by protein phosphatase 1 (PP1). pRb is inactive and reset for activation again.
In response to mitogens, cyclin D accumulates, binds to Cdk4/6, and creates active cyclin D-Cdk4/6 complexes that hypophosphorylated pRb in early G1. Active cyclin D-Cdk4/6 complexes also siphon CKIs from the few cyclin E-Cdk2 complexes that exist. This creates active cyclin E-Cdk2 complexes that initiate the hyperphosphorylation of pRb in late G1
The role of pRb as a cell cycle gatekeeper and tumor suppressor was elucidated through research on DNA tumor viruses.
Adenovirus (E1A), SV40 (large T), and HPV (E7) use unrelated oncogenes, but all of them bind to and sequester hypophosphorylated pRb so that it no longer restricts the cell cycle at the R point.
How does the phosphorylation status of pRb govern transition through the R point of the cell cycle? pRb is a transcriptional regulatory protein. And it acts cooperatively with E2F family transcription factors to control gene expression. Moreover, it is the phosphorylation status of pRb that dictates whether it can associate with E2F.
E2Fs (1-8) normally function along side DP1/2 to activate or repress gene expression.
The activation of gene expression by E2Fs 1/2/3 (with DP1/2) occurs through the recruitment of a histone acetylase. [Recall that histone acetylation reorganizes the chromatin into a configuration conducive for transcription.]
In mid-G1, hypophosphorylated pRb binds to E2Fs 1/2/3-DP1/2 (E2F-DP), displacing the histone acetylase, and recruiting a histone deacetylase instead. This leads to repressed gene expression and a temporary arrest in the cell cycle. By the R point, if mitogens are plentiful and pRb gets hyperphosphorylated, pRb will dissociate from E2F-DP, thus re-establishing the recruitment of histone acetylases and restoration of gene expression for S phase initiation.
Among the many targets of E2F-DP activation is the gene for cyclin E. As cyclin E accumulates, cyclin E-Cdk2 activity increases. Cyclin E-Cdk2 directly promotes the hyper-phosphorylation of pRb. This dissociates pRb from and activates more E2F- DP, thus enabling more cyclin E production
Another target of cyclin E-Cdk2 is p27Kip1. Phosphorylation of p27Kip1 (like that by Akt) translocates it to the cytosol where it is a target for ubiquitylation and degradation at proteasomes. As p27Kip1 is a negative regulator of cyclin E-Cdk2, its destruction acts as another positive feedback or feed-forward loop for further cyclin E-Cdk2 activity.
During the S phase, cyclin A displaces cyclin E on Cdk2. Among the targets of cyclin. A-Cdk2 are E2F-DP. Phosphorylation of E2Fs dissociates the complex, relinquishes its transcriptional activity, and targets E2F for ubiquitylation and degradation at proteasomes.
like cyclin D1, many growth factor signaling pathways converge on Myc regulation. Myc promotes cell proliferation. Indeed, 70% of human cancers overexpress myc (myc, N-myc, L-myc) (i.e., myc is an oncogene). Myc is a basic helix-loop-helix (bHLH) transcription factor that forms heterodimers (e.g., Myc/Max), binds to E-box enhancers on DNA, and induces genes necessary for proliferation.
As a growth suppressor, TGF-β downregulates Myc. TGF-β signaling involves phosphorylation of Smad3, which leads to coupling with the repressive E2Fs 4/5 transcription factors, along with p107, a cousin of pRb. These factors collectively down-regulate transcription of myc, and thereby remove a key regulator of cell cycle progression and proliferation.
Cell fusion experiments explain the ‘power’ of tumor suppressor genes in cell fate determination.
For nearly all human-derived cancer cells, the fusion to a normal cell is a normal phenotype.
Fusion to a normal cell restores lost tumor suppressor genes and, thus, restores normal cell growth.
Normal cell growth is dominant to cancerous growth. For cancers to develop, tumor suppressor genes must be hit twice. Mutation combined with a loss of heterozygosity (LOH) event creates this necessary loss in tumor suppressor gene presence (or expression).
pRb is a gatekeeper, surveying a cell’s environment to ensure adequate nutrients are available for cell proliferation. pRb also recognizes when conditions are unfavorable for proliferation and arrests the cell cycle until they improve
Cells cycle/proliferate when growth factors are present, such that cyclin D accumulates so that it can bind to Cdk4/6. The accumulation of active cycle D-Cdk4/6 complexes can hypo-phosphorylate the gatekeeping tumor suppressor gene retinoblastoma (pRb) leading up to the ‘R point
Hypo-phosphorylation by cyclin D-Cdk4/6 activates pRb and restricts movement through the R point.
Hyper-phosphorylation by cyclin E-Cdk2 inactivates pRb and enables passage through the R point.
Cells cycle past the R point when active cyclin D-Cdk4/6 complexes accumulate and start siphoning off the CKIs p21Cip1 and p27Kip1 from cycle E-Cdk2. This frees up active cycle E-Cdk2 complexes to hyper-phosphorylate pRb.
The phosphorylation status of pRb dictates its association with E2F transcription factors that control, among other things, DNA synthesis (S phase).
When pRb is hypo-phosphorylated, it associates with E2Fs, binds to promoters in conjunction with a histone deacetylase, and represses transcriptional activity at the R point.
When pRb is hyper-phosphorylated, it dissociates from E2Fs, histone acetylase takes its place along promoters, and E2F transcriptional activity is activated to go past the R point.
p53 is also a gatekeeper. It surveys a cell’s genome for damage and then calls in a repair team so that the next generation of cells will function normally. p53 also recognizes when the damage is too great, and, if so, initiates the cellular execution program called apoptosis
TP53 is a tumor suppressor gene. This can be shown experimentally by its ability to restore normal growth in Ras-transformed fibroblasts that previously lacked p53. The normal growth is seen as a loss of foci.
p53 was discovered based on its ability to be targeted/inhibited by tumor virus oncoproteins like E1B (adenovirus), large T antigen (SV40), and E6 (HPV). [Note, tumor virus oncoproteins like E1A (adenovirus), large T antigen (SV40), and E7 (HPV) target/inhibit pRb.]
Deletion of p53 in mice is not embryonically lethal, but there is lower long-term survival in adulthood because of lymphomas and sarcomas. Mutations in TP53 are highly prevalent in human cancers.
The ectopic addition of a p53 mutant construct accentuates the transforming properties of ras mutations even though wild-type (normal) p53 is still present in the fibroblasts.
If not completely deleted, most mutations (splice site, nonsense, frameshift) in tumor suppressor genes lead to gross changes in protein structure. Proteins that cannot be folded correctly often get degraded, and thus the net effect is the same as a deletion.
Unlike APC (and nearly all other tumor suppressors), most of the mutations in TP53 are missense, small point mutations that at most inactivate p53 function. This means that oddly as a tumor suppressor, the impact of TP53 mutations are usually dominant like an oncogene, not recessive like a tumor suppressor
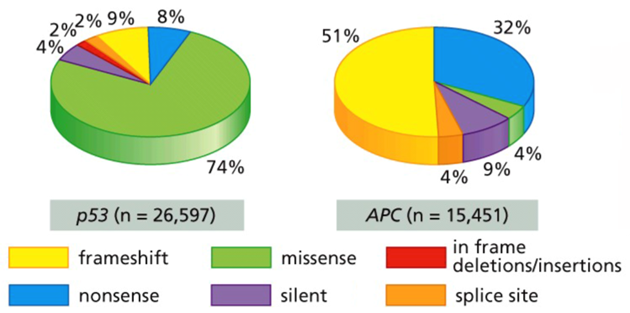
The effect of TP53 mutations in cancer can be explained by the biochemical properties of a functional p53 protein, a transcription factor that exists as a homotetramer.
If there is a deletion (or a mutation that leads to p53 degradation) in a single allele, then the function of the p53 homotetramer will be nearly normal because the other allele is still present and wild-type. If, however, there is a single missense mutation affecting a single allele, then the integrity of the p53 homotetramer will usually be compromised. These mutations have a dominant-negative effect on p53
In most cancers, one allele harbors a missense mutation while the other allele undergoes a LOH event.
Stabilization of wild-type p53 arrests the cell cycle so that DNA damage can be detected and repaired before normal cell functions are restored.
Under severe stress, when DNA damage may be irrecoverable, stabilization of p53 may induce apoptosis.
Irradiating normal thymocytes leads to cell death because of the p53 response.
Cancer cells lacking functional p53 do not suffer this fate. It’s as if the cells are ‘blind’ to the damage present. They also pass the damage on to the next generation of cancer cells.
p53 is mostly regulated at the level of protein stability.
p53 is expressed abundantly, but it turns over quickly under normal growth conditions. Thus, the overall protein levels of p53 are low in cells.
The steady-state degradation of p53 occurs when bound to the protein Mdm2 (mouse double minutes).
Mdm2 supports the ubiquitination and proteasome-mediated degradation of p53. In this way Mdm2 functions as an oncogene.
The transcriptional activation of HDM2 (which encodes Mdm2) is done by p53. This is an example of a negative feedback mechanism through which p53 activity promotes its own destruction.
Loss of DNA integrity by irradiation (double-stranded DNA breaks, DSB) or DNA synthesis inhibitors (single-stranded DNA, ssDNA) induce phosphorylation events that protect p53 against Mdm2-binding, and thus p53 ubiquitination/proteasomal degradation.
Mdm2 is also regulated by phosphorylation.
Chk1 and Chk2 introduce inactivating phosphorylations on Mdm2.
Conversely, activating phosphorylations on Mdm2 are introduced by Akt.
Chk1/2 operates in response to DNA damage; so inactivating Mdm2 or blocking its binding to p53 would stabilize p53 so that the damage can be repaired.
Akt, on the other hand, is a survival factor, an oncogene. It is looking to activate Mdm2 in order to target p53 for degradation so that the cells do not go through apoptosis.
the p53 tumor suppressor often accumulates in cancerous tissue.
The explanation is that most of that p53 is mutated and therefore inactive as a transcription factor.
Thus, Mdm2 no longer accumulates, and p53 cannot be degraded by Mdm2-,dependent p53 ubiquitination.
If that is not enough, Mdm2, like p53, has its own inactivator. Its name is ARF. ARF (aka murine p19ARF and human p14ARF) is an alternative reading frame for the p16INK4A gene locus (called CDKN2A).
Like p16INK4A, ectopic expression of ARF in cells strongly inhibits proliferation, but it only works if p53 is present. ARF functions by sequestering Mdm2 away from p53. This enables p53 accumulation, and cell cycle arrest. ARF is thus p53’s friend, because it is Mdm2’s enemy.
E1A -->ARF: E1A binds to pRb, pRb cannot bind to E2F, E2F drives transcription of ARF
c-Myc --> ARF: c-Myc drives transcription of E2F and cyclin D, cyclin D associates with Cdk4/6, cyclin D-Cdk4/6 promotes the inactivating hyper-phosphorylation of pRb via cyclin E-Cdk2, pRb cannot bind to E2F, E2F drives transcription of ARF
Ras --> ARF: Ras-GTP effects Raf, Raf activates the MAPK pathway, Erk1/2 activates Ets, Ets drives the transcription of cyclin D, cyclin D associates with Cdk4/6, cyclin D-Cdk4/6 promotes the inactivating hyper-phosphorylation of pRb via cyclin E-Cdk2, pRb cannot bind to E2F, E2F drives transcription of ARF
Oncogenes like E1A (adenovirus), myc, and ras increase ARF levels by acting through the E2F transcription factors.
This leads to a sequestering of Mdm2, the upregulation of p53 (because it is no longer degraded), and either cell cycle arrest or apoptosis.
This might seem counterintuitive. But apoptosis is in fact a common response to oncogenic signaling, as it represents a final defense against the abnormal, continuous growth of cancer cells.
If a single ARF allele is inactivated, a LOH event will often inactivate the other. Under these circumstances, the presence of an oncogene (e.g., ras) will not be counterbalanced by apoptosis, tumor cells will grow, and the survival of the mice dramatically declines.
The power of ARF in regulating apoptosis makes it as useful a target for inactivation during cancer development as the inactivation of p53.
In one study, cell stress brought on by treatment with the transcription inhibitor actinomycin led to p53 binding to 1546 of these sites in the human genome.
Bax: increase in pro-apopototic genes
Bcl2: decrease in anti-apoptotic genes
Physical characteristics of the apoptotic program: Blebbing, pyknosis, DNA fragmentation, phagocytosis of apoptotic bodies
A key step in apoptosis is the movement of cytochrome c from the inter-membrane space of the mitochondria, through the VDAC1 channel on the outer membrane, and into the cytosol. Bcl-2 is an anti-apoptotic protein and typically blocks the flow of cytochrome c through this channel.
Bcl-2 is a pro-survival protein and typically blocks the flow of cytochrome c through the VDAC1 channel. Counter-balancing the Bcl-2 family of pro-survival proteins are up to 18 different pro-apoptotic proteins. Some, like Bad, Bax, Bak, and Bid work to keep the VDAC1 channel open. [Bax is upregulated by p53.]
The balance between survival and apoptosis is linked to the concentration, activity, and localization of pro-survival and pro-apoptotic proteins in the cell.
There are many pro-survival and pro-apoptotic proteins because there are many signaling pathways regulating the choice between survival and death.
Once in the cytosol, cytochrome c binds to the protein Apaf-1 to form the apoptosome. The apoptosome converts the initiator caspase 9 from its pro-form to its catalytically active form.
Caspase (cysteine-aspartyl specific protease) 9 is one of 12 family members. Caspase 9 cleaves and activates procaspase 3. Then, caspase 3 cleaves and activates other ‘executioner’ caspases in the caspase cascade. The caspases then cleave ‘death substrates’ (e.g., DNA, cytoskeleton) that number in the 1000’s.
There is also an extrinsic apoptotic program where death signals bind receptors on the cell surface that activate executioner caspases of the intrinsic program
The ligands are members of the tumor necrosis factor family (TNFα, TRAIL, FasL), and each have their own receptor.
The death domain within each receptor (TRADD) binds to an intracellular protein called FADD (Fas-associated death domain) and leads to the assembly of DISC, the death-inducing signaling complex.
This activates the cleavage and activation of procaspases 8 and 10, which converge on the intrinsic caspase cascade.
p53 activates apoptotic programs on multiple levels.

Cancer cells face many challenges (anoxia, DNA damage, activated Ras or Myc (oncogenes), deregulated pRb/E2Fs) during the tumor progression process that threatens their own apoptotic demise. All cancer cells must overcome this apoptosis barrier—it is after all a cancer hallmark. This is why most cancers find a way to inactivate p53.
The inactivation of p53 also leads to a mutability phenotype. If DNA damage isn’t recognized or repaired, then it will be easier for other mutations to be acquired. This is another cancer hallmark. Finally, the inactivation of p53 also leads to a loss in thrombospondin-1 expression, and increased tumor angiogenesis, still another cancer hallmark.
If cancer cells do not inactivate p53, then they can also increase Mdm2 levels or decrease ARF (deletion, promoter methylation) with similar effect.
APAF1 promoter methylation Bax inactivating mutation Bcl-2 overexpression PI3K/Akt pathway activation NFκB pathway activation Caspase 8 deletion TNFR promoter methylation